Indoxylsulfate, a Metabolite of the Microbiome, Has Cytostatic Effects in Breast Cancer via Activation of AHR and PXR Receptors and Induction of Oxidative Stress


 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
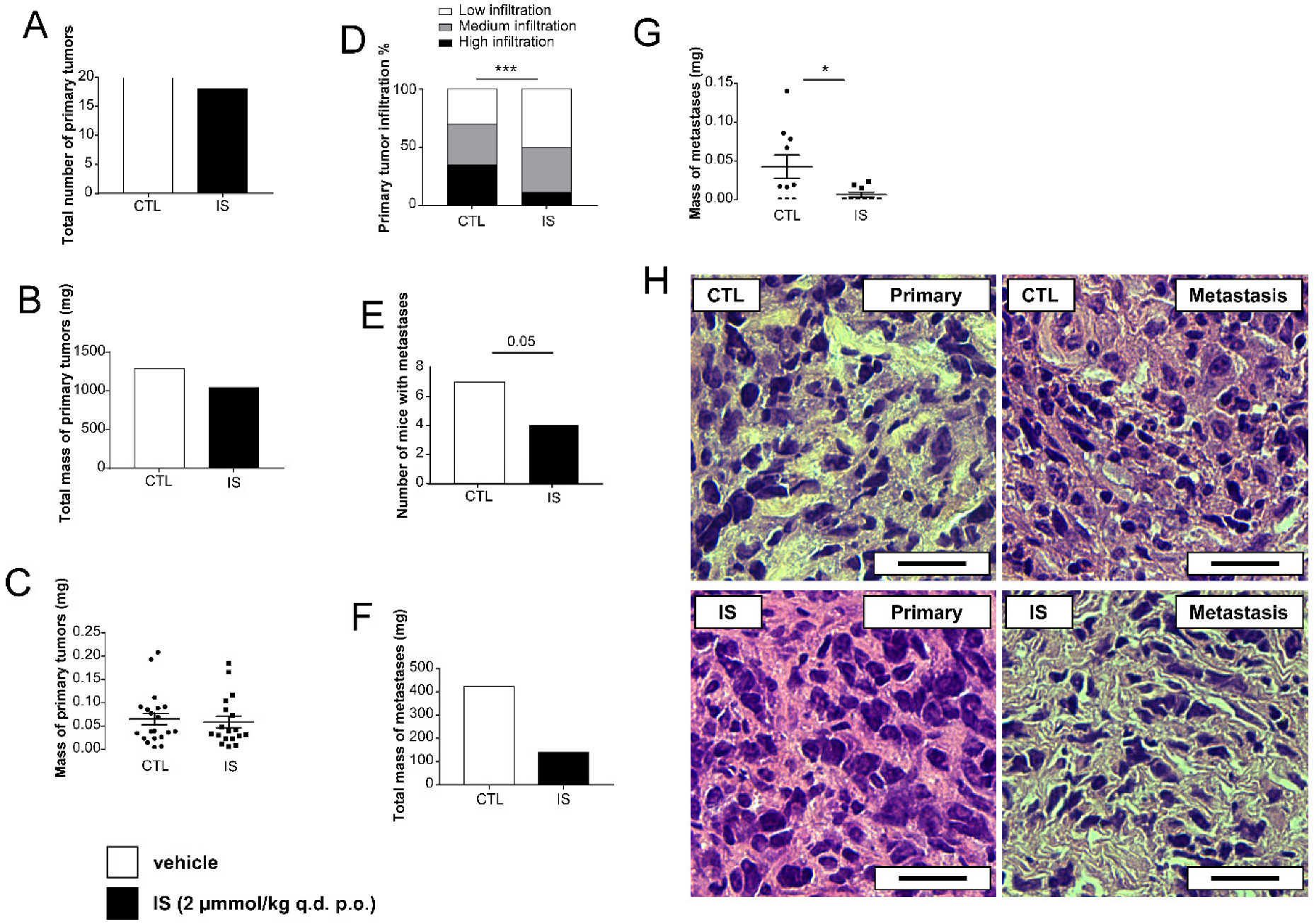
2.1. Indoxyl Sulfate Reduces the Severity of Breast Cancer In Vivo
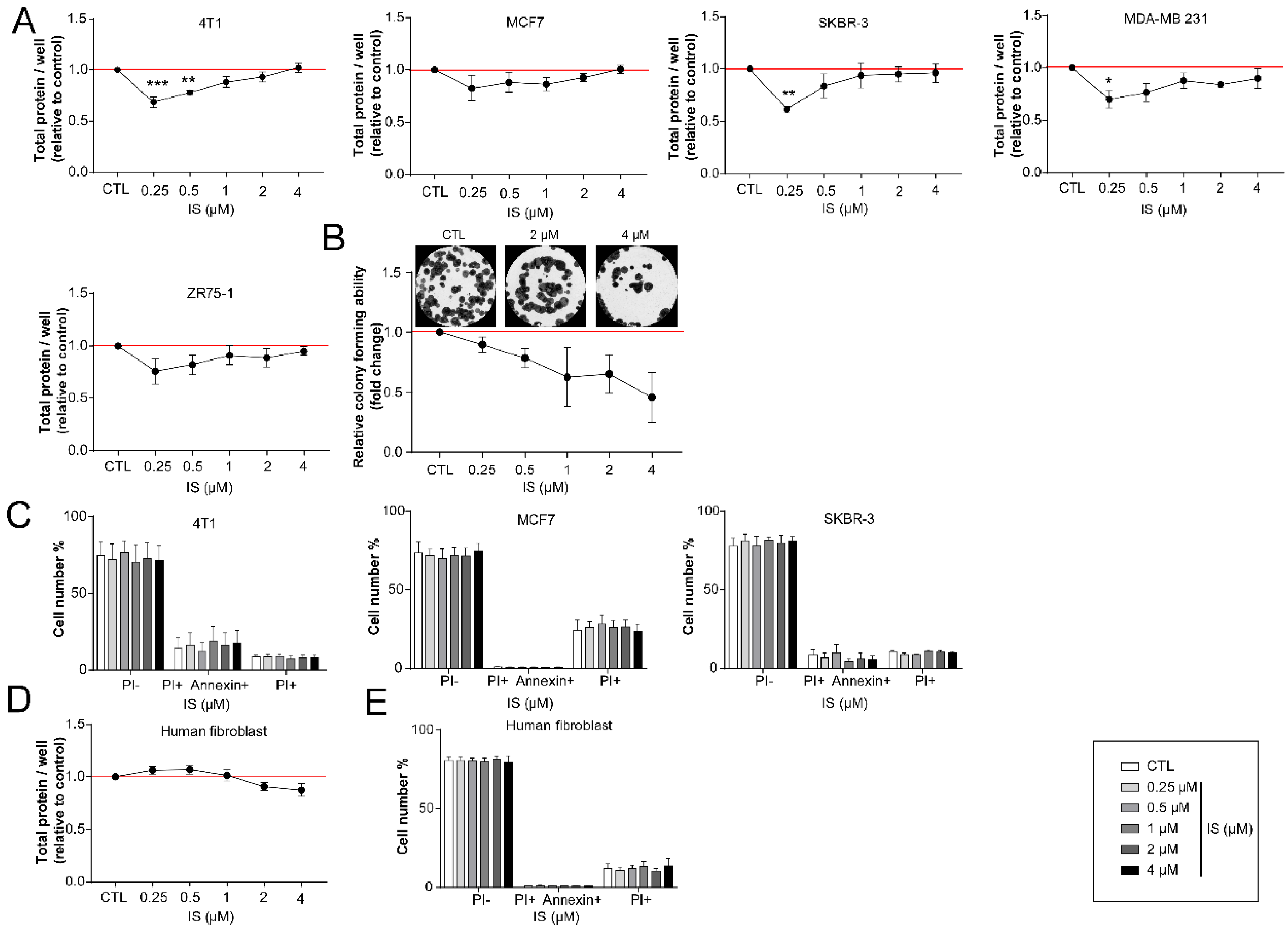
2.2. Indoxyl Sulfate Treatment Inhibits the Proliferation of Breast Cancer Cells
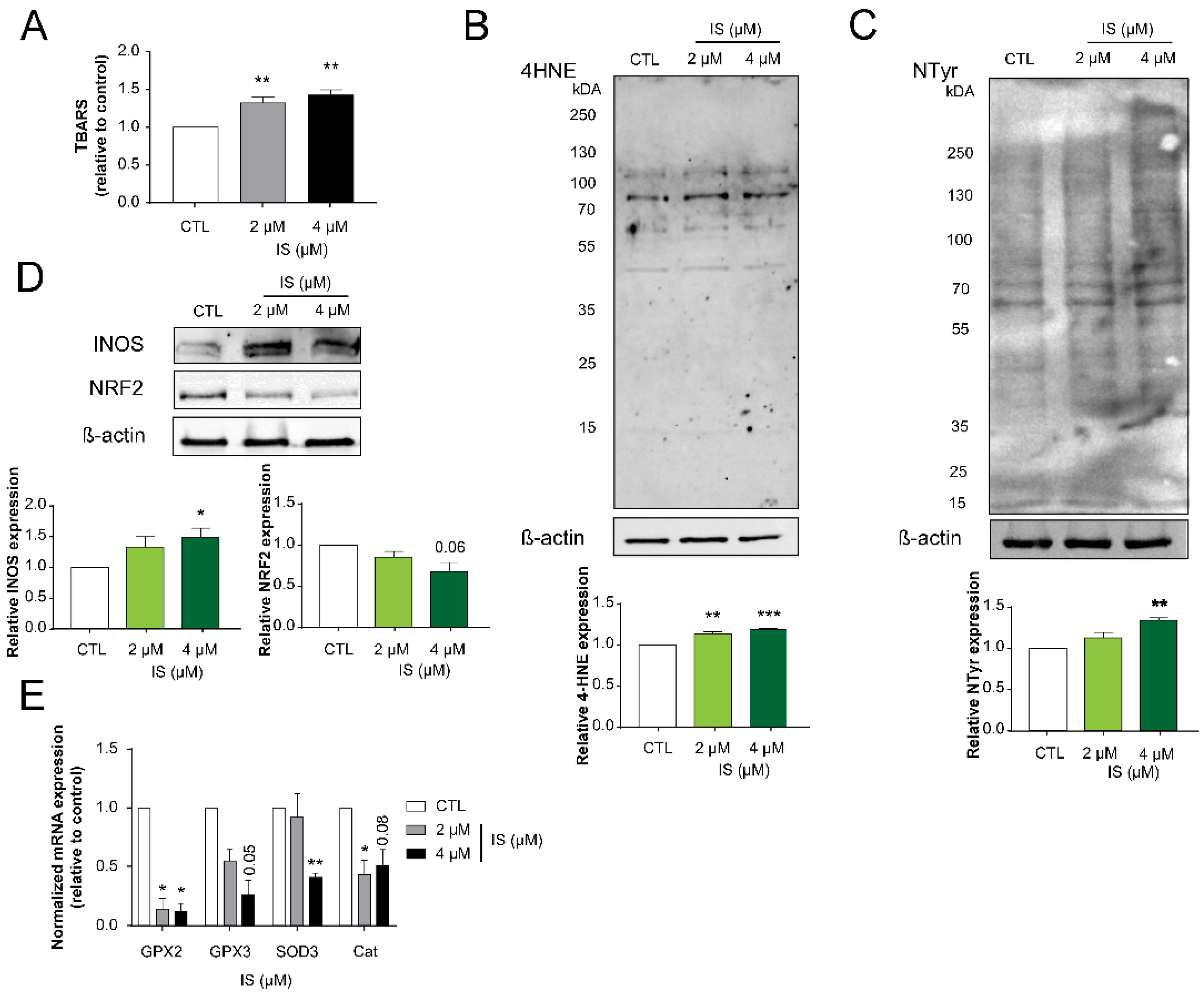
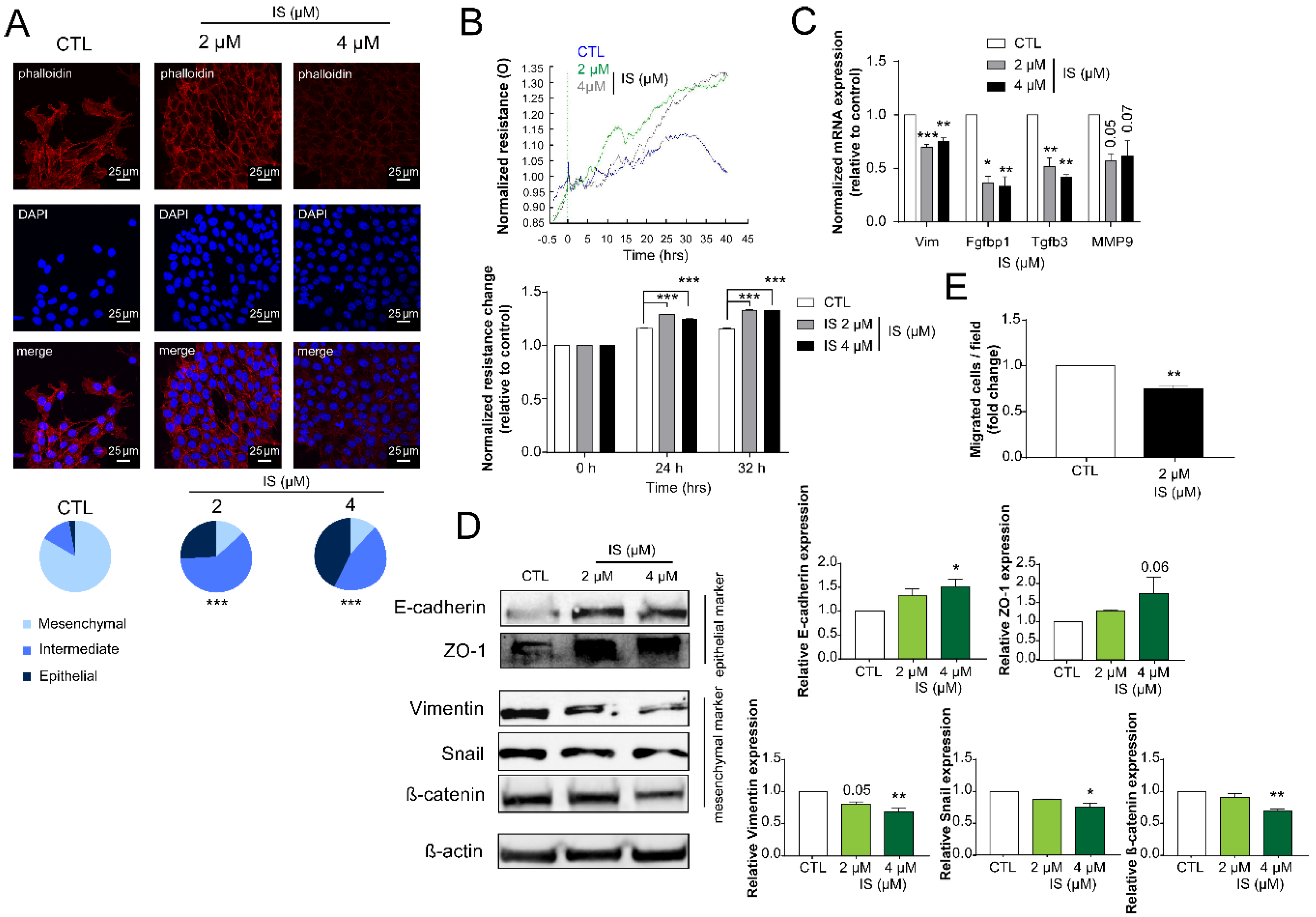
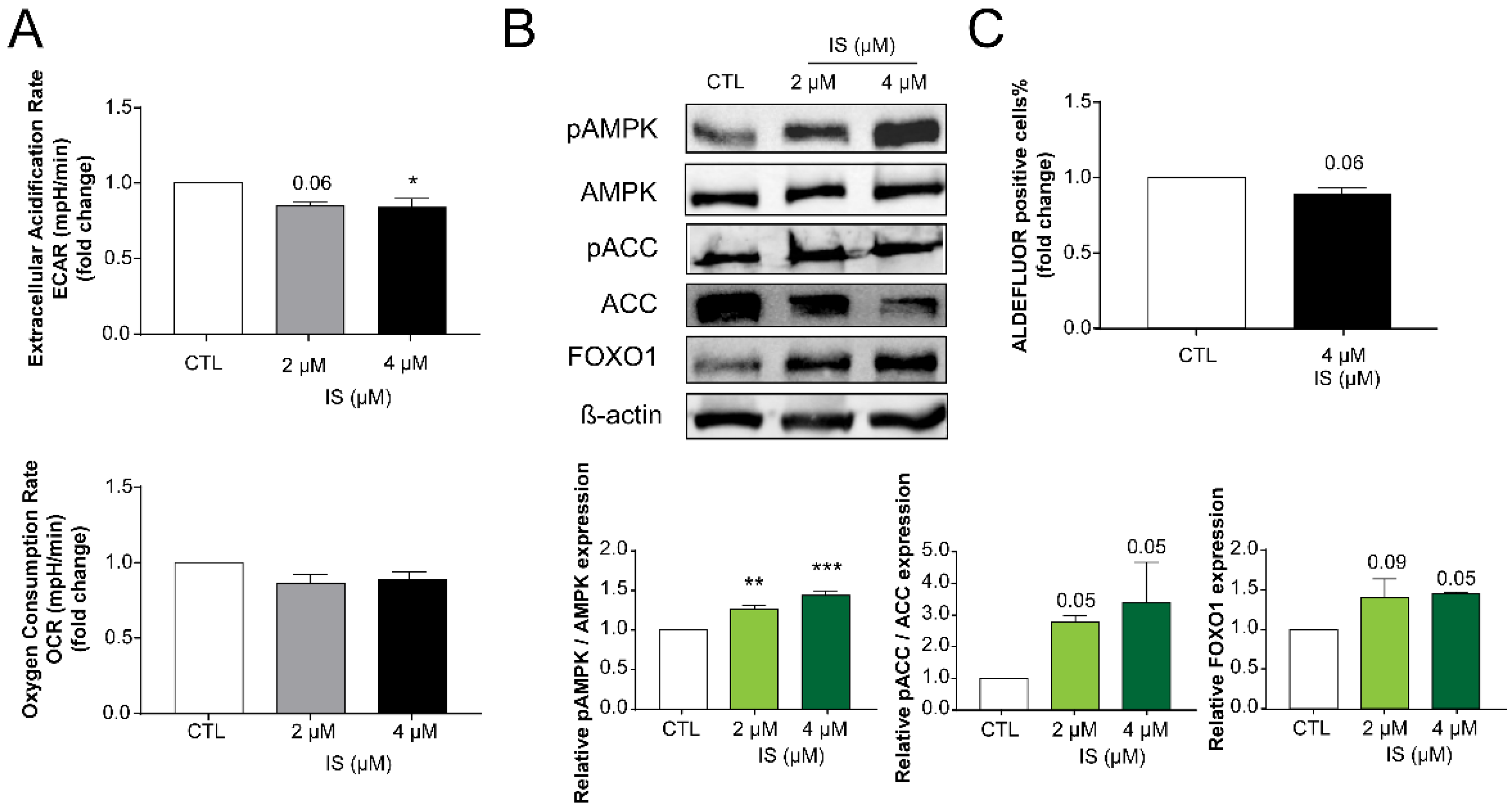
2.3. Indoxyl Sulfate Inhibits Numerous Hallmarks of Cancer
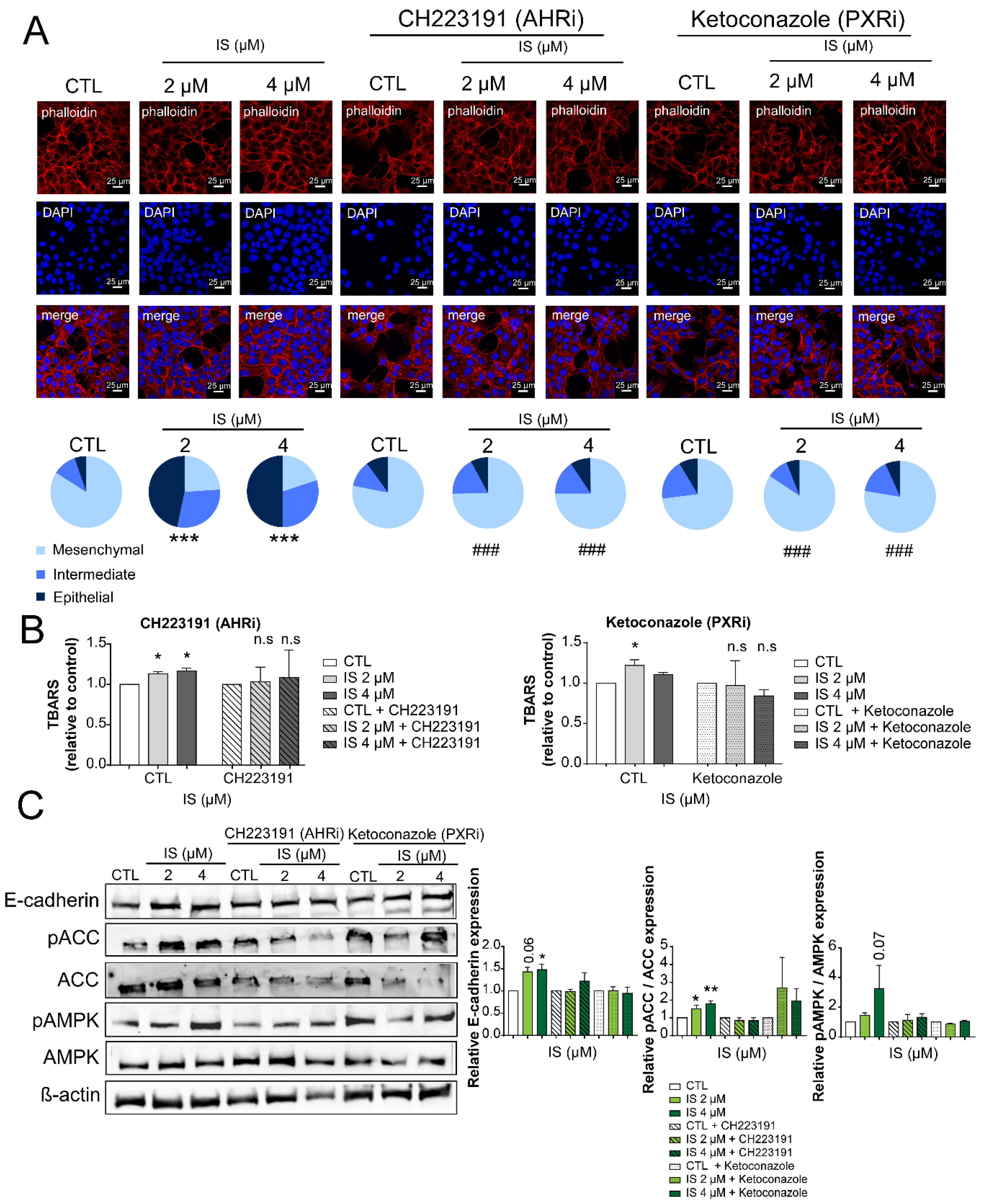
2.4. IS Exerts Its Effects through the AHR and PXR Receptors
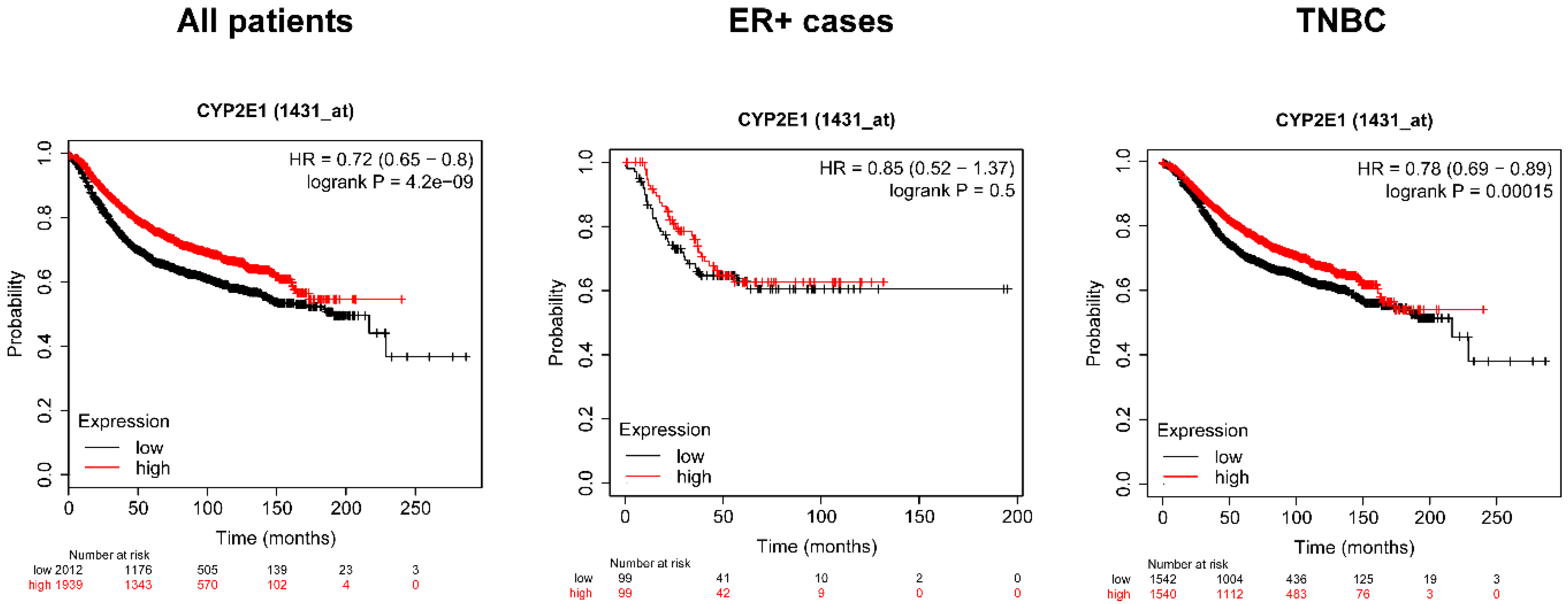
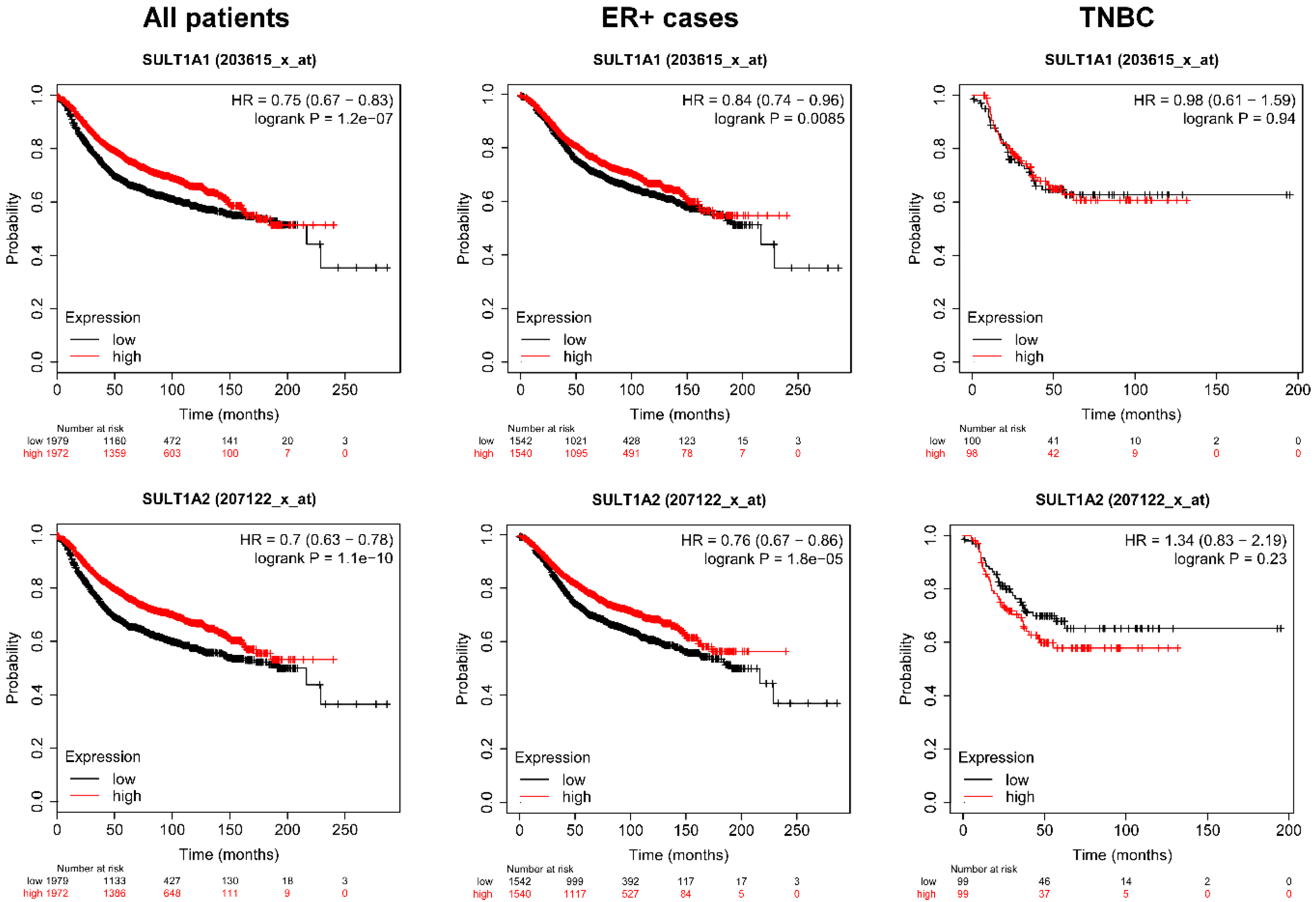
2.5. Higher Expression of the Isoforms of SULT and Cyp2e1 Correlate with Better Survival in Breast Cancer
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. In Vitro Cell Proliferation Assays
4.4. Detection of Cell Death
4.5. Electric Cell-Substrate Impedance Sensing (ECIS)
4.6. Immunocytochemistry
4.7. mRNA Preparation and Quantitation
4.8. Seahorse Metabolic Flux Analysis
4.9. Aldefluor Assay
4.10. SDS-PAGE and Western Blotting
4.11. Determination of Lipid Peroxidation
4.12. Invasion
4.13. Animal Study
4.14. Database Screening
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrman, B.J.; Feigelson, H.S.; Flores, R.; Gail, M.H.; Xu, X.; Ravel, J.; Goedert, J.J. Associations of the fecal microbiome with urinary estrogens and estrogen metabolites in postmenopausal women. J. Clin. Endocrinol. Metab. 2014, 99, 4632–4640. [Google Scholar] [CrossRef] [PubMed]
- Goedert, J.J.; Jones, G.; Hua, X.; Xu, X.; Yu, G.; Flores, R.; Falk, R.T.; Gail, M.H.; Shi, J.; Ravel, J.; et al. Investigation of the association between the fecal microbiota and breast cancer in postmenopausal women: A population-based case-control pilot study. J. Natl. Cancer Inst. 2015, 107, djv147. [Google Scholar] [CrossRef]
- Goedert, J.J.; Hua, X.; Bielecka, A.; Okayasu, I.; Milne, G.L.; Jones, G.S.; Fujiwara, M.; Sinha, R.; Wan, Y.; Xu, X.; et al. Postmenopausal breast cancer and oestrogen associations with the IgA-coated and IgA-noncoated faecal microbiota. Br. J. Cancer 2018, 23, 435. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.S.; Spencer Feigelson, H.; Falk, R.T.; Hua, X.; Ravel, J.; Yu, G.; Flores, R.; Gail, M.H.; Shi, J.; Xu, X.; et al. Mammographic breast density and its association with urinary estrogens and the fecal microbiota in postmenopausal women. PLoS ONE 2019, 14, e0216114. [Google Scholar] [CrossRef]
- Fruge, A.D.; Van der Pol, W.; Rogers, L.Q.; Morrow, C.D.; Tsuruta, Y.; Demark-Wahnefried, W. Fecal Akkermansia muciniphila Is Associated with Body Composition and Microbiota Diversity in Overweight and Obese Women with Breast Cancer Participating in a Presurgical Weight Loss Trial. J. Acad. Nutr. Diet. 2018, 9, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, T.H.; Michel, C.; Bard, J.M.; Dravet, F.; Nazih, H.; Bobin-Dubigeon, C. Intestinal Proportion of Blautia sp. is Associated with Clinical Stage and Histoprognostic Grade in Patients with Early-Stage Breast Cancer. Nutr. Cancer 2017, 69, 267–275. [Google Scholar] [CrossRef]
- Zhu, J.; Liao, M.; Yao, Z.; Liang, W.; Li, Q.; Liu, J.; Yang, H.; Ji, Y.; Wei, W.; Tan, A.; et al. Breast cancer in postmenopausal women is associated with an altered gut metagenome. Microbiome 2018, 6, 136. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Tian, T.; Wei, Z.; Shih, N.; Feldman, M.D.; Peck, K.N.; DeMichele, A.M.; Alwine, J.C.; Robertson, E.S. Distinct Microbial Signatures Associated With Different Breast Cancer Types. Front. Microbiol. 2018, 9, 951. [Google Scholar] [CrossRef] [Green Version]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar]
- Howe, C.; Kim, S.J.; Mitchell, J.; Im, E.; Kim, Y.S.; Kim, Y.S.; Rhee, S.H. Differential expression of tumor-associated genes and altered gut microbiome with decreased Akkermansia muciniphila confer a tumor-preventive microenvironment in intestinal epithelial Pten-deficient mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3746–3758. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Pierre, J.F.; Makowski, L.; Tolley, E.; Lyn-Cook, B.; Lu, L.; Vidal, G.; Starlard-Davenport, A. Distinct microbial communities that differ by race, stage, or breast-tumor subtype in breast tissues of non-Hispanic Black and non-Hispanic White women. Sci. Rep. 2019, 9, 11940. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Wei, Z.; Tan, F.; Peck, K.N.; Shih, N.; Feldman, M.; Rebbeck, T.R.; Alwine, J.C.; Robertson, E.S. Distinct microbiological signatures associated with triple negative breast cancer. Sci. Rep. 2015, 5, 15162. [Google Scholar] [CrossRef] [PubMed]
