Id1 and PD-1 Combined Blockade Impairs Tumor Growth and Survival of KRAS-mutant Lung Cancer by Stimulating PD-L1 Expression and Tumor Infiltrating CD8+ T Cells

 , , , , , , , , , ,
, , , , , , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
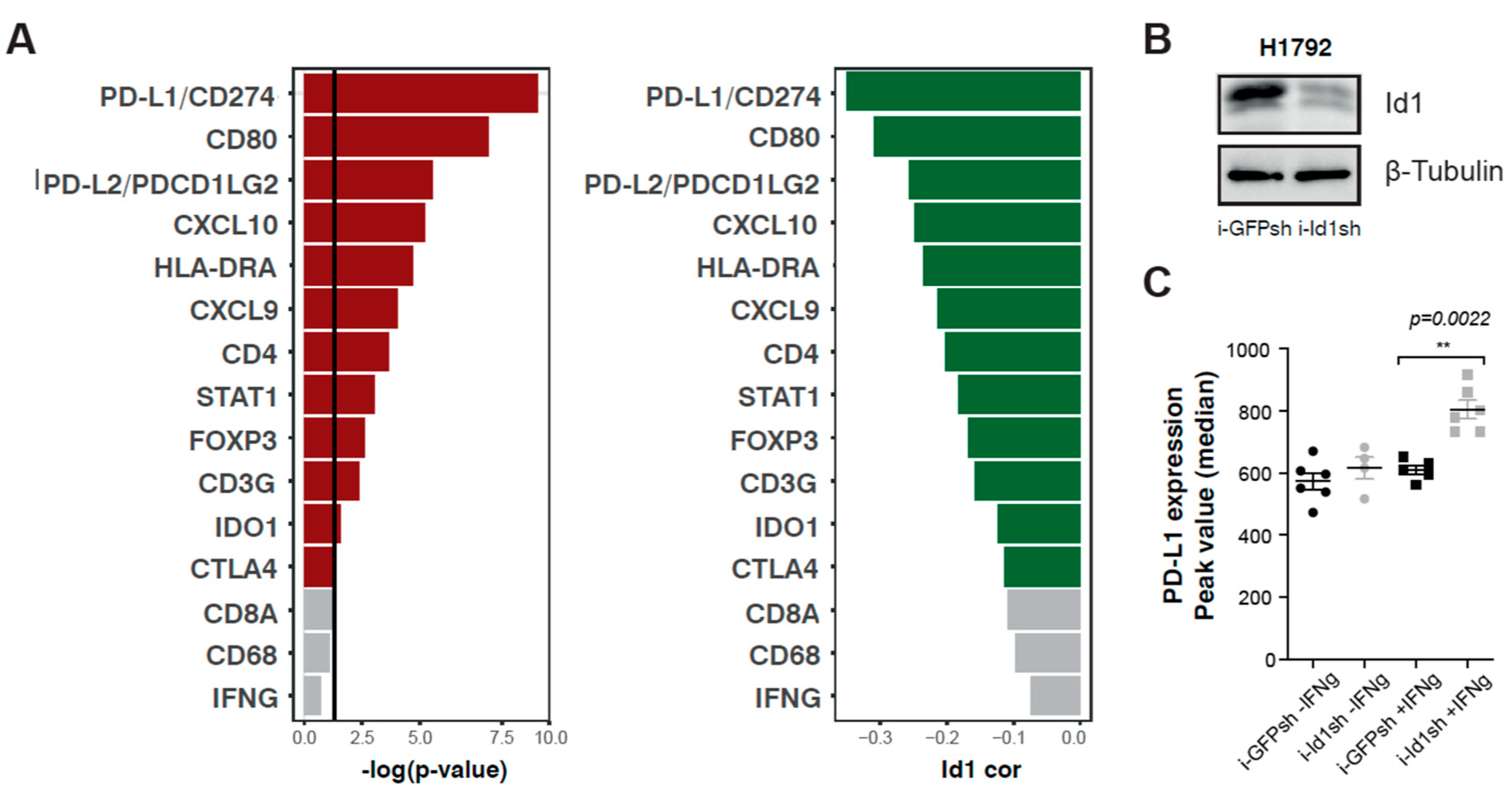
2.1. Id1 Expression is Associated with PD-L1 Expression in LUAD Patients
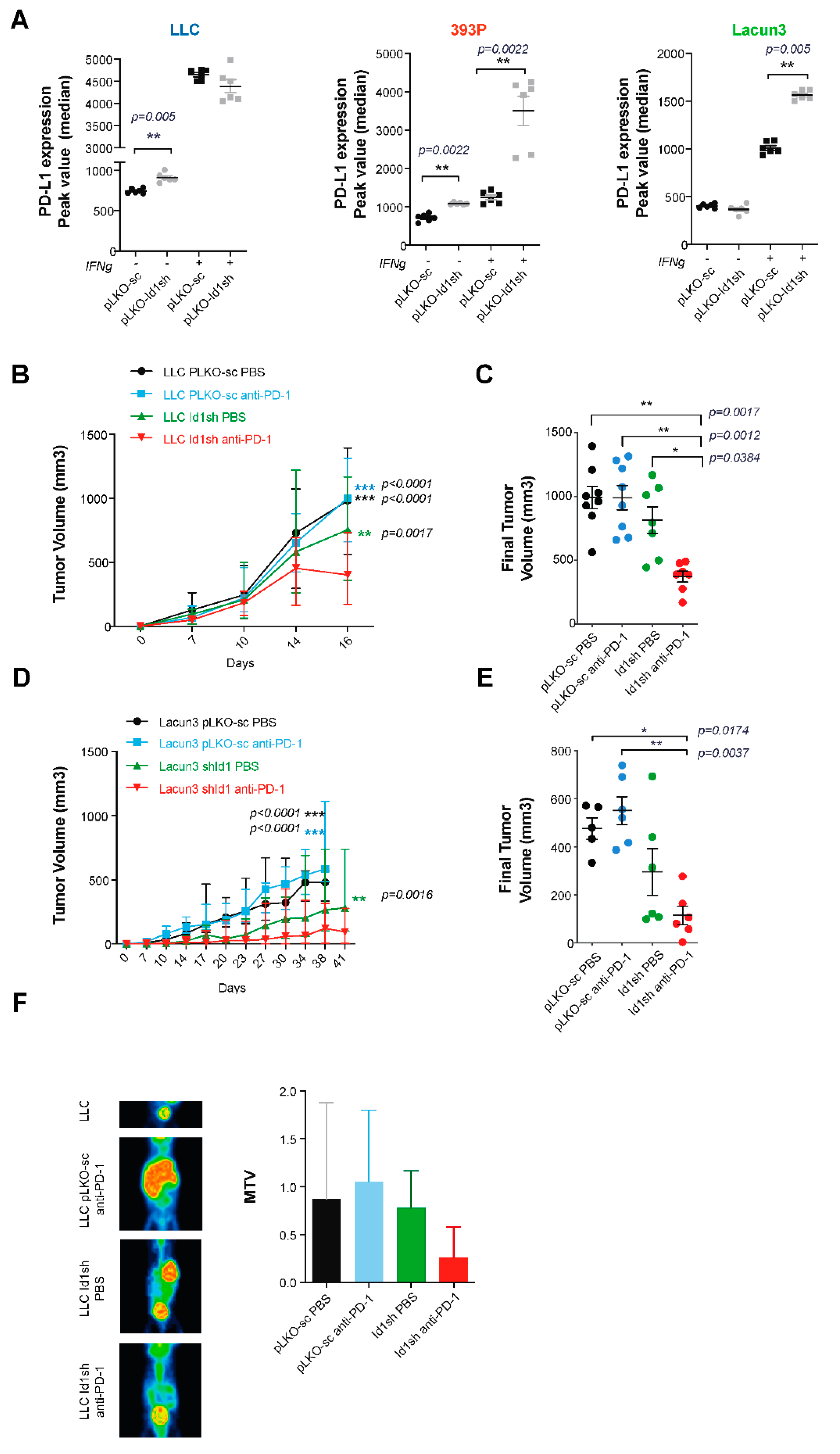
2.2. Up-Regulation of Surface PD-L1 Expression Occurs in Id1-Deficient KRAS Mutant LUAD Cells Exposed to IFN-γ
2.3. Combined Id1 Inhibition in Tumor Cells and PD-1 Blockade Impairs KRAS-Driven LUAD Growth In Vivo
2.4. Combined Id1 Inhibition in Tumor Cells and PD-1 Blockade Impairs Tumor Progression in a Clinically Relevant Metastasis Model
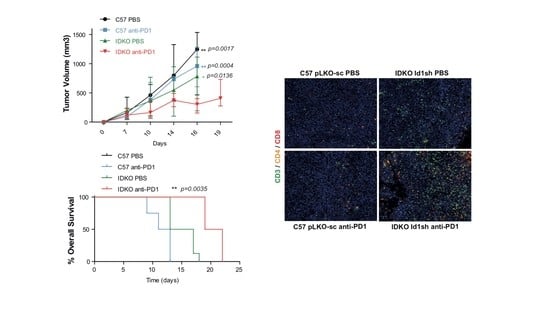
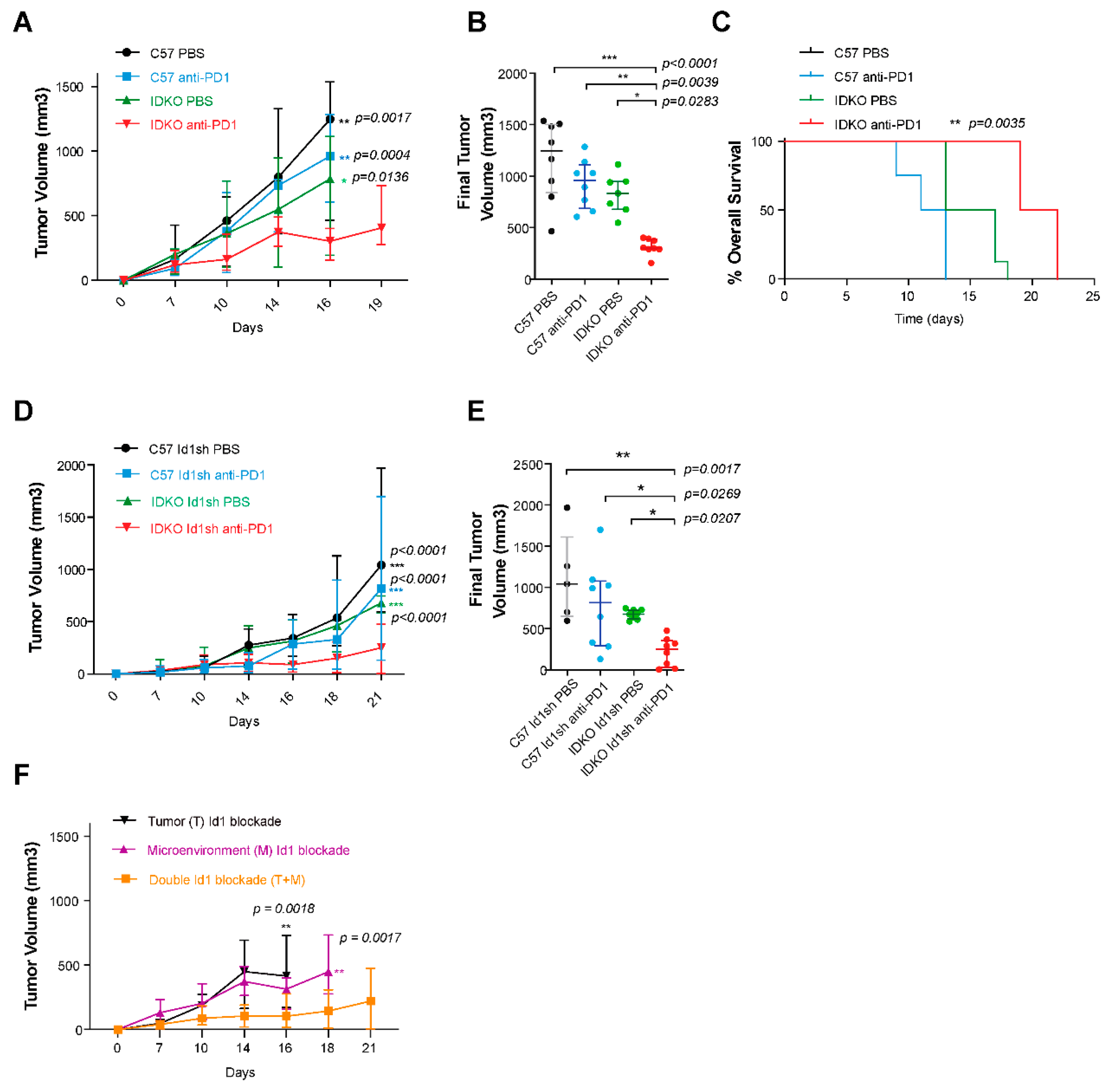
2.5. Id1 Inhibition in the Tumor Microenvironment Potentiates Response to Anti-PD-1 In Vivo and Improves Mouse Overall Survival
2.6. Combined Id1 Inhibition in both Tumor Cells and Tumor Microenvironment and PD-1/PD-L1 Blockade Impairs KRAS-Driven LUAD In Vivo Growth
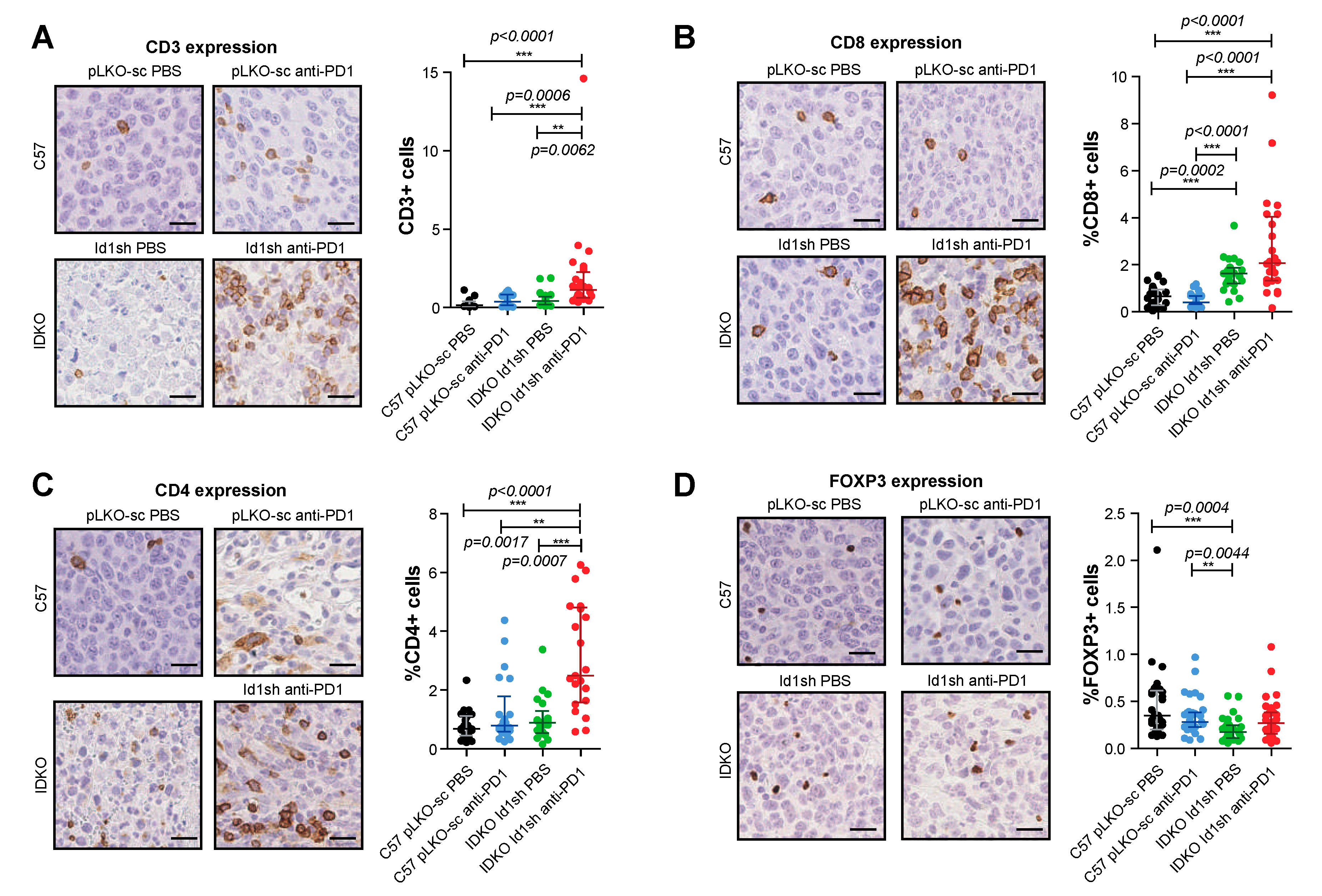
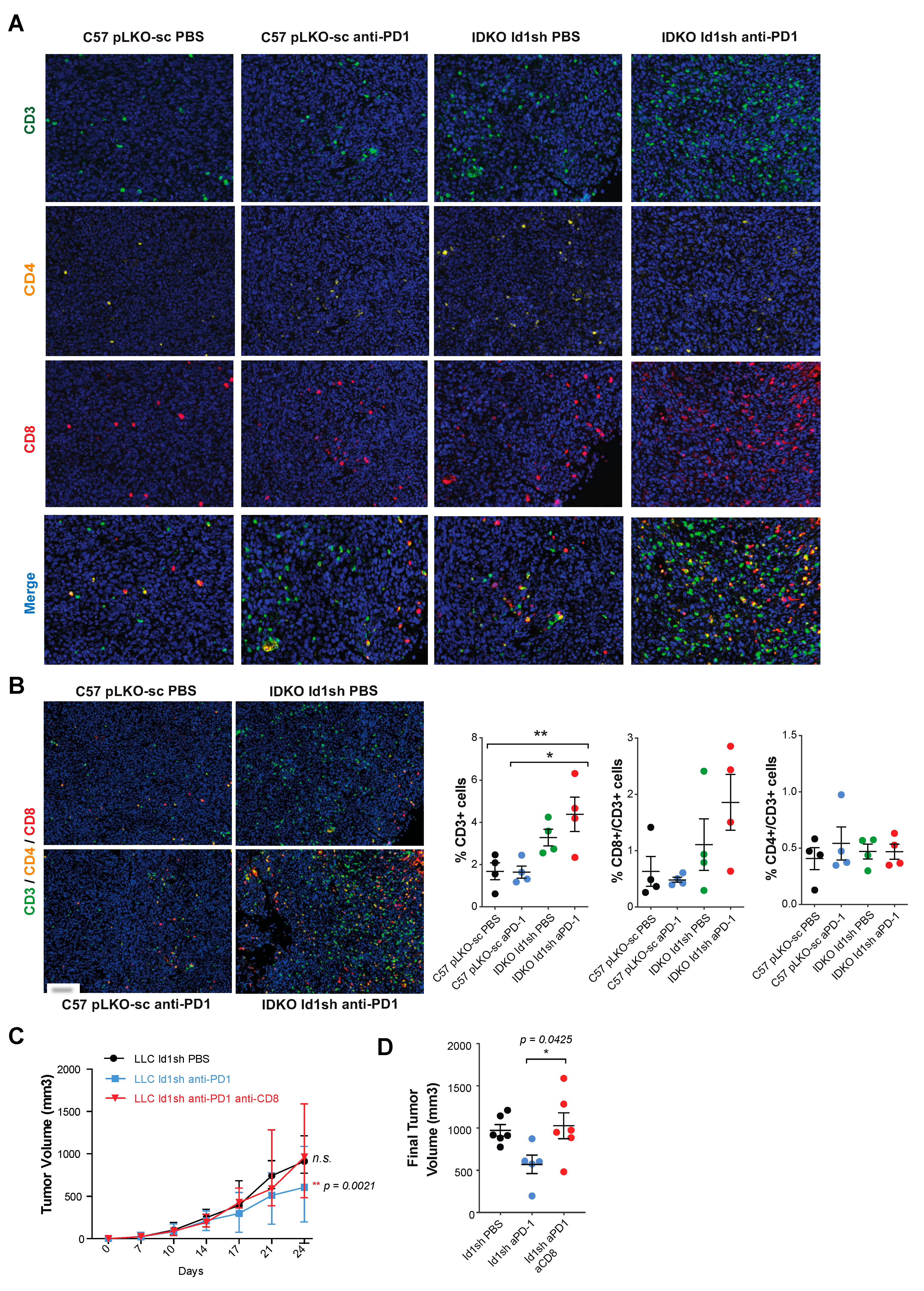
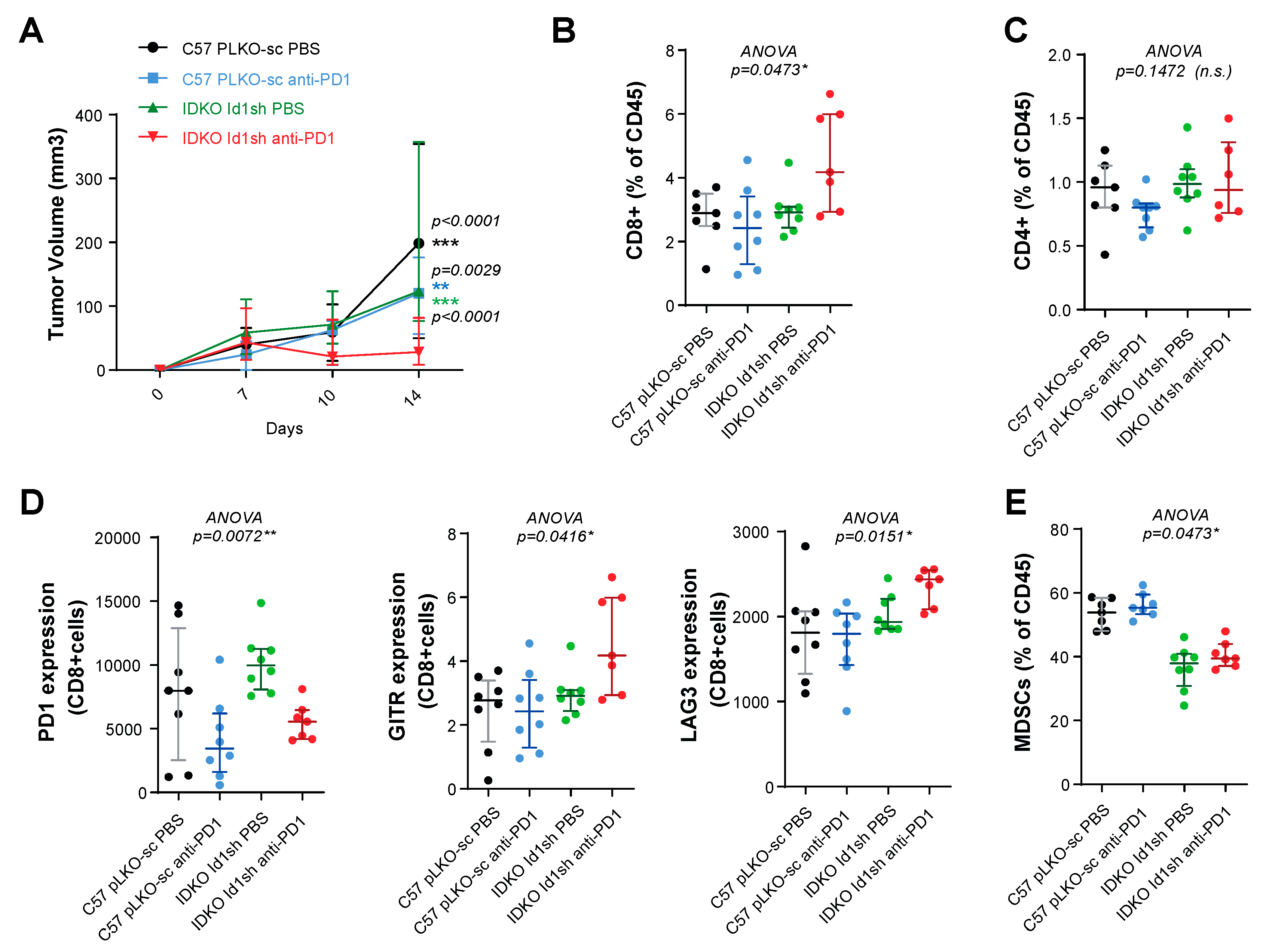
2.7. Combined Inhibition of Id1 and PD-1 Increases the Frequency of Effector T Cells within the Tumor Microenvironment
2.8. The Antitumor Activity of the Combined Blockade May Be Mediated by CD8+ T Cells
3. Discussion
4. Materials and Methods
4.1. Id1-PD-L1 Correlation
4.2. Cell Line Cultures
4.3. Silencing Id1 Expression
4.4. Plasmid and Retroviral Production
4.5. Western Blot Analyses
4.6. Cell Proliferation
4.7. Cell Cycle Assay
4.8. Co-Culture Assay
