Altered Spatial Composition of the Immune Cell Repertoire in Association to CD34+ Blasts in Myelodysplastic Syndromes and Secondary Acute Myeloid Leukemia

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
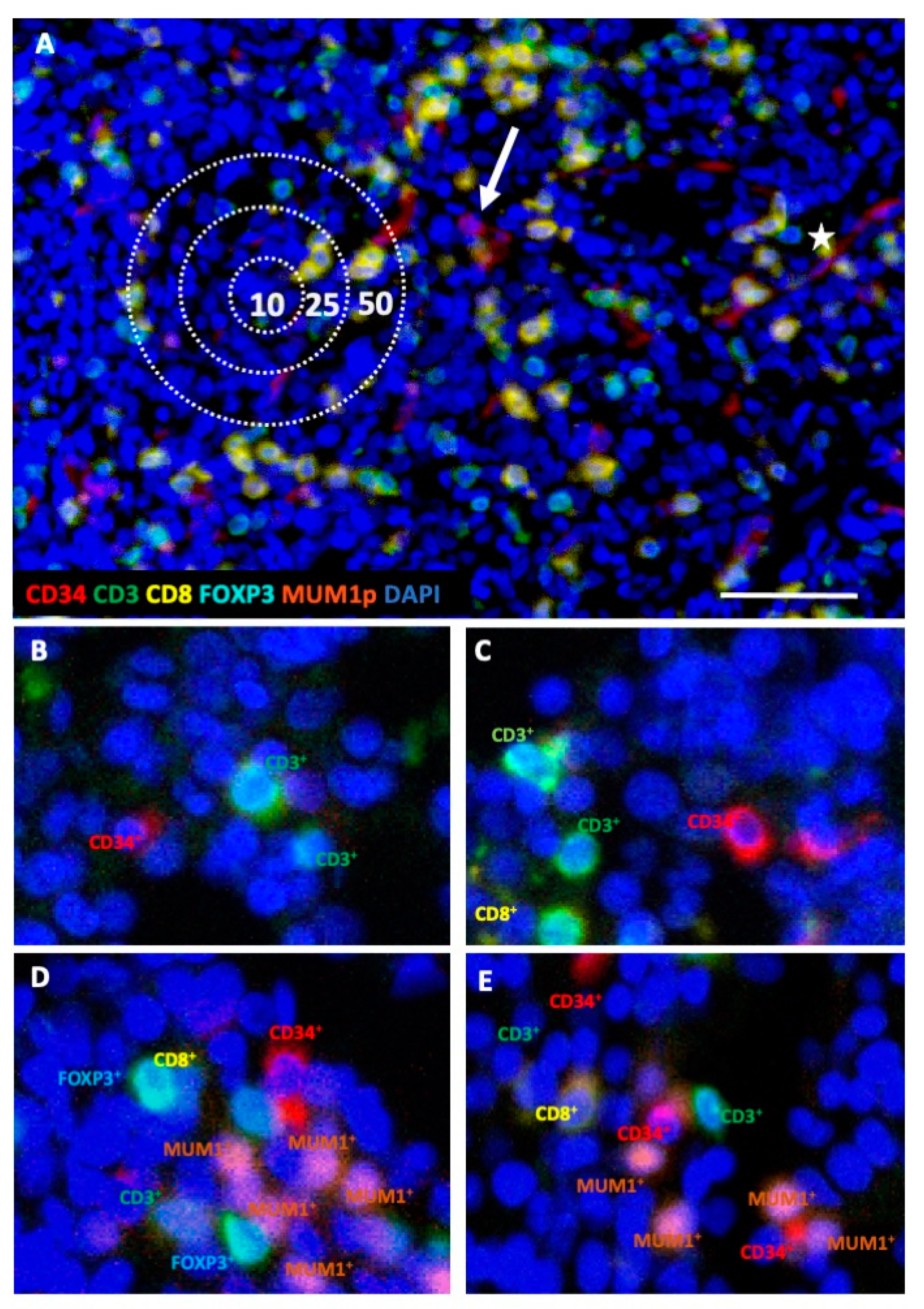
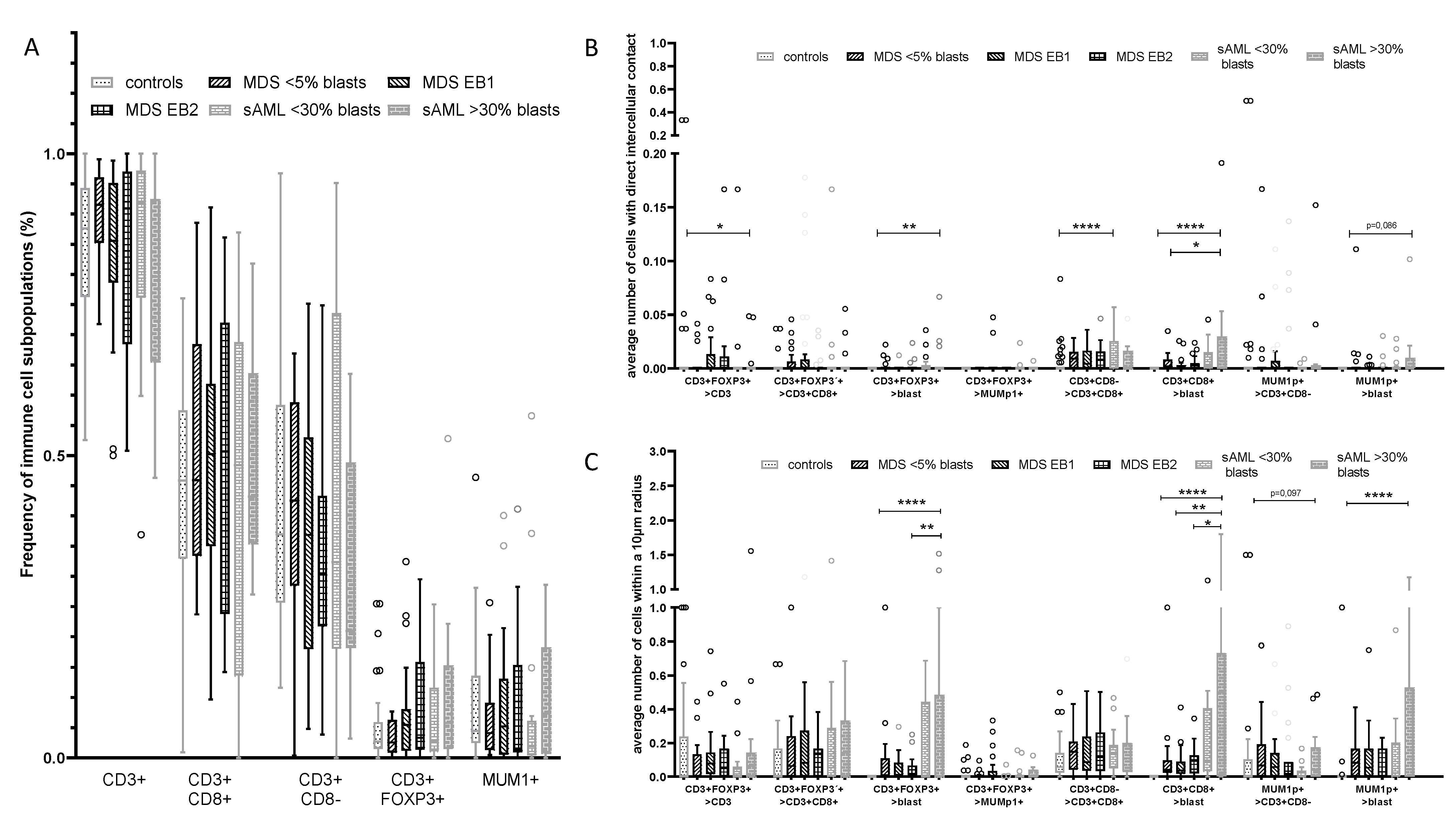
2.1. Differences in the Frequency and Spatial Distribution of Immune Cell Subpopulations to CD34+ HSPC/Blasts in BMB from MDS, sAML, and Control Samples
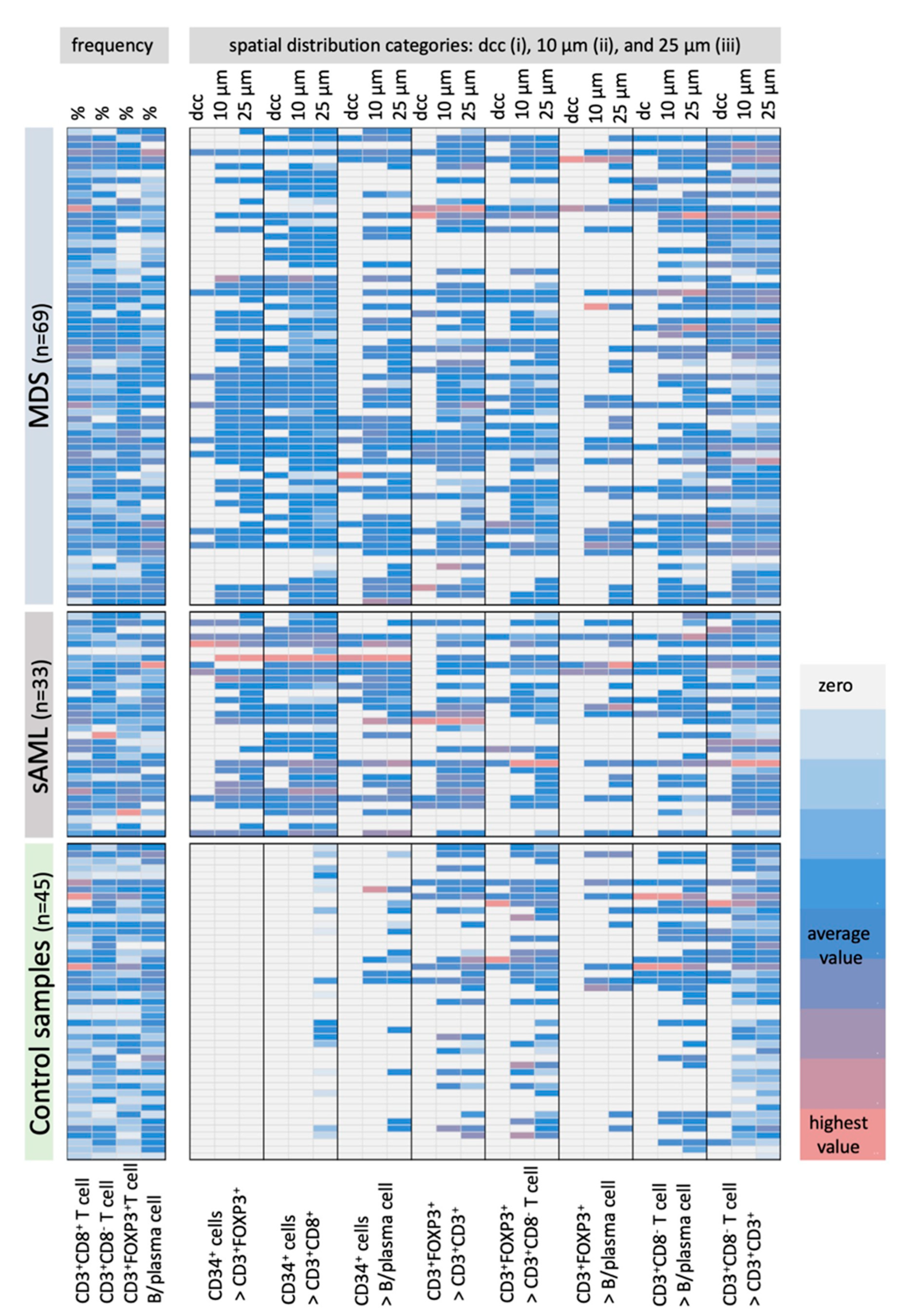
2.2. Differences in the Frequency and Histotopography of Immune Cell Subsets in Spatial Relationship with CD34+ HSPC/Blasts in MDS, sAML, and Control Samples
2.3. Correlation of the Frequency and Histotopography of Immune Cell Subsets to CD34+ Blasts in MDS and sAML
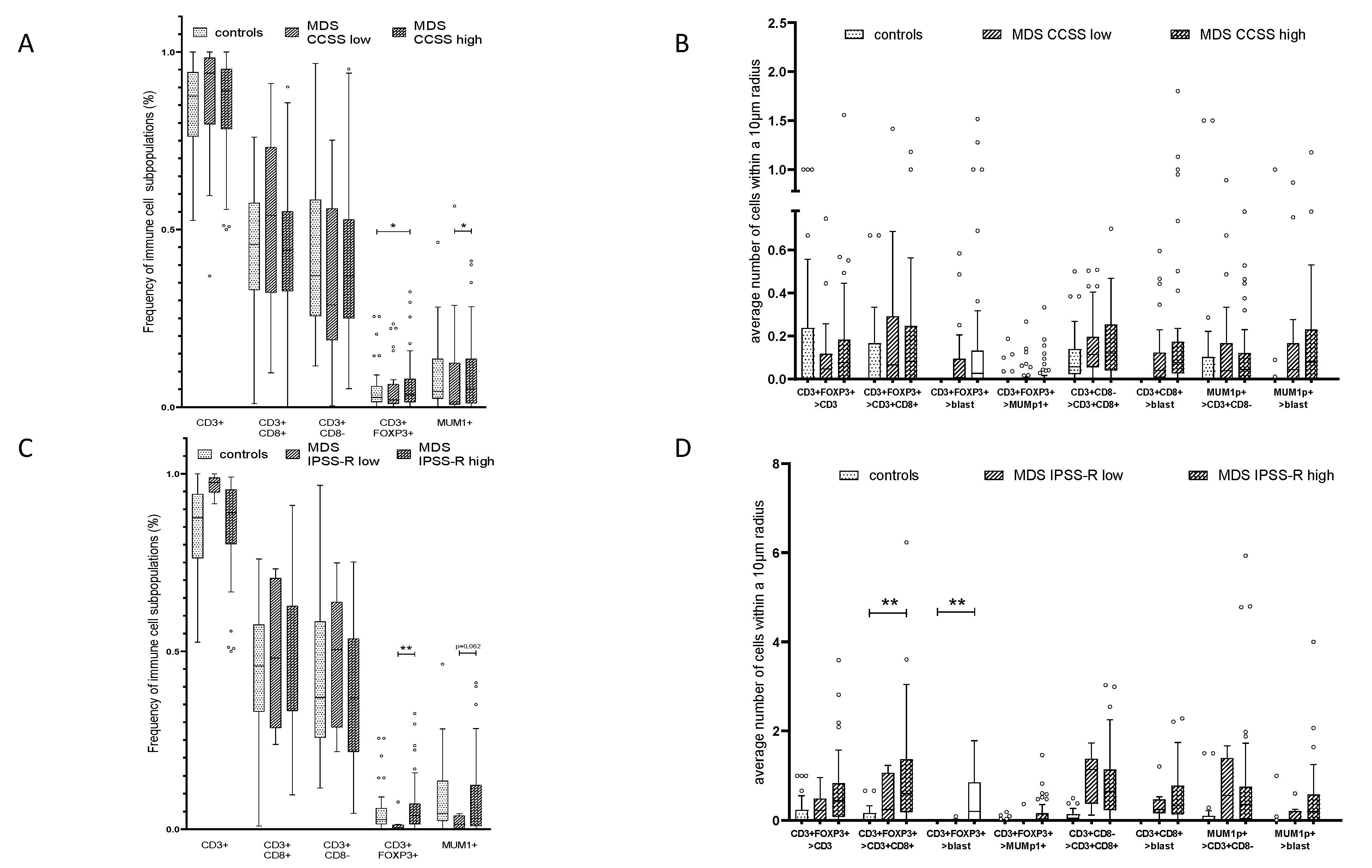
2.4. Effect of the Mutational Status in MDS on the Frequency and Histotopography of the Immune Cell Subsets
2.5. Influence of Cytogenetic Alterations and the Frequency and Histotopography of the Immune Cell Subsets in MDS
2.6. Spatial Distribution of Immune Cell Subsets and CD34+ Blasts in Relation to MDS Progression to sAML
3. Discussion
4. Materials and Methods
4.1. Patient Characteristics
4.2. Study Approval
4.3. Standard Morphological Evaluation of the Bone Marrow
4.4. Multispectral Imaging (MSI)
4.5. Mutational Analysis—Targeted Next Generation Sequencing
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Corey, S.J.; Minden, M.D.; Barber, D.L.; Kantarjian, H.; Wang, J.C.Y.; Schimmer, A.D. Myelodysplastic Syndromes: The Complexity of Stem-Cell Diseases. Nat. Rev. Cancer 2007, 7, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobrial, I.M.; Detappe, A.; Anderson, K.C.; Steensma, D.P. The Bone-Marrow Niche in MDS and MGUS: Implications for AML and MM. Nat. Rev. Clin. Oncol. 2018, 15, 219–233. [Google Scholar] [CrossRef]
- Greenberg, P.L. Molecular and Genetic Features of Myelodysplastic Syndromes. Int. J. Lab. Hematol. 2012, 34, 215–222. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Sole, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Esplin, B.L.; Shimazu, T.; Welner, R.S.; Garrett, K.P.; Nie, L.; Zhang, Q.; Humphrey, M.B.; Yang, Q.; Borghesi, L.A.; Kincade, P.W. Chronic Exposure to a TLR Ligand Injures Hematopoietic Stem Cells. J. Immunol. 2011, 186, 5367–5375. [Google Scholar] [CrossRef] [Green Version]
