Immunotherapeutic Efficacy of Retargeted oHSVs Designed for Propagation in an Ad Hoc Cell Line

 , ,
, ,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Cancer-Selective Oncolytic Herpes Simplex Viruses and Synergism with Check Point Blockade
2. Strategies towards Cancer-Specific and Efficacious oHSVs
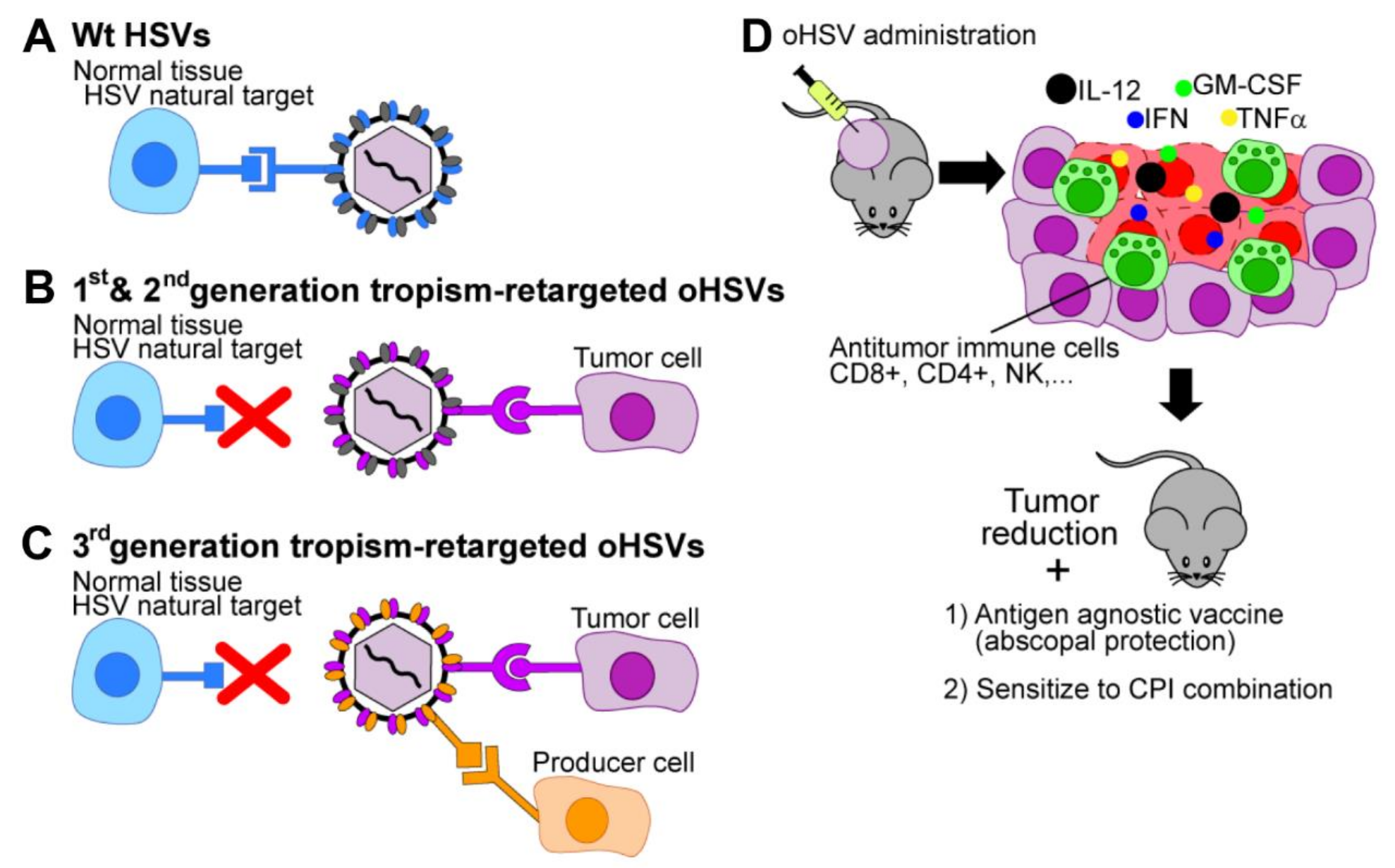
2.1. Tropism Retargeting
2.2. Transcriptional and Post-Transcriptional Retargeting Strategies
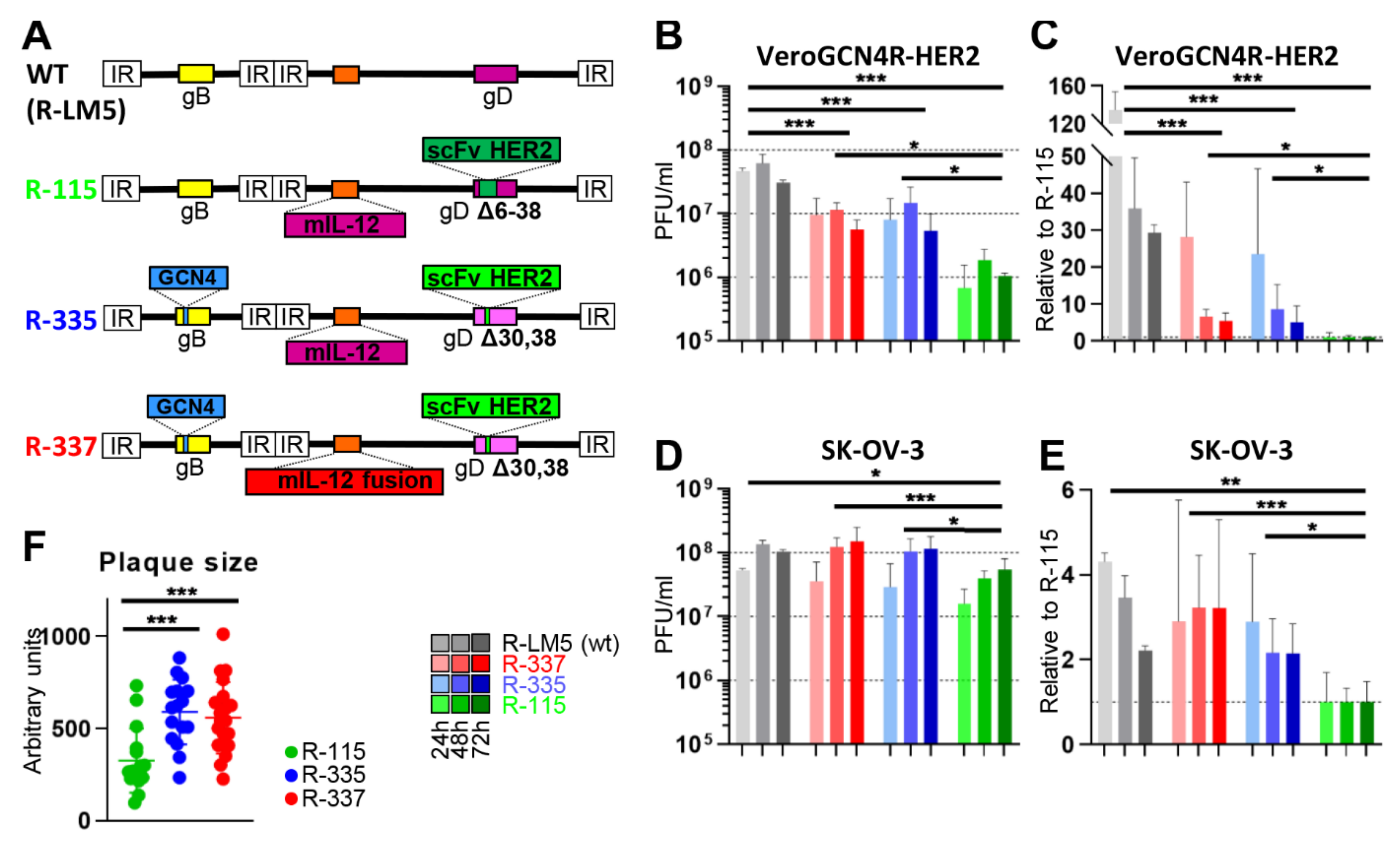
3. Properties of the Tropism-Retargeted oHSVs Generated in Our Laboratory
3.1. Three Generations of Tropism Retargeted oHSVs
3.2. The Retargeted oHSVs are a Platform
3.3. Cultivation of Tropism-Retargeted oHSVs in Non-Cancerous Producer Cells
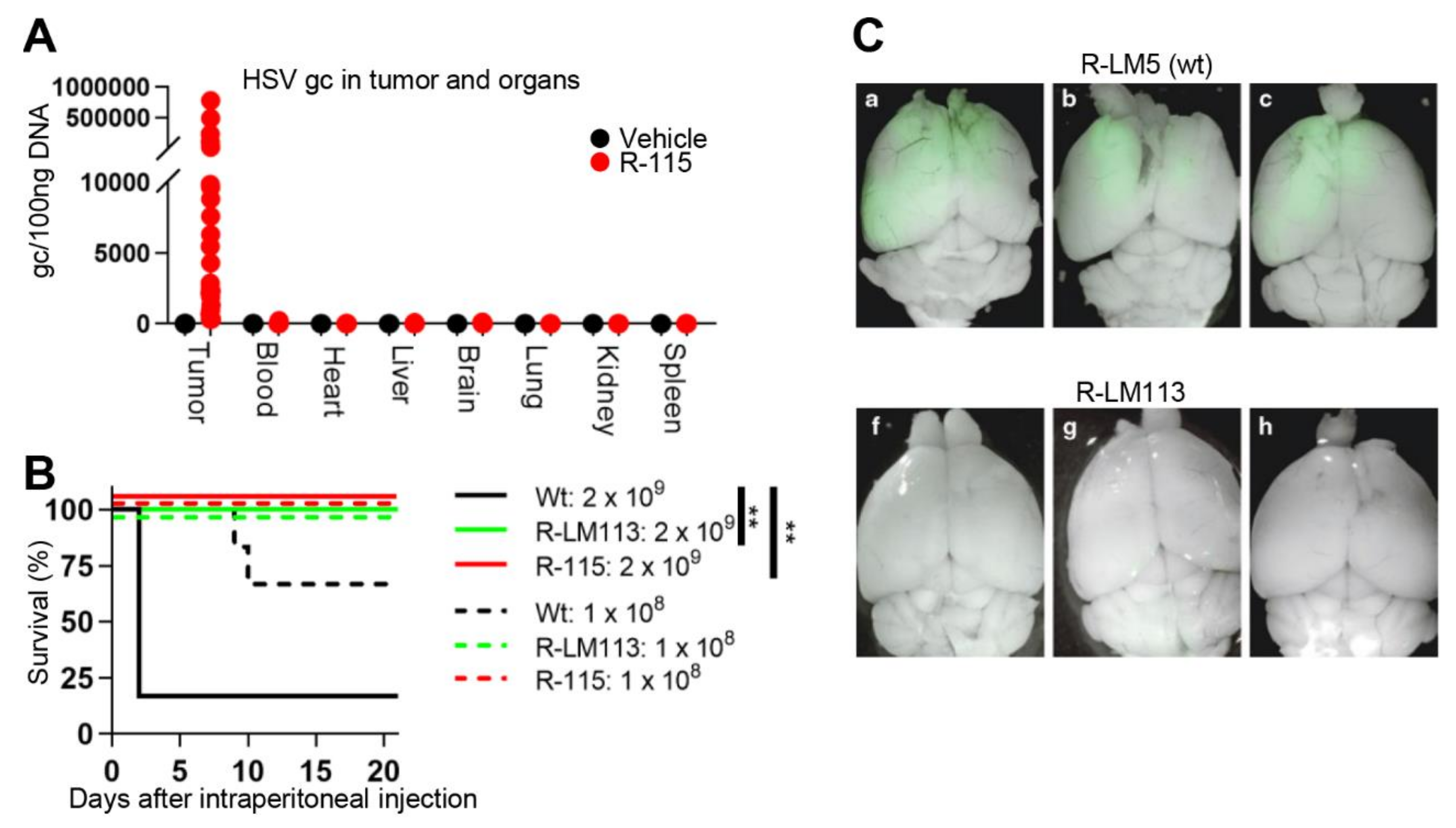
3.4. Safety Profile of Retargeted oHSVs
4. In Vivo Efficacy of Retargeted oHSVs in Immunocompetent Mouse Models
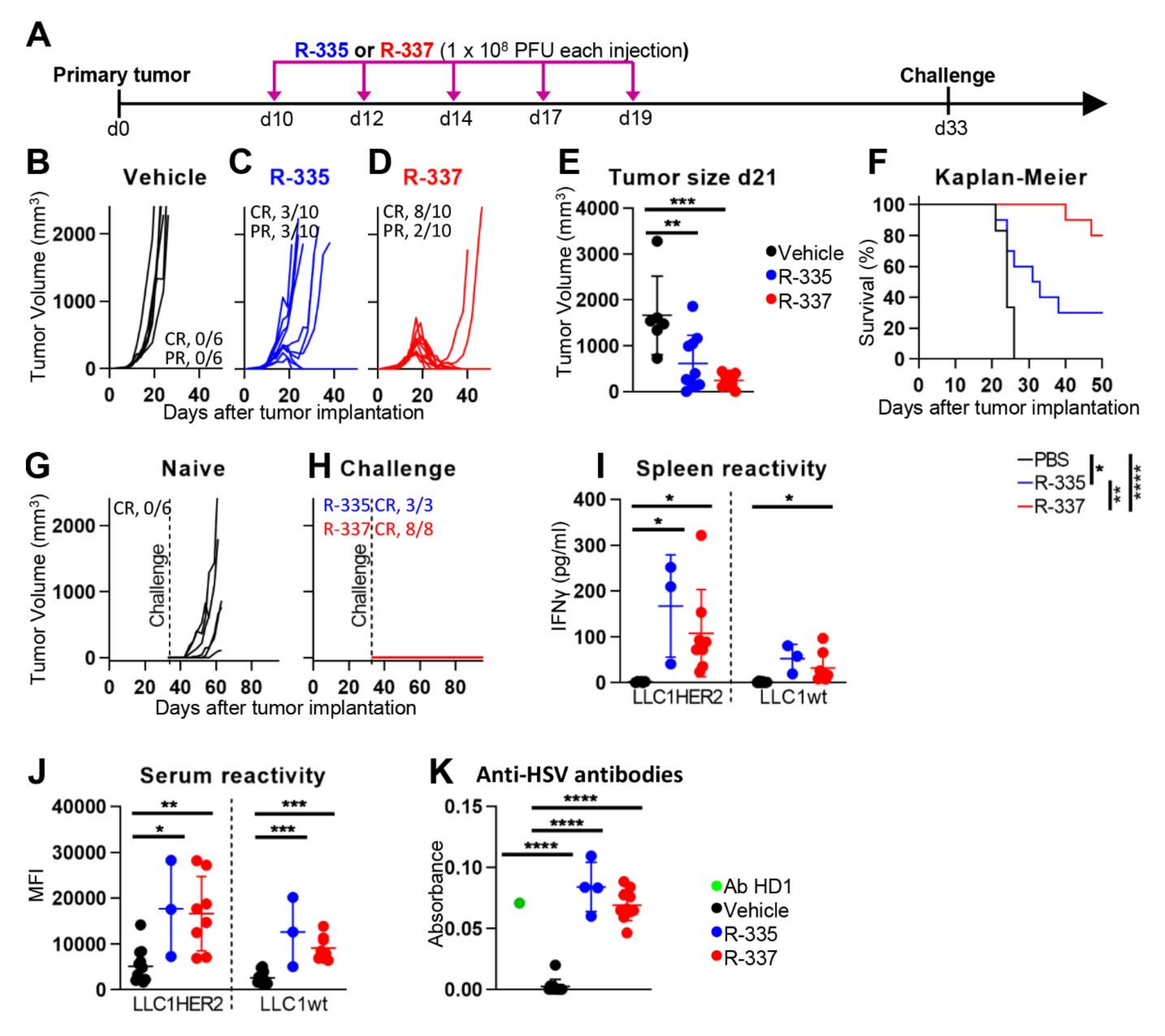
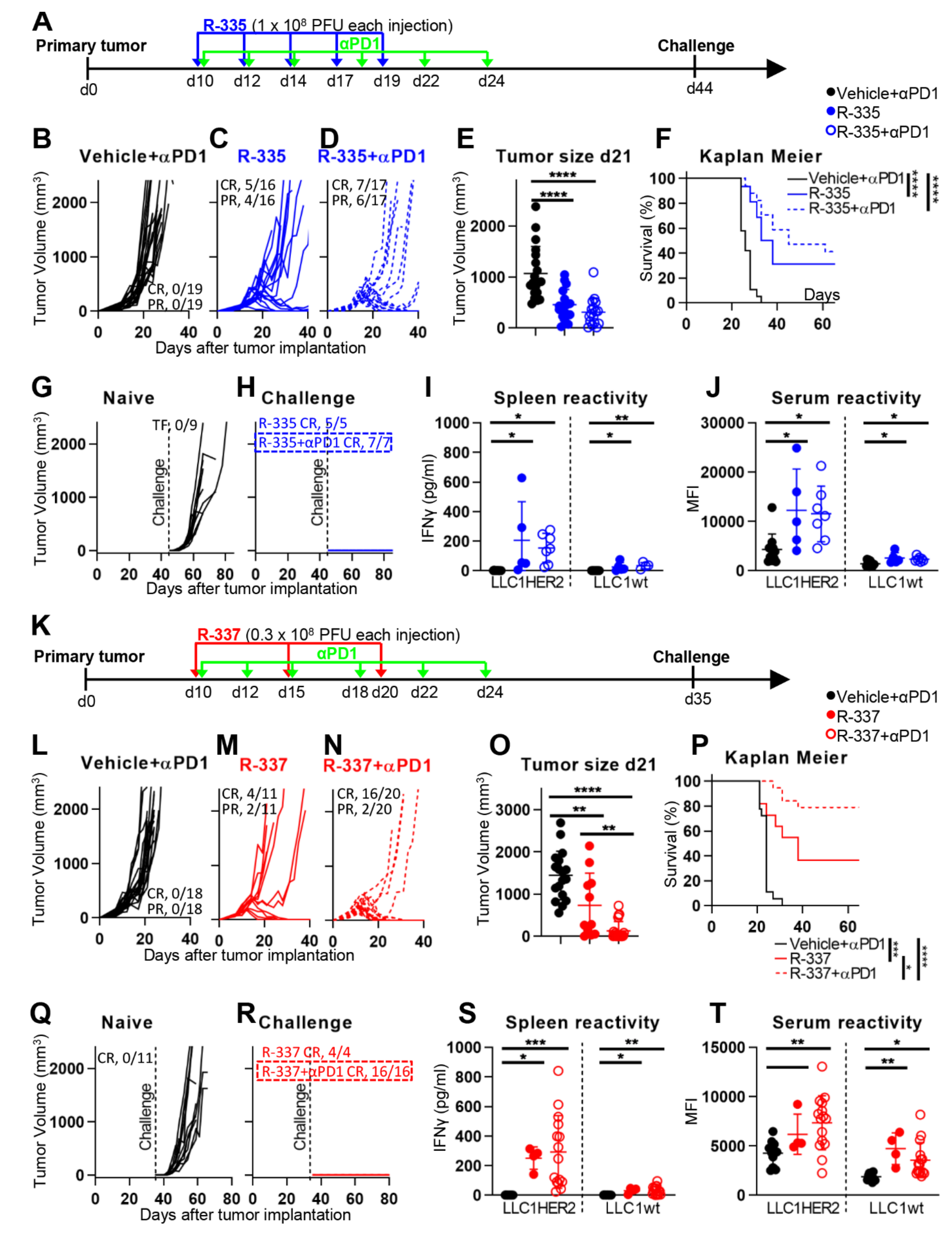
4.1. Efficacy against LLC-1-HER2 Primary Tumors
4.2. Retargeted oHSVs Promote Antigen Agnostic Vaccination Effect against Distant Tumors
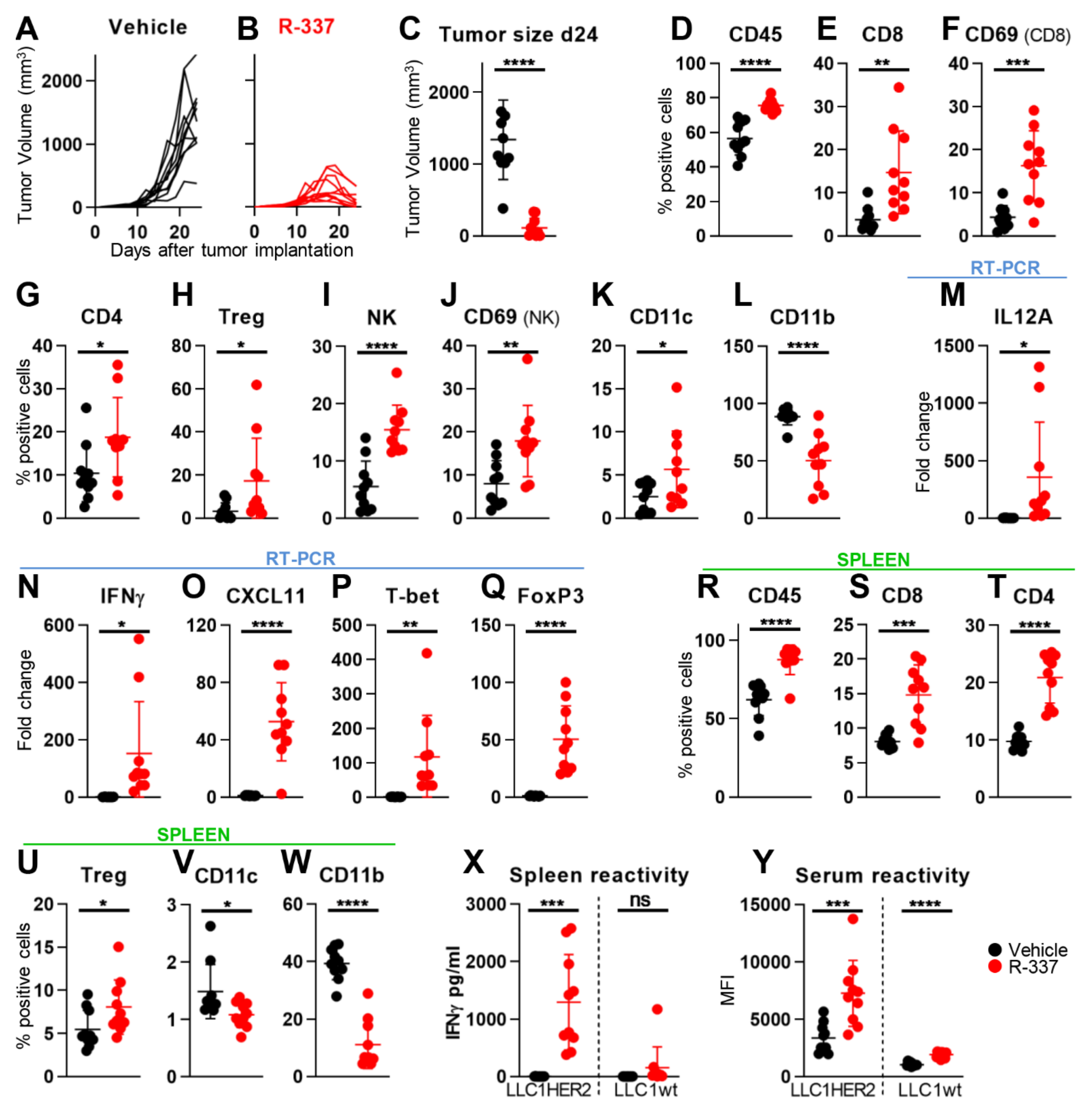
4.3. Retargeted oHSVs Subvert TME Immunosuppression
4.4. The “Immune Heating” of the Tumor Predisposes to Combination Therapy
4.5. Retargeted oHSVs Eradicate High Grade Gliomas (HGG) in Preclinical Models
4.6. Efficacy of Systemically Administered Retargeted oHSVs
4.7. Patents
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef]
- Chambers, R.; Gillespie, G.Y.; Soroceanu, L.; Andreansky, S.; Chatterjee, S.; Chou, J.; Roizman, B.; Whitley, R.J. Comparison of genetically engineered herpes simplex viruses for the treatment of brain tumors in a scid mouse model of human malignant glioma. Proc. Natl. Acad. Sci. USA 1995, 92, 1411–1415. [Google Scholar] [CrossRef] [Green Version]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunol. Ther. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef]
- Patel, M.R.; Kratzke, R.A. Oncolytic virus therapy for cancer: The first wave of translational clinical trials. Transl. Res. 2013, 161, 355–364. [Google Scholar] [CrossRef]