4.9. Murine Models
4.10. 18F-FDG mPET Study
4.11. Immunohistochemistry
4.12. Multiplex Quantification of CD3/CD4/CD8
4.13. Flow Cytometry Analyses
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Ahmedin, J. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellman, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA J. Am. Med. Assoc. 2014, 311, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; Van den Heuvel, M.M.; Ciuleanu, T.-E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; Von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet (London, England) 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abre, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfired, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Doroshow, D.B.; Sanmamed, M.F.; Hastings, K.; Politi, K.; Rimm, D.L.; Chen, L.; Melero, I.; Schalper, K.A.; Herbst, R.S. Immunotherapy in Non-Small Cell Lung Cancer: Facts and Hopes. Clin. Cancer Res. 2019, 25, 4592–4602. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Sznol, M. Diminished but not dead: Chemotherapy for the treatment of NSCLC. Lancet Oncol. 2016, 17, 1464–1465. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Capuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Shekarian, T.; Alcazar, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Combining cancer immunotherapy and targeted therapy. Curr. Opin. Immunol. 2013, 25, 291–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perk, J.; Iavarone, A.; Benezra, R. Id family of helix-loop-helix proteins in cancer. Nat. Rev. Cancer 2005, 5, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Benezra, R.; Iavarone, A. Cancer stem cells The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Publ. Gr. 2014, 14, 77–91. [Google Scholar]