- Xuan, C.; Shamonki, J.M.; Chung, A.; Dinome, M.L.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial dysbiosis is associated with human breast cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, M.F.; Reina-Perez, I.; Astorga, J.M.; Rodriguez-Carrillo, A.; Plaza-Diaz, J.; Fontana, L. Breast Cancer and Its Relationship with the Microbiota. Int. J. Enviorn. Res. Public Health 2018, 15, 1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miko, E.; Vida, A.; Kovacs, T.; Ujlaki, G.; Trencsenyi, G.; Marton, J.; Sari, Z.; Kovacs, P.; Boratko, A.; Hujber, Z.; et al. Lithocholic acid, a bacterial metabolite reduces breast cancer cell proliferation and aggressiveness. Biochim. Biophys. Acta 2018, 1859, 958–974. [Google Scholar] [CrossRef]
- Kovács, T.; Mikó, E.; Vida, A.; Sebő, É.; Toth, J.; Csonka, T.; Boratkó, A.; Ujlaki, G.; Lente, G.; Kovács, P.; et al. Cadaverine, a metabolite of the microbiome, reduces breast cancer aggressiveness through trace amino acid receptors. Sci. Rep. 2019, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.H.; Tseng, C.; Vigen, C.; Yu, Y.; Cozen, W.; Garcia, A.A.; Spicer, D. Gut microbiome associations with breast cancer risk factors and tumor characteristics: A pilot study. Breast Cancer Res. Treat. 2020, 182, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, C.; Cummins, J.; Brackstone, M.; Macklaim, J.M.; Gloor, G.B.; Baban, C.K.; Scott, L.; O’Hanlon, D.M.; Burton, J.P.; Francis, K.P.; et al. Microbiota of human breast tissue. Appl. Environ. Microbiol. 2014, 80, 3007–3014. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.A.; Bashir, M.; Rivas, M.N.; Duvall, K.; Sieling, P.A.; Pieber, T.R.; Vaishampayan, P.A.; Love, S.M.; Lee, D.J. Characterization of the microbiome of nipple aspirate fluid of breast cancer survivors. Sci. Rep. 2016, 6, 28061. [Google Scholar] [CrossRef] [Green Version]
- Hieken, T.J.; Chen, J.; Hoskin, T.L.; Walther-Antonio, M.; Johnson, S.; Ramaker, S.; Xiao, J.; Radisky, D.C.; Knutson, K.L.; Kalari, K.R.; et al. The Microbiome of Aseptically Collected Human Breast Tissue in Benign and Malignant Disease. Sci. Rep. 2016, 6, 30751. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Altemus, J.; Niazi, F.; Green, H.; Calhoun, B.C.; Sturgis, C.; Grobmyer, S.R.; Eng, C. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget 2017, 8, 88122–88138. [Google Scholar] [CrossRef] [Green Version]
- Costantini, L.; Magno, S.; Albanese, D.; Donati, C.; Molinari, R.; Filippone, A.; Masetti, R.; Merendino, N. Characterization of human breast tissue microbiota from core needle biopsies through the analysis of multi hypervariable 16S-rRNA gene regions. Sci. Rep. 2018, 8, 16893. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Chen, B.; Yang, J.; Wang, J.; Zhu, D.; Meng, Q.; Zhang, L. Study of Microbiomes in Aseptically Collected Samples of Human Breast Tissue Using Needle Biopsy and the Potential Role of in situ Tissue Microbiomes for Promoting Malignancy. Front. Oncol. 2018, 8, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkup, B.M.; McKee, A.M.; Madgwick, M.; Price, C.A.; Dreger, S.A.; Makin, K.A.; Caim, S.; Gall, G.L.; Paveley, J.; Leclaire, C.; et al. Antibiotic-induced disturbances of the gut microbiota result in accelerated breast tumour growth via a mast cell-dependent pathway. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kirkup, B.; McKee, A.; Makin, K.; Paveley, J.; Caim, S.; Alcon-Giner, C.; Leclaire, C.; Dalby, M.; Le Gall, G.; Andrusaite, A.; et al. Perturbation of the gut microbiota by antibiotics results in accelerated breast tumour growth and metabolic dysregulation. Biorxiv. 2019. [Google Scholar] [CrossRef]
- Velicer, C.M.; Heckbert, S.R.; Lampe, J.W.; Potter, J.D.; Robertson, C.A.; Taplin, S.H. Antibiotic use in relation to the risk of breast cancer. JAMA 2004, 291, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamim, H.M.; Hanley, J.A.; Hajeer, A.H.; Boivin, J.F.; Collet, J.P. Risk of breast cancer in relation to antibiotic use. Pharm. Drug Saf. 2008, 17, 144–150. [Google Scholar] [CrossRef]
- Sergentanis, T.N.; Zagouri, F.; Zografos, G.C. Is antibiotic use a risk factor for breast cancer? A meta-analysis. Pharm. Drug Saf. 2010, 19, 1101–1107. [Google Scholar] [CrossRef]