- Winter, S.; Shoaie, S.; Kordasti, S.; Platzbecker, U. Integrating the “Immunome” in the Stratification of Myelodysplastic Syndromes and Future Clinical Trial Design. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S.; Kulasekararaj, A.; Al Ali, N.H.; Kordasti, S.; Bart-Smith, E.; Craig, B.M.; Padron, E.; Zhang, L.; Lancet, J.E.; Pinilla-Ibarz, J.; et al. Autoimmune Diseases and Myelodysplastic Syndromes. Am. J. Hematol. 2016, 91, E280–E283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenthøj, A.; Ørskov, A.D.; Hansen, J.W.; Hadrup, S.R.; O’Connell, C.; Grønbæk, K. Immune Mechanisms in Myelodysplastic Syndrome. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, W.-K. Characterization of Gene Expression of CD34+ Cells from Normal and Myelodysplastic Bone Marrow. Blood 2002, 100, 3553–3560. [Google Scholar] [CrossRef] [Green Version]
- Pellagatti, A.; Cazzola, M.; Giagounidis, A.; Perry, J.; Malcovati, L.; Della Porta, M.G.; Jädersten, M.; Killick, S.; Verma, A.; Norbury, C.J.; et al. Deregulated Gene Expression Pathways in Myelodysplastic Syndrome Hematopoietic Stem Cells. Leukemia 2010, 24, 756–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, L.A.; Pfeiffer, R.M.; Landgren, O.; Gadalla, S.; Berndt, S.I.; Engels, E.A. Risks of Myeloid Malignancies in Patients with Autoimmune Conditions. Br. J. Cancer 2009, 100, 822–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fozza, C. The Burden of Autoimmunity in Myelodysplastic Syndromes. Hematol. Oncol. 2018, 36, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, S.Y.; Björkholm, M.; Hultcrantz, M.; Derolf, Å.R.; Landgren, O.; Goldin, L.R. Chronic Immune Stimulation Might Act as a Trigger for the Development of Acute Myeloid Leukemia or Myelodysplastic Syndromes. J. Clin. Oncol. 2011, 29, 2897–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schanz, J.; Tüchler, H.; Solé, F.; Mallo, M.; Luño, E.; Cervera, J.; Granada, I.; Hildebrandt, B.; Slovak, M.L.; Ohyashiki, K.; et al. New Comprehensive Cytogenetic Scoring System for Primary Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia after MDS Derived from an International Database Merge. J. Clin. Oncol. 2012, 30, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, Y.; Gao, S.; Chen, J.; Yu, J.; Zhang, H.; Li, M.; Zhan, X.; Li, W. Immune Dysregulation in Myelodysplastic Syndrome: Clinical Features, Pathogenesis and Therapeutic Strategies. Crit. Rev. Oncol. Hematol. 2018, 122, 123–132. [Google Scholar] [CrossRef]
