Radioresistance in Glioblastoma and the Development of Radiosensitizers

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. History and Current Status of GBM Detection and Imaging Techniques
3. Treatment Options for GBM/History of GBM Treatment
4. Current Status of Radiation Treatment in GBM and Emergence of Radioresistance
5. Mechanisms of GBM Radioresistance
5.1. Tumor Microenvironment
5.2. Hypoxia
5.3. Metabolic Alteration
5.4. Glioma Stem Cells
5.5. GBM Tumor Heterogeneity
5.6. MicroRNAs
5.7. Cell Cycle, DNA Repair and Other Signaling Pathways
6. Radiosensitizers in GBM and Other Cancer Treatment
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kohler, B.A.; Ward, E.; McCarthy, B.J.; Schymura, M.J.; Ries, L.A.G.; Eheman, C.; Jemal, A.; Anderson, R.N.; Ajani, U.A.; Edwards, B.K. Annual Report to the Nation on the Status of Cancer, 1975-2007, Featuring Tumors of the Brain and Other Nervous System. J. Natl. Cancer Inst. 2011, 103, 714–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-Dense Temozolomide for Newly Diagnosed Glioblastoma: A Randomized Phase III Clinical Trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Mandel, J.; Yust-Katz, S.; Patel, A.J.; Cachia, D.; Liu, D.; Park, M.; Yuan, Y.; A Kent, T.; De Groot, J.F. Inability of positive phase II clinical trials of investigational treatments to subsequently predict positive phase III clinical trials in glioblastoma. Neuro Oncol. 2017, 20, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef]
- Westphal, M.; Heese, O.; Steinbach, J.P.; Schnell, O.; Schackert, G.; Mehdorn, H.M.; Schulz, D.; Simon, M.; Schlegel, U.; Senft, C.; et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur. J. Cancer 2015, 51, 522–532. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Pfahler, G. Cerebral skiagraphy: transactions of the American Roentgen Ray Society—5th Annual Meeting. Am. J. Roentgenol. Radium. Ther. Nucl. Med. 1904, 4, 174–186. [Google Scholar]
- Adson, A.W.; Ott, W.O.; Crawford, A.S. A Study of Ventriculography 1. Radiology 1924, 2, 65–73. [Google Scholar] [CrossRef]
- Schwartz, C.W. The Gliomas Roentgenologically Considered. Radiology 1936, 27, 419–432. [Google Scholar] [CrossRef]
- Forestier, J. Actual Technic of Examination of the Spinal Cavities with Lipiodol 1. Radiol. 1928, 11, 481–489. [Google Scholar] [CrossRef]
- Scott, W.G.; Seaman, W.B. Developments in cerebral angiography with rapid serialized X-ray exposures on roll film 9 1/2 inches wide. Radiology 1951, 56, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Hoeffner, E.G.; Mukherji, S.K.; Srinivasan, A.; Quint, D.J. Neuroradiology Back to the Future: Brain Imaging. Am. J. Neuroradiol. 2011, 33, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.B. Brain Tumors in Childrern. Radiology 1952, 58, 688–695. [Google Scholar] [CrossRef]
- Seaman, W.B.; Ter-Pogossian, M.M.; Schwartz, H.G. Localization of Intracranial Neoplasms with Radioactive Isotopes. Radiology 1954, 62, 30–36. [Google Scholar] [CrossRef]
- New, P.F.J.; Scott, W.R.; Schnur, J.A.; Davis, K.R.; Taveras, J.M. Computerized Axial Tomography with the EMI Scanner. Radiology 1974, 110, 109–123. [Google Scholar] [CrossRef]
- Baker, H.L.; Houser, O.W.; Campbell, J.K. National Cancer Institute study: evaluation of computed tomography in the diagnosis of intracranial neoplasms. I. Overall results. Radiology 1980, 136, 91–96. [Google Scholar] [CrossRef]
- Potts, D.G.; Abbott, G.F.; Von Sneidern, J.V. National Cancer Institute study: evaluation of computed tomography in the diagnosis of intracranial neoplasms. III. Metastatic tumors. Radiology 1980, 136, 657–664. [Google Scholar] [CrossRef] [PubMed]
- New, P.F.; Aronow, S.; Hesselink, J.R. National Cancer Institute study: evaluation of computed tomography in the diagnosis of intracranial neoplasms. IV. Meningiomas. Radiology 1980, 136, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Inouye, T.; Suzuki, H.; Machida, T.; Iio, M. Magnetic resonance imaging of brain tumors: measurement of T1. Work in progress. Radiology 1984, 150, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M. History and Evolution of Brain Tumor Imaging: Insights through Radiology. Radiology 2014, 273, 111–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17, iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Whelan, H.T.; Clanton, J.A.; Wilson, R.E.; Tulipan, N.B. Comparison of CT and MRI brain tumor imaging using a canine glioma model. Pediatr. Neurol. 1988, 4, 279–283. [Google Scholar] [CrossRef]
- Alexander, A.L.; Lee, J.E.; Lazar, M.; Field, A.S. Diffusion tensor imaging of the brain. Neurotherapeutics 2007, 4, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Yanagihara, T.K.; Wang, T. Diffusion-weighted imaging of the brain for glioblastoma: Implications for radiation oncology. Appl. Radiat. Oncol. 2014, 5–13. [Google Scholar]
- Schmainda, K.M. Diffusion-weighted MRI as a biomarker for treatment response in glioma. CNS Oncol. 2012, 1, 169–180. [Google Scholar] [CrossRef]
- Guo, A.C.; Cummings, T.J.; Dash, R.C.; Provenzale, J.M. Lymphomas and High-Grade Astrocytomas: Comparison of Water Diffusibility and Histologic Characteristics. Radiology 2002, 224, 177–183. [Google Scholar] [CrossRef]
- Kono, K.; Inoue, Y.; Nakayama, K.; Shakudo, M.; Morino, M.; Ohata, K.; Wakasa, K.; Yamada, R. The role of diffusion-weighted imaging in patients with brain tumors. Am. J. Neuroradiol. 2001, 22, 1081–1088. [Google Scholar] [PubMed]
- Kalpathy-Cramer, J.; Gerstner, E.R.; Emblem, K.; Andronesi, O.C.; Rosen, B. Advanced Magnetic Resonance Imaging of the Physical Processes in Human Glioblastoma. Cancer Res. 2014, 74, 4622–4637. [Google Scholar] [CrossRef] [Green Version]