- Román, M.; López, I.; Guruceaga, E.; Baraibar, I.; Ecay, M.; Collantes, M.; Nadal, E.; Vallejo, A.; Cadenas, S.; Echávarri-de Miguel, M.; et al. Inhibitor of differentiation-1 (Id1) sustains mutant KRAS-driven progression, maintenance, and metastasis of lung adenocarcinoma via regulation of a FOSL1 network. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Papaspyridonos, M.; Matei, I.; Huang, Y.; do Rosario Andre, M.; Brazier-Mitouart, H.; Waite, J.C.; Chan, A.S.; Kalter, J.; Ramos, I.; Wu, Q.; et al. Id1 suppresses anti-tumour immune responses and promotes tumour progression by impairing myeloid cell maturation. Nat. Commun. 2015, 6, 6840. [Google Scholar] [CrossRef]
- Ponz-Sarvisé, M.; Nguewa, P.A.; Pajares, M.J.; Agorreta, A.; Lozano, M.D.; Redrado, M.; Pío, R.; Behrens, C.; Wistuba, I.I.; García-Franco, C.E. Inhibitor of differentiation-1 as a novel prognostic factor in NSCLC patients with adenocarcinoma histology and its potential contribution to therapy resistance. Clin. Cancer Res. 2011, 17, 4155–4166. [Google Scholar] [CrossRef] [Green Version]
- Castañón, E.; Soltermann, A.; López, I.; Román, M.; Ecay, M.; Collantes, M.; Redrado, M.; Baraibar, I.; López-Picazo, J.M.; Rolfo, C.; et al. The inhibitor of differentiation-1 (Id1) enables lung cancer liver colonization through activation of an EMT program in tumor cells and establishment of the pre-metastatic niche. Cancer Lett. 2017, 402, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Hu, J.; Chen, F.; Lecomte, N.; Basnet, H.; David, C.J.; Witkin, M.D.; Allen, P.J.; Leach, S.D.; Hollmann, T.J.; et al. ID1 Mediates Escape from TGFβ Tumor Suppression in Pancreatic Cancer. Cancer Discov. 