- Buijs, P.R.; Verhagen, J.H.; van Eijck, C.H.; van den Hoogen, B.G. Oncolytic viruses: From bench to bedside with a focus on safety. Hum. Vaccines Immunother. 2015, 11, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic viruses and immune checkpoint inhibition: The best of both worlds. Mol. Ther. Oncol. 2019, 13, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, B.A.; Bell, J.C. Oncolytic viruses-immunotherapeutics on the rise. J. Mol. Med. 2016, 94, 979–991. [Google Scholar] [CrossRef]
- Gujar, S.; Bell, J.; Diallo, J.-S. SnapShot: Cancer immunotherapy with oncolytic viruses. Cell 2019, 176, 1240.e1. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Razdan, S.N.; Kuo, H.-C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Vannini, A.; Petrovic, B.; Gatta, V.; Leoni, V.; Pepe, S.; Menotti, L.; Campadelli-Fiume, G.; Gianni, T. Rescue, Purification, and Characterization of a Recombinant HSV Expressing a Transgenic Protein. In Herpes Simplex Virus; Springer: Cham, Switzerland, 2020; pp. 153–168. [Google Scholar]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a phase I clinical trial to evaluate M032, a genetically engineered HSV-1 expressing IL-12, in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Farkaly, T.; Grzesik, P.; Lee, J.; Denslow, A.; Hewett, J.; Bryant, J.; Behara, P.; Goshert, C.; Wambua, D. Design of an interferon-resistant oncolytic HSV-1 incorporating redundant safety modalities for improved tolerability. Mol. Ther. Oncol. 2020, 18, 476–490. [Google Scholar] [CrossRef]
- De Lucia, M.; Cotugno, G.; Bignone, V.; Garzia, I.; Nocchi, L.; Langone, F.; Petrovic, B.; Sasso, E.; Pepe, S.; Froechlich, G.; et al. Retargeted and multi-cytokine armed herpes virus is a potent cancer endovaccine for local and systemic anti-tumor treatment in combination with anti-PD1. Mol. Ther. Oncol. 2020, 19, 253–264. [Google Scholar] [CrossRef]
- Laquerre, S.; Anderson, D.B.; Stolz, D.B.; Glorioso, J.C. Recombinant herpes simplex virus type 1 engineered for targeted binding to erythropoietin receptor-bearing cells. J. Virol. 1998, 72, 9683–9697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorioso, J.C.; Cohen, J.B.; Goins, W.F.; Hall, B.; Jackson, J.W.; Kohanbash, G.; Amankulor, N.; Kaur, B.; Caligiuri, M.A.; Chiocca, E.A.; et al. Oncolytic HSV Vectors and Anti-Tumor Immunity. Curr. Issues Mol. Biol. 2020, 41, 381–468. [Google Scholar] [PubMed]