- Satram-Hoang, S.; Moran, E.M.; Anton-Culver, H.; Burras, R.W.; Heimann, T.M.; Boggio, I.; Dykstra-Long, G.R.; Wood, P.A.; Zulka, R.; Hufnagel, G.; et al. A pilot study of male breast cancer in the Veterans Affairs healthcare system. J. Environ. Pathol. Toxicol. Oncol. 2010, 29, 235–244. [Google Scholar] [CrossRef]
- Wirtz, H.S.; Buist, D.S.; Gralow, J.R.; Barlow, W.E.; Gray, S.; Chubak, J.; Yu, O.; Bowles, E.J.; Fujii, M.; Boudreau, D.M. Frequent antibiotic use and second breast cancer events. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1588–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkrief, A.; Derosa, L.; Kroemer, G.; Zitvogel, L.; Routy, B. The negative impact of antibiotics on outcomes in cancer patients treated with immunotherapy: A new independent prognostic factor? Ann. Oncol. 2019, 30, 1572–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, G.D.; Oestreicher, N.; Chan, J.; Quesenberry, C.P., Jr.; Udaltsova, N.; Habel, L.A. Antibiotics and risk of breast cancer: Up to 9 years of follow-Up of 2.1 million women. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2102–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velicer, C.M.; Heckbert, S.R.; Rutter, C.; Lampe, J.W.; Malone, K. Association between antibiotic use prior to breast cancer diagnosis and breast tumour characteristics (United States). Cancer Causes Control 2006, 17, 307–313. [Google Scholar] [CrossRef]
- Aragon, F.; Carino, S.; Perdigon, G.; de Moreno de LeBlanc, A. The administration of milk fermented by the probiotic Lactobacillus casei CRL 431 exerts an immunomodulatory effect against a breast tumour in a mouse model. Immunobiology 2014, 219, 457–464. [Google Scholar] [CrossRef]
- Hassan, Z.; Mustafa, S.; Rahim, R.A.; Isa, N.M. Anti-breast cancer effects of live, heat-killed and cytoplasmic fractions of Enterococcus faecalis and Staphylococcus hominis isolated from human breast milk. Vitr. Cell Dev. Biol. Animal 2016, 52, 337–348. [Google Scholar] [CrossRef]
- Mendoza, L. Potential effect of probiotics in the treatment of breast cancer. Oncol. Rev. 2019, 13, 422. [Google Scholar] [CrossRef] [Green Version]
- Ranjbar, S.; Seyednejad, S.A.; Azimi, H.; Rezaeizadeh, H.; Rahimi, R. Emerging Roles of Probiotics in Prevention and Treatment of Breast Cancer: A Comprehensive Review of Their Therapeutic Potential. Nutr. Cancer 2019, 71, 1–12. [Google Scholar] [CrossRef]
- Viaud, S.; Daillere, R.; Boneca, I.G.; Lepage, P.; Pittet, M.J.; Ghiringhelli, F.; Trinchieri, G.; Goldszmid, R.; Zitvogel, L. Harnessing the intestinal microbiome for optimal therapeutic immunomodulation. Cancer Res. 2014, 74, 4217–4221. [Google Scholar] [CrossRef] [Green Version]
- Sári, Z.; Mikó, E.; Kovács, T.; Jankó, L.; Csonka, T.; Sebő, E.; Toth, J.; Tóth, D.; Árkosy, P.; Boratkó, A.; et al. Indolepropionic acid, a metabolite of the microbiome, has cytostatic properties in breast cancer by activating AHR and PXR receptors and inducing oxidative stress. Cancers 2020, 12, 2411. [Google Scholar] [CrossRef]
- Buchta Rosean, C.; Bostic, R.R.; Ferey, J.C.M.; Feng, T.Y.; Azar, F.N.; Tung, K.S.; Dozmorov, M.G.; Smirnova, E.; Bos, P.D.; Rutkowski, M.R. Preexisting Commensal Dysbiosis Is a Host-Intrinsic Regulator of Tissue Inflammation and Tumor Cell Dissemination in Hormone Receptor-Positive Breast Cancer. Cancer Res. 2019, 79, 3662–3675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Ayyoub, M.; Routy, B.; Kroemer, G. Microbiome and Anticancer Immunosurveillance. Cell 2016, 165, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergara, D.; Simeone, P.; Damato, M.; Maffia, M.; Lanuti, P.; Trerotola, M. The Cancer Microbiota: EMT and Inflammation as Shared Molecular Mechanisms Associated with Plasticity and Progression. J. Oncol. 2019, 1253727. [Google Scholar] [CrossRef] [PubMed]
- Kovács, P.; Csonka, T.; Kovács, T.; Sári, Z.; Ujlaki, G.; Sipos, A.; Karányi, Z.; Szeőcs, D.; Hegedűs, C.; Uray, K.; et al. Lithocholic acid, a metabolite of the microbiome, increases oxidative stress in breast cancer. Cancers 2019, 11, 1255. [Google Scholar] [CrossRef] [Green Version]
- Chiba, A.; Bawaneh, A.; Velazquez, C.; Clear, K.Y.J.; Wilson, A.S.; Howard-McNatt, M.; Levine, E.A.; Levi-Polyachenko, N.; Yates-Alston, S.A.; Diggle, S.P.; et al. Neoadjuvant Chemotherapy Shifts Breast Tumor Microbiota Populations to Regulate Drug Responsiveness and the Development of Metastasis. Mol. Cancer Res. 2019, 18, 1541–7786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingman, W.V. The Gut Microbiome: A New Player in Breast Cancer Metastasis. Cancer Res. 2019, 79, 3539–3541. [Google Scholar] [CrossRef] [Green Version]