2020, 10, 142–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Wang, H.-C.; Yu, S.; Jin, R.; Tang, H.; Liu, Y.-F.; Ge, Q.; Sun, X.-H.; Zhang, Y. Id1 Expression Promotes T Regulatory Cell Differentiation by Facilitating TCR Costimulation. J. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Dong, J.; Li, B.; Lin, D.; Zhou, Q.; Huang, D. Advances in Targeted Therapy and Immunotherapy for Non-small Cell Lung Cancer Based on Accurate Molecular Typing. Front. Pharmacol. 2019, 10, 230. [Google Scholar] [CrossRef]
- Koopmans, I.; Hendriks, D.; Samplonius, D.F.; Van Ginkel, R.J.; Heskamp, S.; Wiesrstra, P.J.; Bremer, E.; Helfrich, W. A novel bispecific antibody for EGFR-directed blockade of the PD-1/PD-L1 immune checkpoint. Oncoimmunology 2018, 7, e1466016. [Google Scholar] [CrossRef]
- Castañón, E.; Rolfo, C.; Viñal, D.; López, I.; Fusco, J.P.; Santisteban, M.; Martín, P.; Zubiri, L.; Echeveste, J.; IGil-Bazo, I. Impact of epidermal growth factor receptor (EGFR) activating mutations and their targeted treatment in the prognosis of stage IV non-small cell lung cancer (NSCLC) patients harboring liver metastasis. J. Transl. Med. 2015, 13, 257. [Google Scholar] [CrossRef] [Green Version]
- García-Aranda, M.; Redondo, M. Targeting Protein Kinases to Enhance the Response to anti-PD-1/PD-L1 Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2296. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Gao, Z.; Li, X.; Dong, L.; Han, W.; Nie, J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget 2017, 8, 110693–110707. [Google Scholar] [CrossRef] [Green Version]