- Van Dijken, B.R.; Van Laar, P.J.; Smits, M.; Dankbaar, J.W.; Enting, R.H.; Van Der Hoorn, A. Perfusion MRI in treatment evaluation of glioblastomas: Clinical relevance of current and future techniques. J. Magn. Reson. Imaging 2018, 49, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, V.; Parker, G.J.; Barker, G.J.; Birnie, K.; Tofts, P.S.; Miller, D.; Thompson, A. Variations in T1 and T2 relaxation times of normal appearing white matter and lesions in multiple sclerosis. J. Neurol. Sci. 2000, 178, 81–87. [Google Scholar] [CrossRef]
- Østergaard, L. Principles of cerebral perfusion imaging by bolus tracking. J. Magn. Reson. Imaging 2005, 22, 710–717. [Google Scholar] [CrossRef]
- Nelson, S.J. Multivoxel magnetic resonance spectroscopy of brain tumors. Mol. Cancer Ther. 2003, 2, 497–507. [Google Scholar]
- Blüml, S.; Moreno-Torres, A.; Shic, F.; Nguy, C.H.; Ross, B.D. Tricarboxylic acid cycle of glia in the in vivo human brain. NMR Biomed. 2002, 15, 1–5. [Google Scholar] [CrossRef]
- Howe, F.A.; Opstad, K.S. 1H MR spectroscopy of brain tumours and masses. NMR Biomed. 2003, 16, 123–131. [Google Scholar] [CrossRef]
- Kurhanewicz, J.; Vigneron, D.B.; Brindle, K.; Chekmenev, E.Y.; Comment, A.; Cunningham, C.H.; DeBerardinis, R.J.; Green, G.G.; Leach, M.O.; Rajan, S.S.; et al. Analysis of Cancer Metabolism by Imaging Hyperpolarized Nuclei: Prospects for Translation to Clinical Research. Neoplasia 2011, 13, 81–97. [Google Scholar] [CrossRef] [Green Version]
- Pinker, K.; Stadlbauer, A.; Bogner, W.; Gruber, S.; Helbich, T.; Pinker, K. Molecular imaging of cancer: MR spectroscopy and beyond. Eur. J. Radiol. 2012, 81, 566–577. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Esmaeili, M.; Borra, R.J.H.; Emblem, K.; Gerstner, E.R.; Pinho, M.C.; Plotkin, S.R.; Chi, A.S.; Eichler, A.F.; Dietrich, J.; et al. Early changes in glioblastoma metabolism measured by MR spectroscopic imaging during combination of anti-angiogenic cediranib and chemoradiation therapy are associated with survival. NPJ Precis. Oncol. 2017, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Arias-Ramos, N.; Ferrer-Font, L.; Lope-Piedrafita, S.; Mocioiu, V.; Julià-Sapé, M.; Pumarola, M.; Arús, C.; Candiota, A.P. Metabolomics of Therapy Response in Preclinical Glioblastoma: A Multi-Slice MRSI-Based Volumetric Analysis for Noninvasive Assessment of Temozolomide Treatment. Metabolites 2017, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laprie, A.; Ken, S.; Filleron, T.; Lubrano, V.; Vieillevigne, L.; Tensaouti, F.; Catalaa, I.; Boetto, S.; Khalifa, J.; Attal, J.; et al. Dose-painting multicenter phase III trial in newly diagnosed glioblastoma: the SPECTRO-GLIO trial comparing arm A standard radiochemotherapy to arm B radiochemotherapy with simultaneous integrated boost guided by MR spectroscopic imaging. BMC Cancer 2019, 19, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlemmer, H.-P.W.; Pichler, B.J.; Schmand, M.; Burbar, Z.; Michel, C.; Ladebeck, R.; Jattke, K.; Townsend, D.; Nahmias, C.; Jacob, P.K.; et al. Simultaneous MR/PET Imaging of the Human Brain: Feasibility Study. Radiology 2008, 248, 1028–1035. [Google Scholar] [CrossRef] [Green Version]
- Pichler, B.J.; Kolb, A.; Nägele, T.; Schlemmer, H.P. PET/MRI: Paving the Way for the Next Generation of Clinical Multimodality Imaging Applications. J. Nucl. Med. 2010, 51, 333–336. [Google Scholar] [CrossRef] [Green Version]
- Chaddad, A.; Kucharczyk, M.J.; Daniel, P.; Sabri, S.; Jean-Claude, B.J.; Niazi, T.; Abdulkarim, B. Radiomics in Glioblastoma: Current Status and Challenges Facing Clinical Implementation. Front. Oncol. 2019, 9, 374. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Ou, X.; Wang, J.; Guo, W.; Ma, X. Radiomics-Based Machine Learning in Differentiation Between Glioblastoma and Metastatic Brain Tumors. Front. Oncol. 2019, 9, 806. [Google Scholar] [CrossRef] [Green Version]
- Wolbers, J.G. Novel strategies in glioblastoma surgery aim at safe, supra-maximum resection in conjunction with local therapies. Chin. J. Cancer 2014, 33, 8–15. [Google Scholar] [CrossRef]
- Li, Y.M.; Suki, D.; Hess, K.; Sawaya, R. The influence of maximum safe resection of glioblastoma on survival in 1229 patients: Can we do better than gross-total resection? J. Neurosurg. 2016, 124, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Sanai, N.; Polley, M.Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An extent of resection threshold for newly diagnosed glioblastomas. J. Neurosurg. 2011, 115, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C.; De Vleeschouwer, S. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; pp. 197–241. [Google Scholar]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.-D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Suchorska, B.; Weller, M.; Tabatabai, G.; Senft, C.; Hau, P.; Sabel, M.C.; Herrlinger, U.; Ketter, R.; Schlegel, U.; Marosi, C.; et al. Complete resection of contrast-enhancing tumor volume is associated with improved survival in recurrent glioblastoma—results from the DIRECTOR trial. Neuro Oncol. 2016, 18, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Le Rhun, E.; Preusser, M.; Tonn, J.-C.; Roth, P. How we treat glioblastoma. ESMO Open 2019, 4, e000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramatzki, D.; Roth, P.; Rushing, E.; Weller, J.; Andratschke, N.; Hofer, S.; Korol, D.; Regli, L.; Pangalu, A.; Pless, M.; et al. Bevacizumab may improve quality of life, but not overall survival in glioblastoma: An epidemiological study. Ann. Oncol. 2018, 29, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Walker, M.D.; Strike, T.A.; Sheline, G.E. An analysis of dose-effect relationship in the radiotherapy of malignant gliomas. Int. J. Radiat. Oncol. 1979, 5, 1725–1731. [Google Scholar] [CrossRef]