- Zhou, G.; Roizman, B. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 5508–5513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campadelli-Fiume, G.; Petrovic, B.; Leoni, V.; Gianni, T.; Avitabile, E.; Casiraghi, C.; Gatta, V. Retargeting Strategies for Oncolytic Herpes Simplex Viruses. Viruses 2016, 8, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menotti, L.; Cerretani, A.; Campadelli-Fiume, G. A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J. Virol. 2006, 80, 5531–5539. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 2008, 20, 10153–10161. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; Amasio, M.; Avitabile, E.; Cerretani, A.; Forghieri, C.; Gianni, T.; Menotti, L. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev. Med. Virol. 2007, 17, 313–326. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; Collins-McMillen, D.; Gianni, T.; Yurochko, A.D. Integrins as Herpesvirus Receptors and Mediators of the Host Signalosome. Annu Rev. Virol. 2016, 3, 215–236. [Google Scholar] [CrossRef] [Green Version]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Gianni, T.; Salvioli, S.; Chesnokova, L.S.; Hutt-Fletcher, L.M.; Campadelli-Fiume, G. alphavbeta6- and alphavbeta8-integrins serve as interchangeable receptors for HSV gH/gL to promote endocytosis and activation of membrane fusion. PLoS Pathog. 2013, 9, e1003806. [Google Scholar] [CrossRef] [Green Version]
- Rey, F.A. Molecular gymnastics at the herpesvirus surface. EMBO Rep. 2006, 7, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Cocchi, F.; Menotti, L.; Mirandola, P.; Lopez, M.; Campadelli-Fiume, G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 1998, 72, 9992–10002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuzmen, C.; Cairns, T.M.; Atanasiu, D.; Lou, H.; Saw, W.T.; Hall, B.L.; Cohen, J.B.; Cohen, G.H.; Glorioso, J.C. Point mutations in retargeted gD eliminate the sensitivity of EGFR/EGFRvIII-targeted HSV to key neutralizing antibodies. Mol. Ther. Methods Clin. Dev. 2020, 16, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludan, S.R.; Bowie, A.G.; Horan, K.A.; Fitzgerald, K.A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11, 143–154. [Google Scholar] [CrossRef]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Paladino, P.; Collins, S.E.; Mossman, K.L. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS ONE 2010, 5, e10428. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Roizman, B. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc. Natl. Acad. Sci. USA 2014, 111, E611–E617. [Google Scholar] [CrossRef] [Green Version]