- Miko, E.; Kovacs, T.; Sebo, E.; Toth, J.; Csonka, T.; Ujlaki, G.; Sipos, A.; Szabo, J.; Mehes, G.; Bai, P. Microbiome-Microbial Metabolome-Cancer Cell Interactions in Breast Cancer-Familiar, but Unexplored. Cells 2019, 8, 293. [Google Scholar] [CrossRef] [Green Version]
- Miko, E.; Vida, A.; Bai, P. Translational aspects of the microbiome-to be exploited. Cell Biol. Toxicol. 2016, 32, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Putluri, V.; Ambati, C.R.; Dorsey, T.H.; Putluri, N.; Ambs, S. Liver- and Microbiome-derived Bile Acids Accumulate in Human Breast Tumors and Inhibit Growth and Improve Patient Survival. Clin. Cancer Res. 2019, 25, 5972–5983. [Google Scholar] [CrossRef] [Green Version]
- Sári, Z.; Kovács, T.; Csonka, T.; Török, M.; Sebő, É.; Toth, J.; Tóth, D.; Mikó, E.; Kiss, B.; Szeőcs, D.; et al. Fecal expression of E. coli lysine decarboxylase (LdcC) is downregulated in E-cadherin negative lobular breast carcinoma. Physiol. Int. 2020, 107, 349–358. [Google Scholar] [CrossRef]
- Wilmanski, T.; Rappaport, N.; Earls, J.C.; Magis, A.T.; Manor, O.; Lovejoy, J.; Omenn, G.S.; Hood, L.; Gibbons, S.M.; Price, N.D. Blood metabolome predicts gut microbiome alpha-diversity in humans. Nat. Biotechnol. 2019, 37, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMoss, R.D.; Moser, K. Tryptophanase in diverse bacterial species. J. Bacteriol. 1969, 98, 167–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, M.T.; Carlson, J.R. Microbial metabolites of tryptophan in the intestinal tract with special reference to skatole. Am. J. Clin. Nutr. 1979, 32, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.C.; Dinatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H. Immune regulation by microbiome metabolites. Immunology 2018, 154, 220–229. [Google Scholar] [CrossRef]
- Auslander, N.; Yizhak, K.; Weinstock, A.; Budhu, A.; Tang, W.; Wang, X.W.; Ambs, S.; Ruppin, E. A joint analysis of transcriptomic and metabolomic data uncovers enhanced enzyme-metabolite coupling in breast cancer. Sci Rep. 2016, 6, 29662. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Lin, C.C.; Spasojevic, I.; Iversen, E.S.; Chi, J.T.; Marks, J.R. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 2014, 16, 415. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-N.; Wu, I.W.; Huang, Y.-F.; Peng, S.-Y.; Huang, Y.-C.; Ning, H.-C. Measuring serum total and free indoxyl sulfate and p-cresyl sulfate in chronic kidney disease using UPLC-MS/MS. J. Food Drug Anal. 2019, 27, 502–509. [Google Scholar] [CrossRef] [Green Version]
- Thirunavukkarasan, M.; Wang, C.; Rao, A.; Hind, T.; Teo, Y.R.; Siddiquee, A.A.; Goghari, M.A.I.; Kumar, A.P.; Herr, D.R. Short-chain fatty acid receptors inhibit invasive phenotypes in breast cancer cells. PLoS ONE 2017, 12, e0186334. [Google Scholar] [CrossRef]
- Salimi, V.; Shahsavari, Z.; Safizadeh, B.; Hosseini, A.; Khademian, N.; Tavakoli-Yaraki, M. Sodium butyrate promotes apoptosis in breast cancer cells through reactive oxygen species (ROS) formation and mitochondrial impairment. Lipids Health Dis. 2017, 16, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, M.M.; Meier, K.E. Free Fatty Acid Receptors and Cancer: From Nutrition to Pharmacology. Handb. Exp. Pharm. 2017, 236, 233–251. [Google Scholar]
- Yonezawa, T.; Kobayashi, Y.; Obara, Y. Short-chain fatty acids induce acute phosphorylation of the p38 mitogen-activated protein kinase/heat shock protein 27 pathway via GPR43 in the MCF-7 human breast cancer cell line. Cell Signal. 2007, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Bindels, L.B.; Porporato, P.; Dewulf, E.M.; Verrax, J.; Neyrinck, A.M.; Martin, J.C.; Scott, K.P.; Buc Calderon, P.; Feron, O.; Muccioli, G.G.; et al. Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br. J. Cancer 2012, 107, 1337–1344. [Google Scholar] [CrossRef] [Green Version]
- Garrido, P.; Shalaby, A.; Walsh, E.M.; Keane, N.; Webber, M.; Keane, M.M.; Sullivan, F.J.; Kerin, M.J.; Callagy, G.; Ryan, A.E.; et al. Impact of inducible nitric oxide synthase (iNOS) expression on triple negative breast cancer outcome and activation of EGFR and ERK signaling pathways. Oncotarget 2017, 8, 80568–80588. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.; Krishnan, A.; Sivasithambaram, N.D. Significance of twist and iNOS expression in human breast carcinoma. Mol. Cell Biochem. 2016, 412, 41–47. [Google Scholar] [CrossRef]
- Girotti, A.W. Upregulation of nitric oxide in tumor cells as a negative adaptation to photodynamic therapy. Lasers Surg. Med. 2018, 50, 590–598. [Google Scholar] [CrossRef]