- Kotsianidis, I.; Bouchliou, I.; Nakou, E.; Spanoudakis, E.; Margaritis, D.; Christophoridou, A.V.; Anastasiades, A.; Tsigalou, C.; Bourikas, G.; Karadimitris, A.; et al. Kinetics, Function and Bone Marrow Trafficking of CD4+CD25+FOXP3+ Regulatory T Cells in Myelodysplastic Syndromes (MDS). Leukemia 2009, 23, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Kordasti, S.Y.; Afzali, B.; Lim, Z.; Ingram, W.; Hayden, J.; Barber, L.; Matthews, K.; Chelliah, R.; Guinn, B.; Lombardi, G.; et al. IL-17-Producing CD4(+) T Cells, pro-Inflammatory Cytokines and Apoptosis Are Increased in Low Risk Myelodysplastic Syndrome. Br. J. Haematol. 2009, 145, 64–72. [Google Scholar] [CrossRef]
- Mailloux, A.W.; Sugimori, C.; Komrokji, R.S.; Yang, L.; Maciejewski, J.P.; Sekeres, M.A.; Paquette, R.; Loughran, T.P.; List, A.F.; Epling-Burnette, P.K. Expansion of Effector Memory Regulatory T Cells Represents a Novel Prognostic Factor in Lower Risk Myelodysplastic Syndrome. J. Immunol. 2012, 189, 3198–3208. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.R.; Traina, F.; de Campos, P.M.; Pereira, J.K.N.; Machado-Neto, J.A.; da Machado, H.C.; Gilli, S.C.O.; Saad, S.T.O.; Favaro, P. IL10 Inversely Correlates with the Percentage of CD8+ Cells in MDS Patients. Leuk. Res. 2013, 37, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Brück, O.; Dufva, O.; Hohtari, H.; Blom, S.; Turkki, R.; Ilander, M.; Kovanen, P.; Pallaud, C.; Ramos, P.M.; Lähteenmäki, H.; et al. Immune Profiles in Acute Myeloid Leukemia Bone Marrow Associate with Patient Age, T-Cell Receptor Clonality, and Survival. Blood Adv. 2020, 4, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Maratheftis, C.I.; Andreakos, E.; Moutsopoulos, H.M.; Voulgarelis, M. Toll-like Receptor-4 Is Up-Regulated in Hematopoietic Progenitor Cells and Contributes to Increased Apoptosis in Myelodysplastic Syndromes. Clin. Cancer Res. 2007, 13, 1154–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuninaka, N.; Kurata, M.; Yamamoto, K.; Suzuki, S.; Umeda, S.; Kirimura, S.; Arai, A.; Nakagawa, Y.; Suzuki, K.; Kitagawa, M. Expression of Toll-like Receptor 9 in Bone Marrow Cells of Myelodysplastic Syndromes Is down-Regulated during Transformation to Overt Leukemia. Exp. Mol. Pathol. 2010, 88, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Dimicoli, S.; Bueso-Ramos, C.; Chen, R.; Yang, H.; Neuberg, D.; Pierce, S.; Jia, Y.; Zheng, H.; Wang, H.; et al. Toll-like Receptor Alterations in Myelodysplastic Syndrome. Leukemia 2013, 27, 1832–1840. [Google Scholar] [CrossRef] [Green Version]