- Sasso, E.; Froechlich, G.; Cotugno, G.; D’Alise, A.M.; Gentile, C.; Bignone, V.; De Lucia, M.; Petrovic, B.; Campadelli-Fiume, G.; Scarselli, E. Replicative conditioning of Herpes simplex type 1 virus by Survivin promoter, combined to ERBB2 retargeting, improves tumour cell-restricted oncolysis. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Nicoletti, G.; Gatta, V.; Croci, S.; Landuzzi, L.; De Giovanni, C.; Nanni, P.; Lollini, P.L.; Campadelli-Fiume, G. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9039–9044. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Avitabile, E.; Gatta, V.; Malatesta, P.; Petrovic, B.; Campadelli-Fiume, G. HSV as A Platform for the Generation of Retargeted, Armed, and Reporter-Expressing Oncolytic Viruses. Viruses 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatta, V.; Petrovic, B.; Campadelli-Fiume, G. The Engineering of a Novel Ligand in gH Confers to HSV an Expanded Tropism Independent of gD Activation by Its Receptors. PLoS Pathog. 2015, 11, e1004907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic, B.; Gianni, T.; Gatta, V.; Campadelli-Fiume, G. Insertion of a ligand to HER2 in gB retargets HSV tropism and obviates the need for activation of the other entry glycoproteins. PLoS Pathog. 2017, 13, e1006352. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Gatta, V.; Casiraghi, C.; Nicosia, A.; Petrovic, B.; Campadelli-Fiume, G. A Strategy for Cultivation of Retargeted Oncolytic Herpes Simplex Viruses in Non-cancer Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic, B.; Leoni, V.; Gatta, V.; Zaghini, A.; Vannini, A.; Campadelli-Fiume, G. Dual Ligand Insertion in gB and gD of Oncolytic Herpes Simplex Viruses for Retargeting to a Producer Vero Cell Line and to Cancer Cells. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Petrovic, B.; Gianni, T.; Gatta, V.; Campadelli-Fiume, G. Simultaneous Insertion of Two Ligands in gD for Cultivation of Oncolytic Herpes Simplex Viruses in Noncancer Cells and Retargeting to Cancer Receptors. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [Green Version]