- Davila-Gonzalez, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.M.; Kuhn, J.G.; Li, W.F.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; et al. Pharmacological Inhibition of NOS Activates ASK1/JNK Pathway Augmenting Docetaxel-Mediated Apoptosis in Triple-Negative Breast Cancer. Clin. Cancer Res. 2018, 24, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Walsh, E.M.; Keane, M.M.; Wink, D.A.; Callagy, G.; Glynn, S.A. Review of Triple Negative Breast Cancer and the Impact of Inducible Nitric Oxide Synthase on Tumor Biology and Patient Outcomes. Crit. Rev. Oncog. 2016, 21, 333–351. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotgia, F.; Fiorillo, M.; Lisanti, M.P. Hallmarks of the cancer cell of origin: Comparisons with “energetic” cancer stem cells (e-CSCs). Aging 2019, 11, 1065–1068. [Google Scholar] [CrossRef]
- Huang, H.; Wang, H.; Sinz, M.; Zoeckler, M.; Staudinger, J.; Redinbo, M.R.; Teotico, D.G.; Locker, J.; Kalpana, G.V.; Mani, S. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene 2007, 26, 258–268. [Google Scholar] [CrossRef] [Green Version]
- Cancilla, M.T.; He, M.M.; Viswanathan, N.; Simmons, R.L.; Taylor, M.; Fung, A.D.; Cao, K.; Erlanson, D.A. Discovery of an Aurora kinase inhibitor through site-specific dynamic combinatorial chemistry. Bioorg. Med. Chem. Lett. 2008, 18, 3978–3981. [Google Scholar] [CrossRef]
- Lanczky, A.; Nagy, A.; Bottai, G.; Munkacsy, G.; Szabo, A.; Santarpia, L.; Gyorffy, B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar] [CrossRef]
- Goldberg, A.A.; Beach, A.; Davies, G.F.; Harkness, T.A.; Leblanc, A.; Titorenko, V.I. Lithocholic bile acid selectively kills neuroblastoma cells, while sparing normal neuronal cells. Oncotarget 2011, 2, 761–782. [Google Scholar] [CrossRef] [Green Version]
- Sreekanth, V.; Bansal, S.; Motiani, R.K.; Kundu, S.; Muppu, S.K.; Majumdar, T.D.; Panjamurthy, K.; Sengupta, S.; Bajaj, A. Design, synthesis, and mechanistic investigations of bile acid-tamoxifen conjugates for breast cancer therapy. Bioconjug. Chem. 2013, 24, 1468–1484. [Google Scholar] [CrossRef]
- Gafar, A.A.; Draz, H.M.; Goldberg, A.A.; Bashandy, M.A.; Bakry, S.; Khalifa, M.A.; AbuShair, W.; Titorenko, V.I.; Sanderson, J.T. Lithocholic acid induces endoplasmic reticulum stress, autophagy and mitochondrial dysfunction in human prostate cancer cells. PeerJ 2016, 4, e2445. [Google Scholar] [CrossRef]
- Luu, T.H.; Bard, J.M.; Carbonnelle, D.; Chaillou, C.; Huvelin, J.M.; Bobin-Dubigeon, C.; Nazih, H. Lithocholic bile acid inhibits lipogenesis and induces apoptosis in breast cancer cells. Cell Oncol. 2018, 41, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Boursi, B.; Mamtani, R.; Haynes, K.; Yang, Y.X. Recurrent antibiotic exposure may promote cancer formation--Another step in understanding the role of the human microbiota? Eur. J. Cancer 2015, 51, 2655–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, T.M.; Vitolins, M.Z.; Cook, K.L. From the Table to the Tumor: The Role of Mediterranean and Western Dietary Patterns in Shifting Microbial-Mediated Signaling to Impact Breast Cancer Risk. Nutrients 2019, 11, 2565. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, D.G.; Nadal-Serrano, M.; Torrens-Mas, M.; Valle, A.; Oliver, J.; Roca, P. UCP2 inhibition sensitizes breast cancer cells to therapeutic agents by increasing oxidative stress. Free Radic. Biol. Med. 2015, 86, 67–77. [Google Scholar] [CrossRef]
- Smolková, K.; Mikó, E.; Kovács, T.; Leguina-Ruzzi, A.; Sipos, A.; Bai, P. NRF2 in regulating cancer metabolism. Antioxid Redox Signal. 2020. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Kai, K.; Arima, Y.; Kamiya, T.; Saya, H. Breast cancer stem cells. Breast Cancer 2010, 17, 80–85. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Sanchez-Rovira, P.; Ramirez-Tortosa, M.C.; Ramirez-Tortosa, C.L.; Granados-Principal, S.; Lorente, J.A.; Quiles, J.L. Free radicals in breast carcinogenesis, breast cancer progression and cancer stem cells. Biological bases to develop oxidative-based therapies. Crit. Rev. Oncol. Hematol. 2011, 80, 347–368. [Google Scholar] [CrossRef]
- Sotgia, F.; Ozsvari, B.; Fiorillo, M.; De Francesco, E.M.; Bonuccelli, G.; Lisanti, M.P. A mitochondrial based oncology platform for targeting cancer stem cells (CSCs): MITO-ONC-RX. Cell Cycle 2018, 17, 2091–2100. [Google Scholar] [CrossRef] [Green Version]