- Mährle, T.; Akyüz, N.; Fuchs, P.; Bonzanni, N.; Simnica, D.; Germing, U.; Asemissen, A.M.; Jann, J.C.; Nolte, F.; Hofmann, W.-K.; et al. Deep Sequencing of Bone Marrow Microenvironments of Patients with Del(5q) Myelodysplastic Syndrome Reveals Imprints of Antigenic Selection as Well as Generation of Novel T-Cell Clusters as a Response Pattern to Lenalidomide. Haematologica 2019, 104, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone Progenitor Dysfunction Induces Myelodysplasia and Secondary Leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Vaxevanis, C.; Bethmann, D.; Massa, C.; Pazaitis, N.; Wickenhauser, C.; Seliger, B. Multiplex Immunohistochemistry as a Novel Tool for the Topographic Assessment of the Bone Marrow Stem Cell Niche. Methods Enzymol. 2020, 635, 67–79. [Google Scholar] [CrossRef]
- Smith, M.A.; Choudhary, G.S.; Pellagatti, A.; Choi, K.; Bolanos, L.C.; Bhagat, T.D.; Gordon-Mitchell, S.; Von Ahrens, D.; Pradhan, K.; Steeples, V.; et al. U2AF1 Mutations Induce Oncogenic IRAK4 Isoforms and Activate Innate Immune Pathways in Myeloid Malignancies. Nat. Cell Biol. 2019, 21, 640–650. [Google Scholar] [CrossRef]
- Ivy, K.S.; Brent Ferrell, P. Disordered Immune Regulation and Its Therapeutic Targeting in Myelodysplastic Syndromes. Curr. Hematol. Malig. Rep. 2018, 13, 244–255. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Vaxevanis, C.; Heimer, N.; Al-Ali, H.K.; Jaekel, N.; Bachmann, M.; Wickenhauser, C.; Seliger, B. Expression, Regulation and Function of MicroRNA as Important Players in the Transition of MDS to Secondary AML and Their Cross Talk to RNA-Binding Proteins. Int. J. Mol. Sci. 2020, 21, 7140. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Wan, X.; Deng, Z.; Shi, L.; Hao, C.; Zhou, Z.; Zhou, C.; Fang, Y.; Liu, J.; Yang, J.; et al. Epigenetic Regulator CXXC5 Recruits DNA Demethylase Tet2 to Regulate TLR7/9-Elicited IFN Response in PDCs. J. Exp. Med. 2017, 214, 1471–1491. [Google Scholar] [CrossRef] [PubMed]
- Pellagatti, A.; Boultwood, J. Splicing Factor Mutant Myelodysplastic Syndromes: Recent Advances. Adv. Biol. Regul. 2020, 75, 100655. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, L.; Chlon, T.M.; Starczynowski, D.T. Chronic Immune Response Dysregulation in MDS Pathogenesis. Blood 2018, 132, 1553–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.W.; Maciejewski, J.P.; Farzaneh, F.; et al. CD4+CD25high Foxp3+ Regulatory T Cells in Myelodysplastic Syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Qianqiao, Z.; Qi, H.; Feng, X.; Chunkang, C.; Xiao, L. In Vitro Deprivation of CD8(+)CD57(+)T Cells Promotes the Malignant Growth of Bone Marrow Colony Cells in Patients with Lower-Risk Myelodysplastic Syndrome. Exp. Hematol. 2010, 38, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Valent, P.; Greil, R. Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis-Masters of Survival and Clonality? Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Teodorescu, P.; Pasca, S.; Dima, D.; Tomuleasa, C.; Ghiaur, G. Targeting the Microenvironment in MDS: The Final Frontier. Front. Pharmacol. 2020, 11, 1044. [Google Scholar] [CrossRef] [PubMed]