- Uchida, H.; Marzulli, M.; Nakano, K.; Goins, W.F.; Chan, J.; Hong, C.S.; Mazzacurati, L.; Yoo, J.Y.; Haseley, A.; Nakashima, H.; et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol. Ther. 2013, 21, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Uchida, H.; Shiroyama, T.; Okubo, Y.; Suzuki, T.; Ikeda, H.; Yamaguchi, M.; Miyagawa, Y.; Fukuhara, T.; Cohen, J. Development of an oncolytic HSV vector fully retargeted specifically to cellular EpCAM for virus entry and cell-to-cell spread. Gene Ther. 2016, 23, 479–488. [Google Scholar] [CrossRef]
- Hall, B.L.; Leronni, D.; Miyagawa, Y.; Goins, W.F.; Glorioso, J.C.; Cohen, J.B. Generation of an Oncolytic Herpes Simplex Viral Vector Completely Retargeted to the GDNF Receptor GFRα1 for Specific Infection of Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8815. [Google Scholar] [CrossRef]
- Sette, P.; Amankulor, N.; Li, A.; Marzulli, M.; Leronni, D.; Zhang, M.; Goins, W.F.; Kaur, B.; Bolyard, C.; Cripe, T.P. GBM-targeted oHSV armed with matrix metalloproteinase 9 enhances anti-tumor activity and animal survival. Mol. Ther. Oncol. 2019, 15, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannini, A.; Leoni, V.; Campadelli-Fiume, G. Targeted Delivery of IL-12 adjuvants immunotherapy by oncolytic viruses. In Tumor Microenvironment; Birbrair, A., Ed.; Springer: Cham, Switzerland, 2021; Volume 1290. [Google Scholar]
- Menotti, L.; Leoni, V.; Gatta, V.; Petrovic, B.; Vannini, A.; Pepe, S.; Gianni, T.; Campadelli-Fiume, G. oHSV Genome Editing by Means of galK Recombineering. In Herpes Simplex Virus; Springer: Berlin, Germany, 2020; pp. 131–151. [Google Scholar]
- Leoni, V.; Vannini, A.; Gatta, V.; Rambaldi, J.; Sanapo, M.; Barboni, C.; Zaghini, A.; Nanni, P.; Lollini, P.L.; Casiraghi, C.; et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018, 14, e1007209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanni, P.; Gatta, V.; Menotti, L.; De Giovanni, C.; Ianzano, M.; Palladini, A.; Grosso, V.; Dall’ora, M.; Croci, S.; Nicoletti, G.; et al. Preclinical Therapy of Disseminated HER-2(+) Ovarian and Breast Carcinomas with a HER-2-Retargeted Oncolytic Herpesvirus. PLoS Pathog. 2013, 9, e1003155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambini, E.; Reisoli, E.; Appolloni, I.; Gatta, V.; Campadelli-Fiume, G.; Menotti, L.; Malatesta, P. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol. Ther. 2012, 20, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Gatta, V.; Palladini, A.; Nicoletti, G.; Ranieri, D.; Dall’Ora, M.; Grosso, V.; Rossi, M.; Alviano, F.; Bonsi, L.; et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget 2015, 6, 34774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisoli, E.; Gambini, E.; Appolloni, I.; Gatta, V.; Barilari, M.; Menotti, L.; Malatesta, P. Efficacy of HER2 retargeted herpes simplex virus as therapy for high-grade glioma in immunocompetent mice. Cancer Gene Ther. 2012, 19, 788–795. [Google Scholar] [CrossRef]
- Alessandrini, F.; Menotti, L.; Avitabile, E.; Appolloni, I.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 2019, 38, 4467–4479. [Google Scholar] [CrossRef]
- Lechner, M.G.; Karimi, S.S.; Barry-Holson, K.; Angell, T.E.; Murphy, K.A.; Church, C.H.; Ohlfest, J.R.; Hu, P.; Epstein, A.L. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. 2013, 36, 477. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Tateishi, R.; Iwai, M.; Koike, K.; Todo, T. Neoadjuvant use of oncolytic herpes virus G47Δ enhances the antitumor efficacy of radiofrequency ablation. Mol. Ther. Oncol. 2020, 18, 535–545. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, X.; Feng, L.; Yang, Z.; Zhou, L.; Duan, X.; Cheng, S.; Zhang, W.; Liu, B.; Zhang, K. Enhanced therapeutic efficacy of a novel oncolytic herpes simplex virus Type 2 encoding an antibody against programmed cell death 1. Mol. Ther. Oncol. 2019, 15, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic viruses: Priming time for cancer immunotherapy. BioDrugs 2019, 33, 485–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessandrini, F.; Menotti, L.; Apolloni, I.; Avitabile, E.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Il-12 secreting herpes simplex virus retargeted to HER2 as therapy for high grade glioma. Viruses 2018, 10, 352. [Google Scholar]
- Matveeva, O.V.; Shabalina, S.A. Prospects for Using Expression Patterns of Paramyxovirus Receptors as Biomarkers for Oncolytic Virotherapy. Cancers 2020, 12, 3659. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T cell therapy for solid tumors. Ann. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Schmidts, A.; Maus, M.V. Making CAR T cells a solid option for solid tumors. Front. Immunol. 2018, 9, 2593. [Google Scholar] [CrossRef] [Green Version]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor–modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [Green Version]