- Caruso, J.A.; Campana, R.; Wei, C.; Su, C.H.; Hanks, A.M.; Bornmann, W.G.; Keyomarsi, K. Indole-3-carbinol and its N-alkoxy derivatives preferentially target ERalpha-positive breast cancer cells. Cell Cycle 2014, 13, 2587–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajra, S.; Patra, A.R.; Basu, A.; Saha, P.; Bhattacharya, S. Indole-3-Carbinol (I3C) enhances the sensitivity of murine breast adenocarcinoma cells to doxorubicin (DOX) through inhibition of NF-kappabeta, blocking angiogenesis and regulation of mitochondrial apoptotic pathway. Chem. Biol. Interact. 2018, 290, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial beta-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.D. Mechanisms of estrogen carcinogenesis: The role of E2/E1-quinone metabolites suggests new approaches to preventive intervention—A review. Steroids 2015, 99, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Danaceau, J.P.; Anderson, G.M.; McMahon, W.M.; Crouch, D.J. A liquid chromatographic-tandem mass spectrometric method for the analysis of serotonin and related indoles in human whole blood. J. Anal. Toxicol. 2003, 27, 440–444. [Google Scholar] [CrossRef] [Green Version]
- Rosas, H.D.; Doros, G.; Bhasin, S.; Thomas, B.; Gevorkian, S.; Malarick, K.; Matson, W.; Hersch, S.M. A systems-level “misunderstanding”: The plasma metabolome in Huntington’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 756–768. [Google Scholar] [CrossRef]
- Fodor, T.; Szanto, M.; Abdul-Rahman, O.; Nagy, L.; Der, A.; Kiss, B.; Bai, P. Combined Treatment of MCF-7 Cells with AICAR and Methotrexate, Arrests Cell Cycle and Reverses Warburg Metabolism through AMP-Activated Protein Kinase (AMPK) and FOXO1. PLoS ONE 2016, 11, e0150232. [Google Scholar] [CrossRef] [Green Version]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. Bmc Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Szántó, M.; Brunyánszki, A.; Márton, J.; Vámosi, G.; Nagy, L.; Fodor, T.; Kiss, B.; Virag, L.; Gergely, P.; Bai, P. Deletion of PARP-2 induces hepatic cholesterol accumulation and decrease in HDL levels. Biochem. Biophys. Acta Mol. Basis Dis. 2014, 1842, 594–602. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Wu, Z.; Hang, S.; Zhu, W.; Wu, G. Amino acid metabolism in intestinal bacteria and its potential implications for mammalian reproduction. Mol. Hum. Reprod. 2015, 21, 389–409. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, M.; Peiris-Pages, M.; Sanchez-Alvarez, R.; Bartella, L.; Di Donna, L.; Dolce, V.; Sindona, G.; Sotgia, F.; Cappello, A.R.; Lisanti, M.P. Bergamot natural products eradicate cancer stem cells (CSCs) by targeting mevalonate, Rho-GDI-signalling and mitochondrial metabolism. Biochim. Biophys. Acta 2018, 4, 30061–30066. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Marton, J.; Vida, A.; Kis, G.; Bokor, E.; Kun, S.; Gonczi, M.; Docsa, T.; Toth, A.; Antal, M.; et al. Glycogen phosphorylase inhibition improves beta cell function. Br. J. Pharm. 2018, 175, 301–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabley, J.G.; Pacher, P.; Bai, P.; Wallace, R.; Goonesekera, S.; Virag, L.; Southan, G.J.; Szabo, C. Suppression of intestinal polyposis in Apcmin/+ mice by targeting the nitric oxide or poly(ADP-ribose) pathways. Mutat. Res. 2004, 548, 107–116. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Group | Probe | Time (Months) | HR | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 50 | 100 | 150 | 200 | 250 | ||||
| All cancers | CYP2E1 | low | 2012 | 1176 | 505 | 139 | 23 | 3 | 0.72 |
| 1431_at | high | 1939 | 1343 | 570 | 102 | 4 | 0 | (0.65–0.8) | |
| CYP2E1 | low | 1980 | 1177 | 482 | 133 | 16 | 2 | 0.68 | |
| 209975_at | high | 1971 | 1342 | 593 | 108 | 11 | 1 | (0.61–0.76) | |
| CYP2E1 | low | 2091 | 1360 | 633 | 160 | 23 | 2 | 0.96 | |
| 209976_s_at | high | 1860 | 1159 | 442 | 81 | 4 | 1 | (0.86–1.07) | |
| CYP2E1 | low | 1984 | 1171 | 487 | 113 | 12 | 0 | 0.78 | |
| 222100_at | high | 1967 | 1348 | 588 | 128 | 15 | 3 | (0.7–0.87) | |
| SULT1A1 | low | 1979 | 1160 | 472 | 141 | 20 | 3 | 0.75 | |
| 203615_x_at | high | 1972 | 1359 | 603 | 100 | 7 | 0 | (0.67–0.83) | |
| SULT1A1 | low | 1976 | 1150 | 463 | 139 | 22 | 3 | 0.77 | |
| 215299_x_at | high | 1975 | 1369 | 612 | 102 | 5 | 0 | (0.69–0.86) | |
| SULT1A2 | low | 1979 | 1133 | 427 | 130 | 18 | 3 | 0.7 | |
| 207122_x_at | high | 1972 | 1386 | 648 | 111 | 9 | 0 | (0.63–0.78) | |
| SULT1A2 | low | 1976 | 1149 | 440 | 133 | 18 | 3 | 0.73 | |
| 211385_x_at | high | 1975 | 1370 | 635 | 108 | 9 | 0 | (0.66–0.82) | |
| ER+ cases | CYP2E1 | low | 1542 | 1004 | 436 | 125 | 19 | 3 | 0.78 |
| 1431_at | high | 1540 | 1112 | 483 | 76 | 3 | 0 | (0.69–0.89) | |
| CYP2E1 | low | 1542 | 1019 | 430 | 119 | 15 | 2 | 0.74 | |
| 209975_at | high | 1540 | 1097 | 489 | 82 | 7 | 1 | (0.65–0.85) | |