- Oelschlaegel, U.; Westers, T.M.; Mohr, B.; Kramer, M.; Parmentier, S.; Sockel, K.; Thiede, C.; Bornhäuser, M.; Ehninger, G.; van de Loosdrecht, A.A.; et al. Myelodysplastic Syndromes with a Deletion 5q Display a Characteristic Immunophenotypic Profile Suitable for Diagnostics and Response Monitoring. Haematologica 2015, 100, e93–e96. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Klotz, J.; Dunavin, N.; Lu, K.; Koklanaris, E.; Draper, D.; Superata, J.; Chinian, F.; Yu, Q.; Keyvanfar, K.; et al. Cellular Immune Profiling after Sequential Clofarabine and Lenalidomide for High Risk Myelodysplastic Syndromes and Acute Myeloid Leukemia. Leuk. Res. Rep. 2017, 7, 40–44. [Google Scholar] [CrossRef]
- Patel, S.S.; Lipschitz, M.; Pinkus, G.S.; Weirather, J.L.; Pozdnyakova, O.; Mason, E.F.; Inghirami, G.; Hasserjian, R.P.; Rodig, S.J.; Weinberg, O.K. Multiparametric in Situ Imaging of NPM1-Mutated Acute Myeloid Leukemia Reveals Prognostically-Relevant Features of the Marrow Microenvironment. Mod. Pathol. 2020, 33, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Bethmann, D.; Kappler, M.; Ballesteros-Merino, C.; Eckert, A.; Bell, R.B.; Cheng, A.; Bui, T.; Leidner, R.; Urba, W.J.; et al. Multiparametric Immune Profiling in HPV-Oral Squamous Cell Cancer. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Fang, P.; Sharma, A.; Fujimoto, J.; Wistuba, I.; Rao, A.U.K.; Lin, S.H. Spatial Interaction of Tumor Cells and Regulatory T Cells Correlates with Survival in Non-Small Cell Lung Cancer. Lung Cancer Amst. Neth. 2018, 117, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Ema, H.; Suda, T. Two Anatomically Distinct Niches Regulate Stem Cell Activity. Blood 2012, 120, 2174–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and Perivascular Cells Maintain Haematopoietic Stem Cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 Is Required to Resolve Inflammation by Recruiting Hdac2 to Specifically Repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Medyouf, H.; Mossner, M.; Jann, J.-C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic Cells in Patients Reprogram Mesenchymal Stromal Cells to Establish a Transplantable Stem Cell Niche Disease Unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.A.; Lemischka, I.R. “Tie-Ing” down the Hematopoietic Niche. Cell 2004, 118, 139–140. [Google Scholar] [CrossRef]