- Tepper, R.I.; Coffman, R.L.; Leder, P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science 1992, 257, 548–551. [Google Scholar] [PubMed]
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M.; et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J. Exp. Med. 1999, 190, 617–627. [Google Scholar]
- Schietinger, A.; Philip, M.; Liu, R.B.; Schreiber, K.; Schreiber, H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J. Exp. Med. 2010, 207, 2469–2477. [Google Scholar] [PubMed]
- Hung, K.; Hayashi RLafond-Walker, A.; Lowenstein, C.; Pardoll, D.; Levitsky, H. The Central Role of CD4+ T Cells in the Antitumor Immune Response. J. Exp. Med. 1998, 188, 2357–2368. [Google Scholar]
- Corthay, A.; Skovseth, D.K.; Lundin, K.U.; Røsjø, E.; Omholt, H.; Hofgaard, P.O.; Haraldsen, G.; Bogen, B. Primary Antitumor Immune Response Mediated by CD4+ T Cells. Immunity 2005, 22, 371–383. [Google Scholar]
- Curotto de Lafaille, M.A.; Lafaille, J.J. Natural and Adaptive Foxp3+ Regulatory T Cells: More of the Same or a Division of Labor? Immunity 2009, 30, 626–635. [Google Scholar]
- Ronchetti, S.; Zollo, O.; Bruscoli, S.; Agostini, M.; Bianchini, R.; Nocentini, G.; Ayroldi, E.; Riccardi, C. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur. J. Immunol. 2004, 34, 613–622. [Google Scholar]
- Ronchetti, S.; Nocentini, G.; Bianchini, R.; Krausz, L.T.; Migliorati, G.; Riccardi, C. Glucocorticoid-induced TNFR-related protein lowers the threshold of CD28 costimulation in CD8+ T cells. J. Immunol. 2007, 179, 5916–5926. [Google Scholar]
- Qi, Z.; Sun, X.-H. Hyperresponse to T-cell receptor signaling and apoptosis of Id1 transgenic thymocytes. Mol. Cell. Biol. 2004, 24, 7313–7323. [Google Scholar]