- Frankel, S.A.; German, W.J. Glioblastoma multiforme; review of 219 cases with regard to natural history, pathology, diagnostic methods, and treatment. J. Neurosurg. 1958, 15, 489–503. [Google Scholar] [CrossRef]
- Sheline, G.E. Radiation therapy of brain tumors. Cancer 1977, 39, 873–881. [Google Scholar] [CrossRef]
- Yabroff, K.R.; Harlan, L.; Zeruto, C.; Abrams, J.; Mann, B. Patterns of care and survival for patients with glioblastoma multiforme diagnosed during 2006. Neuro Oncol. 2012, 14, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassenko, A.G.; Thiessen, B.; Beattie, B.J.; Malkin, M.G.; Blasberg, R.G. Evaluation of early response to SU101 target-based therapy in patients with recurrent supratentorial malignant gliomas using FDG PET and Gd-DTPA MRI. J. Neurooncol. 2000, 46, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Prados, M.D.; Lamborn, K.R.; Chang, S.; Burton, E.; Butowski, N.; Malec, M.; Kapadia, A.; Rabbitt, J.; Page, M.S.; Fedoroff, A.; et al. Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma1. Neuro Oncol. 2006, 8, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloughesy, T.F.; Kuhn, J.; Robins, H.I.; Abrey, L.; Wen, P.; Fink, K.; Lieberman, F.S.; Mehta, M.; Chang, S.; Yung, A.; et al. Phase I Trial of Tipifarnib in Patients With Recurrent Malignant Glioma Taking Enzyme-Inducing Antiepileptic Drugs: A North American Brain Tumor Consortium Study. J. Clin. Oncol. 2005, 23, 6647–6656. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.M.; Kuhn, J.; Wen, P.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.; Robins, H.I.; Cloughesy, T.; DeAngelis, L.M.; et al. Phase I/pharmacokinetic study of CCI-779 in patients with recurrent malignant glioma on enzyme-inducing antiepileptic drugs. Investig. New Drugs 2004, 22, 427–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreisl, T.N.; Kotliarova, S.; A Butman, J.; Albert, P.S.; Kim, L.; Musib, L.; Thornton, D.; Fine, H.A. A phase I/II trial of enzastaurin in patients with recurrent high-grade gliomas. Neuro Oncol. 2010, 12, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choucair, A.K.; Levin, V.A.; Gutin, P.H.; Davis, R.L.; Silver, P.; Edwards, M.S.B.; Wilson, C.B. Development of multiple lesions during radiation therapy and chemotherapy in patients with gliomas. J. Neurosurg. 1986, 65, 654–658. [Google Scholar] [CrossRef]
- Ammirati, M.; Galicich, J.H.; Arbit, E.; Liao, Y. Reoperation in the treatment of recurrent intracranial malignant gliomas. Neurosurgery 1987, 21, 607–614. [Google Scholar] [CrossRef]
- Loeffler, J.; Alexander, E.; Hochberg, F.H.; Wen, P.Y.; Morris, J.H.; Schoene, W.C.; Siddon, R.L.; Morse, R.H.; Black, P.M. Clinical patterns of failure following stereotactic interstitial irradiation for malignant gliomas. Int. J. Radiat. Oncol. 1990, 19, 1455–1462. [Google Scholar] [CrossRef]
- Dhermain, F.; De Crevoisier, R.; Parker, F.; Cioloca, C.; Kaliski, A.; Beaudre, A.; Lefkopoulos, D.; Armand, J.-P.; Haie-Meder, C. Récidives dans les tumeurs gliales: place de la radiothérapie [Role of radiotherapy in recurrent gliomas]. Bull. Cancer 2004, 91, 883–889. [Google Scholar]
- Combs, S.E.; Debus, J.; Schulz-Ertner, D. Radiotherapeutic alternatives for previously irradiated recurrent gliomas. BMC Cancer 2007, 7, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogh, S.E.; Andrews, D.W.; Glass, J.; Curran, W.; Glass, C.; Champ, C.E.; Evans, J.J.; Hyslop, T.; Pequignot, E.; Downes, B.; et al. Hypofractionated Stereotactic Radiation Therapy: An Effective Therapy for Recurrent High-Grade Gliomas. J. Clin. Oncol. 2010, 28, 3048–3053. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T.; Mulholland, P.; Neyns, B.; Nabors, B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III Randomized Trial Comparing the Efficacy of Cediranib As Monotherapy, and in Combination With Lomustine, Versus Lomustine Alone in Patients With Recurrent Glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.-S.; Lee, J.-I.; Kim, W.S.; Son, M.J.; Lim, D.H.; Kim, S.T.; Park, K.; Kim, J.H.; Eoh, W.; Nam, D.-H. A pilot study of metronomic temozolomide treatment in patients with recurrent temozolomide-refractory glioblastoma. Oncol. Rep. 2006, 16, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab Alone and in Combination With Irinotecan in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zustovich, F.; Lombardi, G.; Della Puppa, A.; Rotilio, A.; Scienza, R.; Pastorelli, D. A phase II study of cisplatin and temozolomide in heavily pre-treated patients with temozolomide-refractory high-grade malignant glioma. Anticancer. Res. 2009, 29, 4275–4279. [Google Scholar]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma. JAMA Oncol. 2020, 6, 1–8. [Google Scholar] [CrossRef]
- Halasz, L.M.; Soltys, S.G.; Breneman, J.C.; Chan, M.D.; Laack, N.N.; Minniti, G.; Kirkpatrick, J. Treatment of Gliomas: A Changing Landscape. Int. J. Radiat. Oncol. 2017, 98, 255–258. [Google Scholar] [CrossRef]
- Briere, T.M.; McAleer, M.F.; Levy, L.B.; Yang, J.N. Sparing of normal tissues with volumetric arc radiation therapy for glioblastoma: single institution clinical experience. Radiat. Oncol. 2017, 12, 79. [Google Scholar] [CrossRef]
- Shaffer, R.; Nichol, A.; Vollans, E.; Fong, M.; Nakano, S.; Moiseenko, V.; Schmuland, M.; Ma, R.; McKenzie, M.; Otto, K. A Comparison of Volumetric Modulated Arc Therapy and Conventional Intensity-Modulated Radiotherapy for Frontal and Temporal High-Grade Gliomas. Int. J. Radiat. Oncol. 2010, 76, 1177–1184. [Google Scholar] [CrossRef]