- Ricca, J.M.; Oseledchyk, A.; Walther, T.; Liu, C.; Mangarin, L.; Merghoub, T.; Wolchok, J.D.; Zamarin, D. Pre-existing Immunity to Oncolytic Virus Potentiates Its Immunotherapeutic Efficacy. Mol. Ther. 2018, 26, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Netea, M.G. Trained Innate Immunity, Epigenetics, and Covid-19. N. Engl. J. Med. 2020, 383, 1078–1080. [Google Scholar] [CrossRef]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2017. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generation of Recombinant | Name of Recombinant | scFv for Retargeting to Tumor Cells Inserted | GCN4 Peptide for Retargeting to Producer Cells Inserted in | Detargeting Strategy, Deletions @ gD | Ref |
|---|---|---|---|---|---|
| 1st | R-LM113 R-115 R-123 | HER2 @ gD | Absent | Δ aa 6–38 | [21,27,42] |
| R-LM249 | HER2 @ gD | Absent | Δ aa 61–218 | [41] | |
| R-611 | EGFR @ gD | Absent | Δ aa 6–38 | [42] | |
| R-613 | EGFRVIII @ gD | Absent | Δ aa 6–38 | [42] | |
| R-593 | PSMA @ gD | Absent | Δ aa 6–38 | [42] | |
| 2nd | R-803 R-809 | HER2 @gH | Absent | No deletion, or Δ aa 6–38 | [43] |
| R-903 R-909 | HER2 @ gB | Absent | No deletion, or Δ aa 6–38 | [44] | |
| 3rd | R-313, R-315 R-317 R-319 | HER2 @ gD | @ gB | Δ aa 6–38 | [46] |
| R-213 | HER2 @ gD | @ gH | Δ aa 6–38 | [45] | |
| R-87 R-89 R-97 R-99 R-99-2 | HER2 @ gD | @ gD | Deletions, various | [47] | |
| R-321, R-335 R-337 | HER2 @ gD | @ gB | Δ aa30 and aa38 | [46] this paper |
| Name of Recombinant | Payload | Preclinical Assays and Main Results | Ref | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tumor | Mice | Efficacy against Primary Tumor | Efficacy against Distant Challenge Tumor | Modifications to TME | Combo with CPI | Route of Administration | ||||
| 1st generation | R-LM249 | None | Human Ovary SK-OV-3 | Immune- deficient | Very high protection. Protection from disseminated metastases. | NT | NT | NT | I.T. I.P. Carrier cells | [41,56,58] |
| R-LM113 | None | Mouse lung (LLC-1-HER2) | Immune-competent, HER2-tolerant | Complete response against primary tumor in 30% mice in early treatment. | High protection | Low modifications | NT | I.T. | [55] | |