- Martínez, V.G.; Hernández-López, C.; Valencia, J.; Hidalgo, L.; Entrena, A.; Zapata, A.G.; Vicente, A.; Sacedón, R.; Varas, A. The canonical BMP signaling pathway is involved in human monocyte-derived dendritic cell maturation. Immunol. Cell Biol. 2011, 89, 610–618. [Google Scholar]
- Newman, J.H.; Augeri, D.J.; Nemoyer, R.; Malhotra, J.; Langenfeld, E.; Chesson, C.B.; Dobias, N.S.; Lee, M.J.; Tarabichi, S.; Jhawar, S.R.; et al. Novel bone morphogenetic protein receptor inhibitor JL5 suppresses tumor cell survival signaling and induces regression of human lung cancer. Oncogene 2018, 37, 3672–3685. [Google Scholar] [CrossRef]
- Melief, J.; Pico de Coaña, Y.; Maas, R.; Fennemann, F.-L.; Wolodarski, M.; Hansson, J.; Kiessling, R. High expression of ID1 in monocytes is strongly associated with phenotypic and functional MDSC markers in advanced melanoma. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Zhuang, Y. Orchestration of Invariant Natural Killer T Cell Development by E and Id Proteins. Crit. Rev. Immunol. 2015, 35, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Volpert, O.V.; Pili, R.; Sikder, H.A.; Nelius, T.; Zaichuk, T.; Morris, C.; Shiflett, C.B.; Devlin, M.K.; Conant, K.; Alani, R.M. Id1 regulates angiogenesis through transcriptional repression of thrombospondin-1. Cancer Cell 2002, 2, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Benezra, R.; Rafii, S.; Lyden, D. The Id proteins and angiogenesis. Oncogene 2001, 20, 8334–8341. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowicz, P.M.; Lima e Silva, R.; Ohnaka, M.; Lee, S.B.; Chin, Y.; Kulukian, A.; Chang, S.H.; Desai, B.; García Escolano, M.; Shah, R.; et al. A Small-Molecule Pan-Id Antagonist Inhibits Pathologic Ocular Neovascularization. Cell Rep. 2019, 29, 62–75.e7. [Google Scholar] [CrossRef] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Bleau, A.-M.; Freire, J.; Pajares, M.J.; Zudaire, I.; Antón, I.; Nistal-Villán, E.; Redrado, M.; Zandueta, C.; Garmendia, I.; Ajona, D.; et al. New syngeneic inflammatory-related lung cancer metastatic model harboring double KRAS/WWOX alterations. Int. J. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | IFNg Signature (Seiwert et al. 2016) | |||||||||||
| CXCL10 | HLA-DRA | CXCL9 | STAT1 | IDO1 | IFNg | |||||||
| cor | p-value | cor | p-value | cor | p-value | cor | p-value | cor | p-value | cor | p-value | |
| Id1 | −0.25 | 0.015 | −0.23 | 0.013 | −0.21 | 0.066 | −0.18 | 0.176 | −0.12 | 0.364 | −0.074 | 0.713 |
| Gene ID | Markers of Immune Cell Populations | |||||||||||
| CD4 | HLA-DRA | CD4 | STAT1 | CD4 | IFNg | |||||||
| cor | p-value | cor | p-value | cor | p-value | cor | p-value | cor | p-value | cor | p-value | |
| Id1 | −0.20 | 0.042 | −0.17 | 0.120 | −0.16 | 0.217 | −0.11 | 0.684 | −0.10 | 0.408 | −0.20 | 0.042 |
| Gene ID | Immune Checkpoints | |||||||||||
| CD274 (PD-L1) | HLA-DRA | CD274 (PD-L1) | STAT1 | CD274 (PD-L1) | IFNg | |||||||
| cor | p-value | cor | p-value | cor | p-value | cor | p-value | cor | p-value | cor | p-value | |
| Id1 | −0.35 | 0.0001 | −0.31 | 0.0003 | −0.26 | 0.007 | −0.113 | 0.568 | −0.12 | 0.364 | −0.074 | 0.713 |
| IHC Marker | C57 pLKO-sc PBS | C57 pLKO-sc anti-PD1 | IDKO Id1sh PBS | IDKO Id1sh anti-PD1 | ||||
|---|---|---|---|---|---|---|---|---|