- Combs, S.E.; Thilmann, C.; Edler, L.; Debus, J.; Schulz-Ertner, D. Efficacy of Fractionated Stereotactic Reirradiation in Recurrent Gliomas: Long-Term Results in 172 Patients Treated in a Single Institution. J. Clin. Oncol. 2005, 23, 8863–8869. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Hall, W.A.; Gerbi, B.J.; Higgins, P.D.; McGuire, W.A.; Clark, H.B. Single dose versus fractionated stereotactic radiotherapy for recurrent high-grade gliomas. Int. J. Radiat. Oncol. 1999, 45, 1133–1141. [Google Scholar] [CrossRef]
- Wang, T.J.C.; Wu, C.-C.; Jani, A.; Estrada, J.; Ung, T.; Chow, D.; Soun, J.E.; Saad, S.; Qureshi, Y.H.; Gartrell, R.; et al. Hypofractionated radiation therapy versus standard fractionated radiation therapy with concurrent temozolomide in elderly patients with newly diagnosed glioblastoma. Pr. Radiat. Oncol. 2016, 6, 306–314. [Google Scholar] [CrossRef]
- Tsien, C.; Moughan, J.; Michalski, J.M.; Gilbert, M.R.; Purdy, J.; Simpson, J.; Kresel, J.J.; Curran, W.J.; Diaz, A.; Mehta, M.; et al. Phase I Three-Dimensional Conformal Radiation Dose Escalation Study in Newly Diagnosed Glioblastoma: Radiation Therapy Oncology Group Trial 98-03. Int. J. Radiat. Oncol. 2009, 73, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordova, J.S.; Shu, H.-K.G.; Liang, Z.; Gurbani, S.S.; Cooper, L.A.D.; Holder, C.A.; Olson, J.J.; Kairdolf, B.; Schreibmann, E.; Neill, S.G.; et al. Whole-brain spectroscopic MRI biomarkers identify infiltrating margins in glioblastoma patients. Neuro Oncol. 2016, 18, 1180–1189. [Google Scholar] [CrossRef]
- Cao, Y.; Tseng, C.-L.; Balter, J.M.; Teng, F.; Parmar, H.A.; Sahgal, A. MR-guided radiation therapy: transformative technology and its role in the central nervous system. Neuro Oncol. 2017, 19, ii16–ii29. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Mazzucco, M.; Mellai, M. The Microenvironment in Gliomas: Phenotypic Expressions. Cancers 2015, 7, 2352–2359. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Mannino, M.; Chalmers, A. Radioresistance of glioma stem cells: Intrinsic characteristic or property of the ‘microenvironment-stem cell unit’? Mol. Oncol. 2011, 5, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Jamal, M.; Rath, B.H.; Tsang, P.S.; Camphausen, K.; Tofilon, P.J. The Brain Microenvironment Preferentially Enhances the Radioresistance of CD133+ Glioblastoma Stem-like Cells. Neoplasia 2012, 14, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Farace, C.; Oliver, J.A.; Melguizo, C.; Alvarez, P.; Bandiera, P.; Rama, A.R.; Malaguarnera, G.; Ortiz, R.; Madeddu, R.; Prados, J. Microenvironmental Modulation of Decorin and Lumican in Temozolomide-Resistant Glioblastoma and Neuroblastoma Cancer Stem-Like Cells. PLoS ONE 2015, 10, e0134111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Lorger, M. Tumor Microenvironment in the Brain. Cancers 2012, 4, 218–243. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; D’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma Stem Cells Microenvironment: The Paracrine Roles of the Niche in Drug and Radioresistance. Stem Cells Int. 2016, 2016, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muz, B.; De La Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis1. Neuro-oncology 2005, 7, 134–153. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C.A. The Concentration of Oxygen Dissolved in Tissues at the Time of Irradiation as a Factor in Radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.L.; Zani, B.; Popov, V.M.; Fratticci, A.; Cerasani, M.; Di Genova, D.; Mancini, M.; Ciccarelli, C.; Ficorella, C.; et al. Hypoxia sustains glioblastoma radioresistance through ERKs/DNA-PKcs/HIF-1α functional interplay. Int. J. Oncol. 2014, 44, 2121–2131. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia Requires Notch Signaling to Maintain the Undifferentiated Cell State. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2 regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Zhou, C.; Xu, L.; Xiao, H. Hypoxia Enhances Stemness of Cancer Stem Cells in Glioblastoma: An In Vitro Study. Int. J. Med. Sci. 2013, 10, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heddleston, J.M.; Li, Z.; E McLendon, R.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta 2016, 1866, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-H.; Lee, C.-H.; Liang, J.-A.; Yu, C.-Y.; Shyu, W.-C. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol. Rep. 2010, 24, 1629–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérard, M.; Corroyer-Dulmont, A.; Lesueur, P.; Collet, S.; Chérel, M.; Bourgeois, M.; Stefan, D.; Limkin, E.J.; Perrio, C.; Guillamo, J.-S.; et al. Hypoxia Imaging and Adaptive Radiotherapy: A State-of-the-Art Approach in the Management of Glioma. Front. Med. 2019, 6, 117. [Google Scholar] [CrossRef] [Green Version]
- McGee, M.C.; Hamner, J.B.; Williams, R.F.; Rosati, S.F.; Sims, T.L.; Ng, C.Y.; Gaber, M.W.; Calabrese, C.; Wu, J.; Nathwani, A.C.; et al. Improved Intratumoral Oxygenation Through Vascular Normalization Increases Glioma Sensitivity to Ionizing Radiation. Int. J. Radiat. Oncol. 2010, 76, 1537–1545. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Venneti, S.; Thompson, C.B. Metabolic Reprogramming in Brain Tumors. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 515–545. [Google Scholar] [CrossRef]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, C.J.; Tran, A.N.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim Biophys Acta Rev. Cancer 2018, 1869, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Duman, C.; Yaqubi, K.; Hoffmann, A.; Acikgöz, A.A.; Korshunov, A.; Bendszus, M.; Herold-Mende, C.; Liu, H.-K.; Alfonso, J. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab. 2019, 30, 274–289.e5. [Google Scholar] [CrossRef]