| CYP2E1 | low | 1622 | 1125 | 531 | 130 | 19 | 2 | 0.94 | |
| 209976_s_at | high | 1460 | 991 | 388 | 71 | 3 | 1 | (0.83–1.07 | |
| CYP2E1 | low | 1606 | 1052 | 451 | 106 | 13 | 1 | 0.82 | |
| 222100_at | high | 1476 | 1064 | 468 | 95 | 9 | 2 | (0.72–0.93) | |
| SULT1A1 | low | 1542 | 1021 | 428 | 123 | 15 | 3 | 0.84 | |
| 203615_x_at | high | 1540 | 1095 | 491 | 78 | 7 | 0 | (0.74–0.96) | |
| SULT1A1 | low | 1542 | 1009 | 421 | 123 | 17 | 3 | 0.87 | |
| 215299_x_at | high | 1540 | 1107 | 498 | 78 | 5 | 0 | (0.76–0.98) | |
| SULT1A2 | low | 1542 | 999 | 392 | 117 | 17 | 3 | 0.76 | |
| 207122_x_at | high | 1540 | 1117 | 527 | 84 | 5 | 0 | (0.67–0.86) | |
| SULT1A2 | low | 1541 | 1004 | 390 | 111 | 16 | 3 | 0.81 | |
| 211385_x_at | high | 1541 | 1112 | 529 | 90 | 6 | 0 | (0.71–0.92) | |
| Triple negative cases | CYP2E1 | low | 99 | 41 | 10 | 2 | 0 | 0.85 | |
| 1431_at | high | 99 | 42 | 9 | 0 | 0 | (0.52–1.37) | ||
| CYP2E1 | low | 100 | 46 | 6 | 2 | 0 | 0.96 | ||
| 209975_at | high | 98 | 37 | 13 | 0 | 0 | (0.59–1.55) | ||
| CYP2E1 | low | 99 | 33 | 5 | 0 | 0 | 0.8 | ||
| 209976_s_at | high | 99 | 50 | 14 | 2 | 0 | (0.49–1.3) | ||
| CYP2E1 | low | 99 | 42 | 12 | 2 | 0 | 1.17 | ||
| 222100_at | high | 99 | 41 | 7 | 0 | 0 | (0.72–1.9) | ||
| SULT1A1 | low | 100 | 41 | 10 | 2 | 0 | 0.98 | ||
| 203615_x_at | high | 98 | 42 | 9 | 0 | 0 | (0.61–1.59) | ||
| SULT1A1 | low | 99 | 40 | 5 | 2 | 0 | 1.18 | ||
| 215299_x_at | high | 99 | 43 | 14 | 0 | 0 | (0.72–1.91) | ||
| SULT1A2 | low | 99 | 46 | 14 | 2 | 0 | 1.34 | ||
| 207122_x_at | high | 99 | 37 | 5 | 0 | 0 | (0.83–2.19) | ||
| SULT1A2 | low | 99 | 42 | 12 | 2 | 0 | 1.06 | ||
| 211385_x_at | high | 99 | 41 | 7 | 0 | 0 | (0.66–1.72) | ||
| Patient Group | CYP2E1 (1431_at) | CYP2E1 (209975_at) | CYP2E1 (209976_s_at) | CYP2E1 (222100_at) | ||||
|---|---|---|---|---|---|---|---|---|
| HR | p-Value | HR | p-Value | HR | p-Value | HR | p-Value | |
| All breast Cancers n = 3951 | 0.72 | *** | 0.68 | *** | 0.96 | 0.460 | 0.78 | *** |
| ER(+), PR(+), n = 577 | 1.29 | 0.170 | 1.02 | 0.920 | 0.76 | 0.140 | 0.84 | 0.340 |
| ER(−), PR(−), n = 298 | 1.06 | 0.770 | 1.23 | 0.300 | 1.05 | 0.820 | 1.04 | 0.830 |
| ER(−), PR(−), HER2(−) n = 198 | 0.80 | 0.500 | 0.96 | 0.860 | 0.80 | 0.370 | 1.17 | 0.520 |
| ER(+), Luminal A, n = 1933 | 0.76 | ** | 0.68 | *** | 0.99 | 0.920 | 0.81 | * |
| ER(+), Luminal A, Grade 1, n = 267 | 0.97 | 0.920 | 1.38 | 0.300 | 0.81 | 0.520 | 1.27 | 0.450 |
| ER(+), Luminal B, n = 1149 | 0.79 | * | 0.81 | 0.030 | 0.87 | 0.160 | 0.74 | ** |
| ER(+), Luminal B, Grade1 n = 56 | 1.46 | 0.530 | 1.04 | 0.950 | 0.83 | 0.770 | 1.24 | 0.720 |
| Grade1, n = 345 | 1.02 | 0.940 | 1.15 | 0.590 | 0.76 | 0.310 | 1.22 | 0.450 |
| Grade2, n = 901 | 1.13 | 0.330 | 1.13 | 0.310 | 0.94 | 0.640 | 0.74 | * |
| Grade3, n = 903 | 0.93 | 0.54 | 0.94 | 0.56 | 1.11 | 0.330 | 0.85 | 0.16 |
| Basal subtype, n = 618 | 0.59 | *** | 0.62 | *** | 0.89 | 0.360 | 0.91 | 0.470 |
| Luminal A, n = 1933 | 0.76 | ** | 0.68 | *** | 0.99 | 0.920 | 0.81 | * |
| Luminal B, n = 1149 | 0.79 | * | 0.81 | * | 0.87 | 0.160 | 0.74 | ** |
| ER(+), HER2(+), n = 156 | 1.25 | 0.470 | 1.69 | 0.093 | 1.14 | 0.670 | 1.26 | 0.460 |
| ER(−), HER2(+), n = 96 | 1.37 | 0.320 | 1.58 | 0.150 | 1.78 | 0.068 | 0.81 | 0.510 |
| ER(+), PR(+),Lymph(+) n = 344 | 1.41 | 0.120 | 1.05 | 0.810 | 0.80 | 0.310 | 1.02 | 0.930 |
| ER(+), PR(+),Lymph(−) n = 228 | 0.81 | 0.560 | 1.06 | 0.870 | 0.76 | 0.400 | 0.44 | * |
| ER(−), PR(−),Lymph(+) n = 127 | 1.26 | 0.390 | 1.08 | 0.780 | 1.16 | 0.580 | 1.21 | 0.470 |
| ER(−), PR(−),Lymph(−) n = 167 | 1.00 | 1.000 | 1.11 | 0.740 | 0.96 | 0.900 | 1.32 | 0.370 |
| ER(+), Luminal A, Grade 2, n = 567 | 0.91 | 0.580 | 1.13 | 0.460 | 1.01 | 0.970 | 0.89 | 0.480 |
| ER(+), Luminal B, Grade2 n = 253 | 0.65 | 0.051 | 0.82 | 0.360 | 0.46 | ** | 0.59 | * |
| ER(+) n = 3082 | 0.78 | *** | 0.74 | *** | 0.94 | 0.3382 | 0.82 | ** |
| Gene | SULT1A1 (203615_x_at) | SULT1A1 (215299_x_at) | SULT1A2 (207122_x_at) | SULT1A2 (211385_x_at) | ||||
|---|---|---|---|---|---|---|---|---|
| Patient Group | HR (Hazard Ratio) | p-Value (Log Rank Test) | HR (Hazard Ratio) | p-Value (Log Rank Test) | HR (Hazard Ratio) | p-Value (Log Rank Test) | HR (Hazard Ratio) | p-Value (Log Rank Test) |
| All breast Cancers n = 3951 | 0.72 | *** | 0.77 | *** | 0.70 | *** | 0.73 | *** |
| ER(+), PR(+), n = 577 | 1.16 | 0.430 | 1.19 | 0.330 | 1.18 | 0.370 | 1.22 | 0.270 |
| ER(−), PR(−), n = 298 | 1.37 | 0.120 | 1.42 | 0.085 | 1.34 | 0.140 | 1.24 | 0.280 |
| ER(−), PR(−), HER2(−) n = 198 | 0.98 | 0.940 | 1.18 | 0.510 | 1.34 | 0.230 | 1.06 | 0.810 |