| R-LM113 | None | Human high grade glioma AND Murine high grade glioma | Immune-deficient AND Immune-competent | Doubling in survival time AND Doubling in survival time | NT | NT | NT | I.C. | [57,59] | |
| R-115 | mIL-12 | LLC-1-HER2 | Immune-competent, HER2-tolerant | Complete response against primary tumor in 70% mice in early treatment. | High protection | Infiltration by immune cells. Increased cytokines | I.T. | [55] | ||
| R-115 | mIL-12 | Murine high grade glioma (HGG-HER2) | Immune-competent | Complete eradication in 30% mice in late treatment | High protection | Increased infiltration by CD4 CD8 | I.C. | [60] | ||
| R-123 | mIL-12 + mGM-CSF | LLC-1-HER2 | Immune-competent, HER2-tolerant | Complete response against primary tumor in 40% mice in late treatment. | NT | Full protection | I.T. I.V. | [21] | ||
| 3rd generation | R-335 | mIL-12 | LLC-1-HER2 | Immune-competent, HER2-tolerant | Complete response in 30% mice in late treatment | High protection | NT | Low potentiation | I.T. | This paper |
| R-337 | mIL-12 fusion protein | LLC-1-HER2 | Immune-competent, HER2-tolerant | Complete response in 80% mice in late treatment | High protection | Increased infiltration by immune cells; increased expression of cytokines | Potentiation | I.T. | This paper | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vannini, A.; Leoni, V.; Sanapo, M.; Gianni, T.; Giordani, G.; Gatta, V.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G. Immunotherapeutic Efficacy of Retargeted oHSVs Designed for Propagation in an Ad Hoc Cell Line. Cancers 2021, 13, 266. https://doi.org/10.3390/cancers13020266
Vannini A, Leoni V, Sanapo M, Gianni T, Giordani G, Gatta V, Barboni C, Zaghini A, Campadelli-Fiume G. Immunotherapeutic Efficacy of Retargeted oHSVs Designed for Propagation in an Ad Hoc Cell Line. Cancers. 2021; 13(2):266. https://doi.org/10.3390/cancers13020266
Chicago/Turabian StyleVannini, Andrea, Valerio Leoni, Mara Sanapo, Tatiana Gianni, Giorgia Giordani, Valentina Gatta, Catia Barboni, Anna Zaghini, and Gabriella Campadelli-Fiume. 2021. "Immunotherapeutic Efficacy of Retargeted oHSVs Designed for Propagation in an Ad Hoc Cell Line" Cancers 13, no. 2: 266. https://doi.org/10.3390/cancers13020266