| Median | Interquartile Range | Median | Interquartile Range | Median | Interquartile Range | Median | Interquartile Range | |
| CD3 | 0.158 | 0.089–0.388 | 0.368 | 0.147–0.840 | 0.413 | 0.205–0.711 | 1.136 | 0.626–2.267 |
| CD8 | 0.655 | 0.283–0.949 | 0.403 | 0.320–0.674 | 1.638 | 1.202–1.870 | 2.072 | 1.321–4.045 |
| CD4 | 0.680 | 0.448–1.128 | 0.788 | 0.588–1.783 | 0.887 | 0.539–1.297 | 2.490 | 1.580–4.810 |
| FOXP3 | 0.35 | 0.2–0.615 | 0.280 | 0.230–0.385 | 0.175 | 0.113–0.248 | 0.270 | 0.155–0.385 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baraibar, I.; Roman, M.; Rodríguez-Remírez, M.; López, I.; Vilalta, A.; Guruceaga, E.; Ecay, M.; Collantes, M.; Lozano, T.; Alignani, D.; et al. Id1 and PD-1 Combined Blockade Impairs Tumor Growth and Survival of KRAS-mutant Lung Cancer by Stimulating PD-L1 Expression and Tumor Infiltrating CD8+ T Cells. Cancers 2020, 12, 3169. https://doi.org/10.3390/cancers12113169
Baraibar I, Roman M, Rodríguez-Remírez M, López I, Vilalta A, Guruceaga E, Ecay M, Collantes M, Lozano T, Alignani D, et al. Id1 and PD-1 Combined Blockade Impairs Tumor Growth and Survival of KRAS-mutant Lung Cancer by Stimulating PD-L1 Expression and Tumor Infiltrating CD8+ T Cells. Cancers. 2020; 12(11):3169. https://doi.org/10.3390/cancers12113169
Chicago/Turabian StyleBaraibar, Iosune, Marta Roman, María Rodríguez-Remírez, Inés López, Anna Vilalta, Elisabeth Guruceaga, Margarita Ecay, María Collantes, Teresa Lozano, Diego Alignani, and et al. 2020. "Id1 and PD-1 Combined Blockade Impairs Tumor Growth and Survival of KRAS-mutant Lung Cancer by Stimulating PD-L1 Expression and Tumor Infiltrating CD8+ T Cells" Cancers 12, no. 11: 3169. https://doi.org/10.3390/cancers12113169