- Oliva, C.R.; Halloran, B.; Hjelmeland, A.B.; Vazquez, A.; Bailey, S.M.; Sarkaria, J.N.; Griguer, C.E. Griguer, IGFBP6 controls the expansion of chemoresistant glioblastoma through paracrine IGF2/IGF-1R signaling. Cell Commun. Signal 2018, 16, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, C.R.; Markert, T.; Ross, L.J.; White, E.L.; Rasmussen, L.; Zhang, W.; Everts, M.; Moellering, D.; Bailey, S.M.; Suto, M.J.; et al. Identification of Small Molecule Inhibitors of Human CytochromecOxidase That Target Chemoresistant Glioma Cells. J. Biol. Chem. 2016, 291, 24188–24199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, C.R.; Zhang, W.; Langford, C.; Suto, M.J.; Griguer, C.E. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget 2017, 8, 37568–37583. [Google Scholar] [CrossRef]
- Oliva, C.R.; Moellering, D.; Gillespie, G.Y.; Griguer, C.E. Acquisition of Chemoresistance in Gliomas Is Associated with Increased Mitochondrial Coupling and Decreased ROS Production. PLoS ONE 2011, 6, e24665. [Google Scholar] [CrossRef] [Green Version]
- Oliva, C.R.; Nozell, S.E.; Diers, A.; McClugage, S.G.; Sarkaria, J.N.; Markert, J.M.; Darley-Usmar, V.M.; Bailey, S.M.; Gillespie, G.Y.; Landar, A.; et al. Acquisition of Temozolomide Chemoresistance in Gliomas Leads to Remodeling of Mitochondrial Electron Transport Chain. J. Biol. Chem. 2010, 285, 39759–39767. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Cell Biol. 2011, 192, 313–326. [Google Scholar] [CrossRef]
- Vartanian, A.; Agnihotri, S.; Wilson, M.R.; Burrell, K.E.; Tonge, P.D.; Alamsahebpour, A.; Jalali, S.; Taccone, M.S.; Mansouri, S.; Golbourn, B.; et al. Targeting hexokinase 2 enhances response to radio-chemotherapy in glioblastoma. Oncotarget 2016, 7, 69518–69535. [Google Scholar] [CrossRef] [Green Version]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.T.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef] [PubMed]
- Wahl, D.R.; Dresser, J.; Wilder-Romans, K.; Parsels, J.D.; Zhao, S.G.; Davis, M.; Zhao, L.; Kachman, M.; Wernisch, S.; Burant, C.F.; et al. Glioblastoma Therapy Can Be Augmented by Targeting IDH1-Mediated NADPH Biosynthesis. Cancer Res. 2016, 77, 960–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, W.-C.; Chiou, S.-H.; Huang, C.-Y.; Chiang, S.-F.; Yang, C.-L.; Sudhakar, J.N.; Lin, T.-Y.; Chiang, I.-P.; Shen, C.-C.; Cheng, W.-Y.; et al. Mitochondrial protein ATPase family, AAA domain containing 3A correlates with radioresistance in glioblastoma. Neuro Oncol. 2013, 15, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Rico, M.A.; Calvo-Vidal, M.N.; Villalonga-Planells, R.; Martínez-Soler, F.; Gimenez-Bonafe, P.; Navarro-Sabaté, À.; Tortosa, A.; Bartrons, R.; Manzano, A. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother. Oncol. 2011, 101, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Rosen, J.M.; Jordan, C.T. The Increasing Complexity of the Cancer Stem Cell Paradigm. Science 2009, 324, 1670–1673. [Google Scholar] [CrossRef] [Green Version]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner, E.M.; Kornblum, I.H. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, E.V.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Gallia, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; DiMeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; MacSwords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin Alpha 6 Regulates Glioblastoma Stem Cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Kim, Y.; Joo, K.M.; Jin, J.; Nam, D.H. Cancer Stem Cells and Their Mechanism of Chemo-Radiation Resistance. Int. J. Stem Cells 2009, 2, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanzani, E.; Martínez-Soler, F.; Mateos, T.M.; Vidal, N.; Villanueva, A.; Pujana, M.A.; Serra-Musach, J.; De La Iglesia, N.; Giménez-Bonafé, P.; Tortosa, A. Radioresistance of mesenchymal glioblastoma initiating cells correlates with patient outcome and is associated with activation of inflammatory program. Oncotarget 2017, 8, 73640–73653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Bertoni, H.; Lal, B.; Li, A.; Caplan, M.; Guerrero-Cazares, H.; Eberhart, C.G.; Quiñones-Hinojosa, A.; Gläß, M.; Scheffler, B.; Laterra, J.; et al. DNMT-dependent suppression of microRNA regulates the induction of GBM tumor-propagating phenotype by Oct4 and Sox2. Oncogene 2014, 34, 3994–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.; Riggi, N.; Chi, A.S.; Cahill, D.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [Green Version]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; Depinho, R.A.; et al. Olig2-Regulated Lineage-Restricted Pathway Controls Replication Competence in Neural Stem Cells and Malignant Glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Veselská, R.; Kuglik, P.; Cejpek, P.; Svachova, H.; Neradil, J.; Loja, T.; Relichova, J. Nestin expression in the cell lines derived from glioblastoma multiforme. BMC Cancer 2006, 6, 32. [Google Scholar] [CrossRef]
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martínez-Saez, E.; Prudkin, L.; et al. TGF-β Receptor Inhibitors Target the CD44high/Id1high Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Stamenkovic, I.; Yu, Q. CD44 attenuates activation of the hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res. 2010, 70, 2455–2464. [Google Scholar] [CrossRef] [Green Version]