- Chen, S.; Zambetti, N.A.; Bindels, E.M.J.; Kenswill, K.; Mylona, A.M.; Adisty, N.M.; Hoogenboezem, R.M.; Sanders, M.A.; Cremers, E.M.P.; Westers, T.M.; et al. Massive Parallel RNA Sequencing of Highly Purified Mesenchymal Elements in Low-Risk MDS Reveals Tissue-Context-Dependent Activation of Inflammatory Programs. Leukemia 2016, 30, 1938–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the Haematopoietic Stem Cell Niche and Control of the Niche Size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, P.P. Galectins as Regulators of Cell Survival in the Leukemia Niche. Adv. Biol. Regul. 2019, 71, 41–54. [Google Scholar] [CrossRef]
- Sallman, D.A.; Komrokji, R.; Vaupel, C.; Cluzeau, T.; Geyer, S.M.; McGraw, K.L.; Al Ali, N.H.; Lancet, J.; McGinniss, M.J.; Nahas, S.; et al. Impact of TP53 Mutation Variant Allele Frequency on Phenotype and Outcomes in Myelodysplastic Syndromes. Leukemia 2016, 30, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of Myeloid Derived Suppressor Cells Correlates with Number of T Regulatory Cells and Disease Progression in Myelodysplastic Syndrome. Oncoimmunology 2016, 5, e1062208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. TP53 Abnormalities Correlate with Immune Infiltration and Associate with Response to Flotetuzumab Immunotherapy in AML. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 Allelic State for Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.-A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 Mutations in Myelodysplastic Syndromes and Secondary AML Confer an Immunosuppressive Phenotype. Blood 2020. [Google Scholar] [CrossRef]
- Weltgesundheitsorganisation. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. In World Health Organization Classification of Tumours, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; ISBN 978-92-832-4494-3. [Google Scholar]
- Wickenhauser, C.; Bethmann, D.; Feng, Z.; Jensen, S.M.; Ballesteros-Merino, C.; Massa, C.; Steven, A.; Bauer, M.; Kaatzsch, P.; Pazaitis, N.; et al. Multispectral Fluorescence Imaging Allows for Distinctive Topographic Assessment and Subclassification of Tumor-Infiltrating and Surrounding Immune Cells. Methods Mol. Biol. Clifton NJ 2019, 1913, 13–31. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | MDS | sAML | Controls | |
|---|---|---|---|---|
| Number of samples | 69 | 33 | 45 | |
| Age | 68.2 (42–86) | 68.9 (47–82) | 59.6 (39–84) | |
| Sex | male | 39 | 18 | 24 |
| female | 30 | 15 | 21 | |
| Leukocyte count in peripheral blood (×109/L) | 4.5 (0.8–46.2) | 5.6 (0.1–30.0) | 8.1 (3.8–9.7) | |