| ER(+), Luminal A, n = 1933 | 0.86 | 0.092 | 0.93 | 0.399 | 0.78 | ** | 0.70 | ** |
| ER(+), Luminal A, Grade 1, n = 267 | 0.83 | 0.560 | 1.59 | 0.150 | 1.01 | 0.960 | 1.13 | 0.700 |
| ER(+), Luminal B, n = 1149 | 0.84 | 0.075 | 0.83 | 0.064 | 0.68 | *** | 0.84 | 0.078 |
| ER(+), Luminal B, Grade1 n = 56 | 0.91 | 0.870 | 0.89 | 0.850 | 1.31 | 0.660 | 1.43 | 0.550 |
| Grade1, n = 345 | 0.94 | 0.810 | 1.32 | 0.300 | 1.17 | 0.560 | 1.14 | 0.630 |
| Grade2, n = 901 | 0.96 | 0.730 | 1.09 | 0.500 | 0.84 | 0.170 | 0.91 | 0.430 |
| Grade3, n = 903 | 1.02 | 0.860 | 1.00 | 1.000 | 0.94 | 0.58 | 1.04 | 0.74 |
| Basal subtype, n = 618 | 0.72 | * | 0.85 | 0.210 | 0.73 | * | 0.75 | * |
| Luminal A, n = 1933 | 0.86 | 0.092 | 0.93 | 0.400 | 0.78 | ** | 0.77 | ** |
| Luminal B, n = 1149 | 0.84 | 0.075 | 0.83 | 0.064 | 0.68 | *** | 0.84 | 0.078 |
| ER(+), HER2(+), n = 156 | 1.24 | 0.490 | 1.13 | 0.690 | 0.96 | 0.900 | 1.18 | 0.600 |
| ER(−), HER2(+), n = 96 | 1.47 | 0.230 | 1.40 | 0.300 | 1.31 | 0.390 | 1.05 | 0.880 |
| ER(+), PR(+),Lymph(+) n = 344 | 1.29 | 0.240 | 1.23 | 0.340 | 1.26 | 0.290 | 1.21 | 0.380 |
| ER(+), PR(+),Lymph(−) n=228 | 0.89 | 0.720 | 0.87 | 0.680 | 1.08 | 0.820 | 1.33 | 0.380 |
| ER(−), PR(−),Lymph(+) n = 127 | 1.54 | 0.110 | 1.22 | 0.470 | 1.32 | 0.310 | 1.25 | 0.400 |
| ER(−), PR(−),Lymph(−) n = 167 | 1.41 | 0.270 | 1.10 | 0.760 | 1.39 | 0.290 | 1.12 | 0.720 |
| ER(+), Luminal A, Grade 2, n = 567 | 1.12 | 0.500 | 1.51 | * | 0.92 | 0.620 | 1.06 | 0.720 |
| ER(+), Luminal B, Grade2 n = 253 | 0.68 | 0.076 | 0.63 | * | 0.78 | 0.240 | 0.76 | 0.200 |
| ER(+) n = 3082 | 0.84 | ** | 0.87 | * | 0.76 | *** | 0.81 | *** |
| Gene Symbol | Murine Forward Primer (5′-3′) | Murine Reverse Primer (5′-3′) |
| CAT | CCTTCAAGTTGGTTAATGCAGA | CAAGTTTTTGATGCCCTGGT |
| VIM | CTCCAGAGAGAGGAAGCCGAAAG | CCTGGATCTCTTCATCGTGCAGT |
| FgfBp1 | CAAGGTCCAAGAAGCTGTCTCCA | AGCTCCAAGATTCCCCACAGAAC |
| Tgfb3 | GGCGTCTCAAGAAGCAAAAGGAT | CCTTAGGTTCGTGGACCCATTTC |
| MMP9 | CATTCGCGTGGATAAGGAGT | ACCTGGTTCACCTCATGGTC |
| GPX2 | GTTCTCGGCTTCCCTTGC | TTCAGGATCTCCTCGTTCTGA |
| GPX3 | GGCTTCCCTTCCAACCAA | CCCACCTGGTCGAACATACT |
| SOD3 | CTCTTGGGAGAGCCTGACA | GCCAGTAGCAAGCCGTAGAA |
| Cyclophilin A | TGGAGAGCACCAAGACAGACA | TGCCGGAGTCGACAATGAT |
| 36B4 | AGATTCGGGATATGCTGTTGG | AAAGCCTGGAAGAAGGAGGTC |
| Gene Symbol | Human Forward Primer (5′-3′) | Human Reverse Primer (5′-3′) |
| AHR | TTGAACCATCCCCATACCCCAC | GAGGTTCTGGCTGGCACTGATA |
| PXR | AGTGAAGGTTCCCGAGGACATG | TTGTCACAGAGCATACCCAGCA |
| Cyclophilin A | GTCTCCTTTGAGCTGTTTGCAGAC | CTTGCCACCAGTGCCATTATG |
| 36B4 | CCATTGAAATCCTGAGTGATGTG | GTCGAACACCTGCTGGATGAC |
| Antibody | Dilution | Vendor |
|---|---|---|
| 4-HNE | 1:1000 | Abcam (ab46545) |
| Nitrotyrosine | 1:1000 | Millipore (06-284) |
| iNOS | 1:1000 | Novus (NB300-605) |
| NRF2 | 1:1000 | Abcam (ab31163) |
| Phospho-AMPKα (Thr172) | 1:1000 | Cell Signaling (#2535) |
| AMPKα | 1:1000 | Cell Signaling (#5832) |
| Phospho-ACC (Ser79) | 1:1000 | Cell Signaling (#3661) |
| ACC | 1:1000 | Cell Signaling (#3676) |
| FOXO1 | 1:1000 | Cell Signaling (#9454) |
| E-cadherin | 1:1000 | Cell Signaling (#3195) |
| ZO1 | 1:1000 | Cell Signaling (#8193) |
| Vimentin | 1:1000 | Cell Signaling (#5741) |
| Snail | 1:1000 | Cell Signaling (#3879) |
| β-Catenin | 1:1000 | Sigma-Aldrich (C7082) |
| β-Actin | 1:20000 | Sigma-Aldrich (A3854) |
| Anti-rabbit IgG, HRP-linked antibody | 1:2000 | Cell Signaling (#7074) |
| Anti-Mouse IgG, Peroxidase antibody | 1:2000 | Sigma-Aldrich (A9044) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sári, Z.; Mikó, E.; Kovács, T.; Boratkó, A.; Ujlaki, G.; Jankó, L.; Kiss, B.; Uray, K.; Bai, P. Indoxylsulfate, a Metabolite of the Microbiome, Has Cytostatic Effects in Breast Cancer via Activation of AHR and PXR Receptors and Induction of Oxidative Stress. Cancers 2020, 12, 2915. https://doi.org/10.3390/cancers12102915
Sári Z, Mikó E, Kovács T, Boratkó A, Ujlaki G, Jankó L, Kiss B, Uray K, Bai P. Indoxylsulfate, a Metabolite of the Microbiome, Has Cytostatic Effects in Breast Cancer via Activation of AHR and PXR Receptors and Induction of Oxidative Stress. Cancers. 2020; 12(10):2915. https://doi.org/10.3390/cancers12102915
Chicago/Turabian StyleSári, Zsanett, Edit Mikó, Tünde Kovács, Anita Boratkó, Gyula Ujlaki, Laura Jankó, Borbála Kiss, Karen Uray, and Péter Bai. 2020. "Indoxylsulfate, a Metabolite of the Microbiome, Has Cytostatic Effects in Breast Cancer via Activation of AHR and PXR Receptors and Induction of Oxidative Stress" Cancers 12, no. 10: 2915. https://doi.org/10.3390/cancers12102915