- Ogden, A.T.; Waziri, A.E.; Lochhead, R.A.; Fusco, D.; Lopez, K.; Ellis, J.A.; Kang, J.; Assanah, M.; McKhann, G.M.; Sisti, M.B.; et al. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery 2008, 62, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, D.S.T.; Hu, B.; Ho, Y.W.; Sauvé, C.-E.G.; Bristow, C.A.; Wang, Q.; Multani, A.S.; Chen, P.; Nezi, L.; Jiang, S.; et al. PAF promotes stemness and radioresistance of glioma stem cells. Proc. Natl. Acad. Sci. USA 2017, 114, E9086–E9095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nature 2012, 14, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Long, L.; Wang, L.; Tan, C.; Fei, X.; Chen, L.; Huang, Q.; Liang, Z.-Q. Knockdown of Cathepsin L promotes radiosensitivity of glioma stem cells both in vivo and in vitro. Cancer Lett. 2016, 371, 274–284. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D. Glioblastoma Heterogeneity and Cancer Cell Plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.-I.; Iwama, T.; Park, D.M. The Evidence of Glioblastoma Heterogeneity. Sci. Rep. 2015, 5, 7979. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Castellino, R.C.; Durden, D.L. Mechanisms of Disease: the PI3K–Akt–PTEN signaling node—an intercept point for the control of angiogenesis in brain tumors. Nat. Clin. Pr. Neurol. 2007, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Yaes, R.J. Tumor heterogeneity, tumor size, and radioresistance. Int. J. Radiat. Oncol. 1989, 17, 993–1005. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, J.R.; Yung, W.K.; Shapiro, W.R. Isolation, karyotype, and clonal growth of heterogeneous subpopulations of human malignant gliomas. Cancer Res. 1981, 41, 2349–2359. [Google Scholar]
- Zalcberg, I.R.; Golgher, D. Molecular Genetics of Glioblastomas. Neuroimaging Clin. N. Am. 2015, 25, 97–103. [Google Scholar]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Thon, N.; Kreth, S.; Kreth, F.W. Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. OncoTargets Ther. 2013, 6, 1363–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1andIDH2Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Moore, L.M.; Li, X.; Yung, W.K.; Zhang, W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro Oncol. 2013, 15, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 Mutations as Molecular Signature and Predictive Factor of Secondary Glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pietsch, T.; et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 2010, 120, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, B.A.; Heaphy, C.M.; De Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Marie, S.K.N.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Yin, N.; Xie, T.; Zhang, H.; Chen, J.; Yu, J.; Liu, F. IDH1-R132H mutation radiosensitizes U87MG glioma cells via epigenetic downregulation of TIGAR. Oncol. Lett. 2019, 19, 1322–1330. [Google Scholar] [CrossRef] [Green Version]
- Pegg, E.A. Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990, 50, 6119–6129. [Google Scholar]
- Yang, P.; Zhang, W.; Wang, Y.; Peng, X.; Chen, B.; Qiu, X.; Li, G.; Li, S.; Wu, C.; Yao, K.; et al. IDH mutation and MGMT promoter methylation in glioblastoma: results of a prospective registry. Oncotarget 2015, 6, 40896–40906. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Verbaan, D.; Lamba, S.; Zanon, C.; Jeuken, J.W.; Boots-Sprenger, S.H.; Wesseling, P.; Hulsebos, T.J.; Troost, D.; Van Tilborg, A.A.; et al. The combination of IDH1 mutations and MGMT methylation status predicts survival in glioblastoma better than either IDH1 or MGMT alone. Neuro Oncol. 2014, 16, 1263–1273. [Google Scholar] [CrossRef]
- Bartel, B. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spizzo, R.; Nicoloso, M.S.; Croce, C.M.; Calin, G.A. SnapShot: MicroRNAs in Cancer. Cell 2009, 137, 586–586.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, B.; Robles, A.I.; Harris, C.C. Genetic variation in microRNA networks: the implications for cancer research. Nat. Rev. Cancer 2010, 10, 389–402. [Google Scholar] [CrossRef]
- Banelli, B.; Forlani, A.; Allemanni, G.; Morabito, A.; Pistillo, M.P.; Romani, M. MicroRNA in Glioblastoma: An Overview. Int. J. Genom. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Sana, J.; Busek, P.; Fadrus, P.; Besse, A.; Radova, L.; Vecera, M.; Reguli, S.; Sromova, L.S.; Hilser, M.; Lipina, R.; et al. Identification of microRNAs differentially expressed in glioblastoma stem-like cells and their association with patient survival. Sci. Rep. 2018, 8, 2836. [Google Scholar] [CrossRef] [Green Version]
- Møller, H.G.; Rasmussen, A.P.; Andersen, H.H.; Johnsen, K.B.; Henriksen, M.; Duroux, M. A Systematic Review of MicroRNA in Glioblastoma Multiforme: Micro-modulators in the Mesenchymal Mode of Migration and Invasion. Mol. Neurobiol. 2012, 47, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Ma, L.; Wu, M.; Zhang, G.; Jin, C.; Guo, Y.; Liu, R. miR-124 radiosensitizes human glioma cells by targeting CDK4. J. Neurooncol. 2013, 114, 263–274. [Google Scholar] [CrossRef]
- Toraih, E.A.; El-Wazir, A.; Abdallah, H.Y.; Tantawy, M.A.; Fawzy, M.S. Deregulated MicroRNA Signature Following Glioblastoma Irradiation. Cancer Control. 2019, 26, 1–8. [Google Scholar] [CrossRef]
- Li, W.; Guo, F.; Wang, P.; Hong, S.; Zhang, C. miR-221/222 confers radioresistance in glioblastoma cells through activating Akt independent of PTEN status. Curr. Mol. Med. 2014, 14, 185–195. [Google Scholar] [CrossRef]