| Patients with (auto)immune disorders | 10 | 2 | 0 | |
| Autoimmune thyroiditis (Hashimoto thyroiditis, Graves’ disease) | 5 | 1 | - | |
| autoimmune cholangitis | 1 | - | - | |
| rheumatoid arthritis | 3 | - | - | |
| Myasthenia gravis | 1 | 1 | - | |
| MDS | MDS-SLD, MDS-RS-SLD, MDS-MLD, MDS-RS-MLD | 22 | - | - |
| MDS-EB1 | 26 | - | - | |
| MDS-EB2 | 21 | - | - | |
| CCSS | 1—loss of Y chromosome, del(11q) | 7 | 2 | - |
| 2—normal karyotype, del(5q), del(12p), del(20q) | 27 | 5 | - | |
| 3—del(7q), gain of chromosome 8 and 19, isochromosome 17q | 17 | 10 | - | |
| 4—gain of chromosome 3 and 7, complex (>2) | 1 | 7 | - | |
| 5—complex (>3) | 17 | 9 | - | |
| IPSS-R | 1—very low risk | 2 | - | - |
| 2—low risk | 6 | - | - | |
| 3—intermediate risk | 20 | - | - | |
| 4—high risk | 26 | - | - | |
| 5—very high risk | 15 | - | - | |
| Treatment | patients without specified treatment prior to BMB extraction | 35 of 69 | - | - |
| HMA treatment prior to BMB extraction | 17 of 69 | 6 of 33 | - | |
| ASCT treatment following BMB extraction | 23 of 69 | 9 of 33 | - | |
| MDS patients with progress to sAML in the course of the disease (regardless treatment) | 18 of 69 | - | - | |
| untreated MDS patients with progress to sAML in the course of the disease (without prior HMA treatment and without ASCT in the course of the disease) | 6 of 35 | - | - | |
| Patient Numbers | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | ||
| function | gene | (n/%) | ||||||||||||||||||||||||||||
| cohesin | STAG2 | 0 / 0.0 | ||||||||||||||||||||||||||||
| DNA repair | TP53 | 10 / 35.7 | * | * | * | * | * | * | * | * | * | * | ||||||||||||||||||
| chromatin | ASXL1 | 3 / 10.7 | * | * | * | |||||||||||||||||||||||||
| modification | EZH2 | 2 / 7.1 | * | * | ||||||||||||||||||||||||||
| DNA | DNMT3A | 2 / 7.1 | * | * | ||||||||||||||||||||||||||
| methylation | IDH1 | 1 / 3.6 | * | |||||||||||||||||||||||||||
| IDH2 | 2 / 7.1 | * | * | |||||||||||||||||||||||||||
| TET2 | 3 / 10.7 | * | * | * | ||||||||||||||||||||||||||
| RNA splicing | SRSF2 | 2 / 7.1 | * | * | ||||||||||||||||||||||||||
| SF3B1 | 1 / 3.6 | * | ||||||||||||||||||||||||||||
| U2AF1 | 0 / 0.0 | |||||||||||||||||||||||||||||
| signal | ZRSR2 | 0 / 0.0 | ||||||||||||||||||||||||||||
| transduction | BRAF | 1 / 3.6 | * | |||||||||||||||||||||||||||
| CALR | 0 / 0.0 | |||||||||||||||||||||||||||||
| CBL | 2 / 7.1 | * | * | |||||||||||||||||||||||||||
| ETKN1 | 0 / 0.0 | |||||||||||||||||||||||||||||
| CSFR3 | 0 / 0.0 | |||||||||||||||||||||||||||||