- Moskwa, P.; Zinn, P.O.; Choi, Y.E.; Shukla, S.A.; Fendler, W.; Chen, C.C.; Lu, J.; Golub, T.R.; Hjelmeland, A.; Chowdhury, D. A functional screen identifies miRs that induce radioresistance in glioblastomas. Mol. Cancer Res. 2014, 12, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marampon, F.; Megiorni, F.; Camero, S.; Crescioli, C.; McDowell, H.P.; Sferra, R.; Vetuschi, A.; Pompili, S.; Ventura, L.; De Felice, F.; et al. HDAC4 and HDAC6 sustain DNA double strand break repair and stem-like phenotype by promoting radioresistance in glioblastoma cells. Cancer Lett. 2017, 397, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kowalski-Chauvel, A.; Modesto, A.; Gouazé-Andersson, V.; Baricault, L.; Gilhodes, J.; Delmas, C.; Lemarié, A.; Toulas, C.; Cohen-Jonathan-Moyal, E.; Seva, C. Alpha-6 integrin promotes radioresistance of glioblastoma by modulating DNA damage response and the transcription factor Zeb1. Cell Death Dis. 2018, 9, 872. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; McEllin, B.; Camacho, C.V.; Tomimatsu, N.; Sirasanagandala, S.; Nannepaga, S.; Hatanpaa, K.J.; Mickey, B.; Madden, C.; Maher, E.; et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009, 69, 4252–4259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golding, S.E.; Morgan, R.N.; Adams, B.R.; Hawkins, A.J.; Povirk, L.F.; Valerie, K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol. Ther. 2009, 8, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, C.R.; Markert, T.; Gillespie, G.Y.; Griguer, C.E. Nuclear-encoded cytochrome c oxidase subunit 4 regulates BMI1 expression and determines proliferative capacity of high-grade gliomas. Oncotarget 2015, 6, 4330–4344. [Google Scholar] [CrossRef] [Green Version]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; Raychaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 Oncogene/Stem Cell Renewal Factor by MicroRNA-128 Inhibits Glioma Proliferation and Self-Renewal. Cancer Res. 2008, 68, 9125–9130. [Google Scholar] [CrossRef] [Green Version]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef]
- Jacobs, J.J.L.; Kieboom, K.; Marino, S.; A DePinho, R.; Van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef]
- Facchino, S.; Abdouh, M.; Chatoo, W.; Bernier, G. BMI1 Confers Radioresistance to Normal and Cancerous Neural Stem Cells through Recruitment of the DNA Damage Response Machinery. J. Neurosci. 2010, 30, 10096–10111. [Google Scholar] [CrossRef]
- Han, X.; Xue, X.; Zhou, H.; Zhang, G. A molecular view of the radioresistance of gliomas. Oncotarget 2017, 8, 100931–100941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolós, V.; Blanco, M.; Medina, V.; Aparicio, G.; Díaz-Prado, S.; Grande, E.; Blanco, M. Notch signalling in cancer stem cells. Clin. Transl. Oncol. 2009, 11, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitoshi, S.; Alexson, T.; Tropepe, V.; Donoviel, D.; Elia, A.J.; Nye, J.S.; Conlon, R.A.; Mak, T.W.; Bernstein, A.; Van Der Kooy, D. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 2002, 16, 846–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitoshi, S.; Seaberg, R.M.; Koscik, C.; Alexson, T.; Kusunoki, S.; Kanazawa, I.; Tsuji, S.; Van Der Kooy, D. Primitive neural stem cells from the mammalian epiblast differentiate to definitive neural stem cells under the control of Notch signaling. Genes Dev. 2004, 18, 1806–1811. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wakeman, T.P.; Latha, J.D.; Hjelmeland, A.B.; Wang, X.-F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch Promotes Radioresistance of Glioma Stem Cells. Stem Cells 2009, 28, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowska, A.; Symons, M. Signaling determinants of glioma cell invasion. Adv. Exp. Med. Biol. 2013, 986, 121–141. [Google Scholar]
- Li, H.-F.; Kim, J.-S.; Waldman, T. Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells. Radiat. Oncol. 2009, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Mehta, M.; Khan, A.J.; Danish, S.; Haffty, B.G.; Sabaawy, H.E. Radiosensitization of Primary Human Glioblastoma Stem-like Cells with Low-Dose AKT Inhibition. Mol. Cancer Ther. 2015, 14, 1171–1180. [Google Scholar] [CrossRef] [Green Version]
- Matsutani, T.; Nagai, Y.; Mine, S.; Murai, H.; Saeki, N.; Iwadate, Y. Akt/protein kinase B overexpression as an accurate prognostic marker in adult diffuse astrocytoma. Acta Neurochir. (Wien) 2009, 151, 263–268. [Google Scholar] [CrossRef]
- Chakravarti, A.; Zhai, G.; Suzuki, Y.; Sarkesh, S.; Black, P.M.; Muzikansky, A.; Loeffler, J.S. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J. Clin. Oncol. 2004, 22, 1926–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chautard, E.; Loubeau, G.; Tchirkov, A.; Chassagne, J.; Vermot-Desroches, C.; Morel, L.; Verrelle, P. Akt signaling pathway: a target for radiosensitizing human malignant glioma. Neuro Oncol. 2010, 12, 434–443. [Google Scholar] [PubMed] [Green Version]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of Phosphatidylinositol-3-OH Kinase/Akt Signaling Impairs DNA Repair in Glioblastoma Cells following Ionizing Radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-A.; Liu, B.-H.; Shao, L.-M.; Guo, Z.-T.; Yang, Q.; Wu, L.-Q.; Ji, B.-W.; Zhu, X.-N.; Zhang, S.-Q.; Li, C.-J.; et al. LRIG1 enhances the radiosensitivity of radioresistant human glioblastoma U251 cells via attenuation of the EGFR/Akt signaling pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 3580–3590. [Google Scholar] [PubMed]
- Malik, U.; Javed, A. LRIGs: A Prognostically Significant Family with Emerging Therapeutic Competence against Cancers. Curr. Cancer Drug Targets 2016, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- McCabe, N.; Hanna, C.; Walker, S.M.; Gonda, D.; Li, J.; Wikstrom, K.; Savage, K.I.; Butterworth, K.T.; Chen, C.; Harkin, D.P.; et al. Mechanistic Rationale to Target PTEN-Deficient Tumor Cells with Inhibitors of the DNA Damage Response Kinase ATM. Cancer Res. 2015, 75, 2159–2165. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Benitez, J.A.; Li, J.; Miki, S.; De Albuquerque, C.P.; Galatro, T.; Orellana, L.; Zanca, C.; Reed, R.; Boyer, A.; et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell 2019, 35, 816. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.K.; Ye, Z.; Darras, B.T. Frequency of p53 tumor suppressor gene mutations in human primary brain tumors. Neurosurgery 1993, 33, 824–830. [Google Scholar] [PubMed]