| FLT3 | 1 / 3.6 | * | ||||||||||||||||||||||||||||
| GNAS | 0 / 0.0 | |||||||||||||||||||||||||||||
| HRAS | 0 / 0.0 | |||||||||||||||||||||||||||||
| JAK2 | 2 / 7.1 | * | * | |||||||||||||||||||||||||||
| KIT | 0 / 0.0 | |||||||||||||||||||||||||||||
| KRAS | 0 / 0.0 | |||||||||||||||||||||||||||||
| MPL | 0 / 0.0 | |||||||||||||||||||||||||||||
| NRAS | 0 / 0.0 | |||||||||||||||||||||||||||||
| STAT3 | 0 / 0.0 | |||||||||||||||||||||||||||||
| transcription | CEBPA | 1 / 3.6 | * | |||||||||||||||||||||||||||
| factors | IKZF1 | 0 / 0.0 | ||||||||||||||||||||||||||||
| NPM1 | 1 / 3.6 | * | ||||||||||||||||||||||||||||
| SETBP1 | 3 / 10.7 | * | * | * | ||||||||||||||||||||||||||
| RUNX1 | 1 / 5.0 | |||||||||||||||||||||||||||||
| total number of mutations/patient | 0 | 2 | 1 | 1 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 3 | 1 | 4 | 1 | 2 | 1 | 1 | 0 | 0 | 3 | 0 | 3 | 1 | ||
| CCSS | 2 | 2 | 5 | 2 | 2 | 3 | 3 | 2 | 2 | 5 | 5 | 5 | 5 | 2 | 3 | 3 | 1 | 3 | 2 | 3 | 3 | 5 | 5 | 3 | 5 | 2 | 3 | 3 | ||
| blast count (%) | 4 | 3 | 7 | 11 | 4 | 7 | 4 | 3 | 3 | 14 | 8 | 3 | 7 | 8 | 9 | 7 | 4 | 4 | 4 | 4 | 9 | 18 | 17 | 8 | 11 | 19 | 14 | 16 | ||
| IPSS-R group | 2 | 3 | 4 | 2 | 3 | 4 | 4 | 3 | 1 | 5 | 5 | 5 | 5 | 2 | 4 | 4 | 3 | 4 | 3 | 4 | 5 | 5 | 5 | 3 | 5 | 5 | 4 | 5 | ||
| progress to sAML | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no | yes | yes | yes | yes | yes | yes | yes | yes | Yes | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, M.; Vaxevanis, C.; Al-Ali, H.K.; Jaekel, N.; Naumann, C.L.H.; Schaffrath, J.; Rau, A.; Seliger, B.; Wickenhauser, C. Altered Spatial Composition of the Immune Cell Repertoire in Association to CD34+ Blasts in Myelodysplastic Syndromes and Secondary Acute Myeloid Leukemia. Cancers 2021, 13, 186. https://doi.org/10.3390/cancers13020186
Bauer M, Vaxevanis C, Al-Ali HK, Jaekel N, Naumann CLH, Schaffrath J, Rau A, Seliger B, Wickenhauser C. Altered Spatial Composition of the Immune Cell Repertoire in Association to CD34+ Blasts in Myelodysplastic Syndromes and Secondary Acute Myeloid Leukemia. Cancers. 2021; 13(2):186. https://doi.org/10.3390/cancers13020186
Chicago/Turabian StyleBauer, Marcus, Christoforos Vaxevanis, Haifa Kathrin Al-Ali, Nadja Jaekel, Christin Le Hoa Naumann, Judith Schaffrath, Achim Rau, Barbara Seliger, and Claudia Wickenhauser. 2021. "Altered Spatial Composition of the Immune Cell Repertoire in Association to CD34+ Blasts in Myelodysplastic Syndromes and Secondary Acute Myeloid Leukemia" Cancers 13, no. 2: 186. https://doi.org/10.3390/cancers13020186