- Chen, P.; Iavarone, A.; Fick, J.; Edwards, M.; Prados, M.; Israel, M.A. Constitutional p53 mutations associated with brain tumors in young adults. Cancer Genet. Cytogenet. 1995, 82, 106–115. [Google Scholar] [CrossRef]
- Shu, H.-K.G.; Kim, M.M.; Chen, P.; Furman, F.; Julin, C.M.; Israel, M.A. The intrinsic radioresistance of glioblastoma-derived cell lines is associated with a failure of p53 to induce p21BAX expression. Proc. Natl. Acad. Sci. USA 1998, 95, 14453–14458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef] [Green Version]
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor Tyrosine Kinase BMX Maintains Self-Renewal and Tumorigenic Potential of Glioblastoma Stem Cells by Activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesanakurti, D.; Chetty, C.; Maddirela, D.R.; Gujrati, M.; Rao, J.S. Essential role of cooperative NF-κB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene 2012, 32, 5144–5155. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-P.; Chang, Y.-L.; Huang, P.-I.; Chiou, G.-Y.; Tseng, L.-M.; Chiou, S.-H.; Chen, M.-H.; Chen, M.-T.; Shih, Y.-H.; Chang, C.-H.; et al. Resveratrol suppresses tumorigenicity and enhances radiosensitivity in primary glioblastoma tumor initiating cells by inhibiting the STAT3 axis. J. Cell. Physiol. 2011, 227, 976–993. [Google Scholar] [CrossRef]
- Xie, B.; Zhang, L.; Hu, W.; Fan, M.; Jiang, N.; Duan, Y.; Jing, D.; Xiao, W.; Fragoso, R.C.; Lam, K.S.; et al. Dual blockage of STAT3 and ERK1/2 eliminates radioresistant GBM cells. Redox Biol. 2019, 24, 101189. [Google Scholar] [CrossRef]
- Raychaudhuri, P.; Park, H.J. FoxM1: a master regulator of tumor metastasis. Cancer Res. 2011, 71, 4329–4333. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Fernández, M.; Medema, R. Novel functions of FoxM1: from molecular mechanisms to cancer therapy. Front. Oncol. 2013, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, K.H.; Kim, D.G.; Cho, H.J.; Kim, Y.; Rheey, J.; Shin, K.; Seo, Y.J.; Choi, Y.-S.; Lee, J.-I.; et al. FoxM1 Promotes Stemness and Radio-Resistance of Glioblastoma by Regulating the Master Stem Cell Regulator Sox2. PLoS ONE 2015, 10, e0137703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maachani, U.B.; Shankavaram, U.; Kramp, T.; Tofilon, P.J.; Camphausen, K.; Tandle, A.T. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget 2016, 7, 77365–77377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventero, M.P.; Fuentes-Baile, M.; Quereda, C.; Perez-Valeciano, E.; Alenda, C.; Garcia-Morales, P.; Esposito, D.; Dorado, P.; Manuel Barbera, V.; Saceda, M. Radiotherapy resistance acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE 2019, 14, e0212581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, M.; Pallini, R.; Luongo, G.; Cenci, T.; Lucantoni, C.; LaRocca, L.M. Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int. J. Cancer 2008, 123, 2955–2960. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Jin, X.; Jeon, H.-Y.; Joo, K.M.; Kim, J.K.; Jin, J.; Kim, S.H.; Kang, B.G.; Beck, S.; Lee, S.J.; Park, A.-K.; et al. Frizzled 4 Regulates Stemness and Invasiveness of Migrating Glioma Cells Established by Serial Intracranial Transplantation. Cancer Res. 2011, 71, 3066–3075. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Magnoni, L.; Miracco, C.; Mori, E.; Tosi, P.; Pirtoli, L.; Tini, P.; Oliveri, G.; Cosci, E.; Bakker, A. β-catenin and Gli1 are prognostic markers in glioblastoma. Cancer Biol. Ther. 2011, 11, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Tompa, M.; Kalovits, F.; Nagy, A.; Kalman, B. Contribution of the Wnt Pathway to Defining Biology of Glioblastoma. Neuro Molecular Med. 2018, 20, 437–451. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.-K.; Ahn, S.H.; Lee, J.; Nam, D.-H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. 2015, 96, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Mccord, M.; Mukouyama, Y.-S.; Gilbert, M.R.; Jackson, S. Targeting WNT Signaling for Multifaceted Glioblastoma Therapy. Front. Cell. Neurosci. 2017, 11, 318. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kim, K.H.; Lee, H.; Yang, H.; Kim, D.; Kang, W.; Jin, J.; Joo, K.M.; Lee, J.; Nam, D.-H. Wnt activation is implicated in glioblastoma radioresistance. In Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research, Chicago, IL, USA, 31 March–4 April 2012. Abstract nr 3458. [Google Scholar]
- Dong, Z.; Zhou, L.; Han, N.; Zhang, M.; Lyu, X. Wnt/β-catenin pathway involvement in ionizing radiation-induced invasion of U87 glioblastoma cells. Strahlenther Onkol. 2015, 191, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Barnett, G.C.; West, C.M.L.; Dunning, A.M.; Elliott, R.M.; Coles, C.E.; Pharoah, P.D.P.; Burnet, N.G. Normal tissue reactions to radiotherapy: towards tailoring treatment dose by genotype. Nat. Rev. Cancer 2009, 9, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Med Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Marie-Egyptienne, D.T.; Lohse, I.; Hill, R. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett. 2013, 341, 63–72. [Google Scholar] [CrossRef]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Lalla, R.V.; Treister, N.; Sollecito, T.; Schmidt, B.; Patton, L.L.; Mohammadi, K.; Hodges, J.S.; Brennan, M.T. OraRad Study Group Oral complications at 6 months after radiation therapy for head and neck cancer. Oral Dis. 2017, 23, 1134–1143. [Google Scholar] [CrossRef]
- Shenoy, M.A.; Singh, B.B. Chemical Radiosensitizers in Cancer Therapy. Cancer Investig. 1992, 10, 533–551. [Google Scholar] [CrossRef]
- Wang, H.; Mu, X.; He, H.; Zhang, X.-D. Cancer Radiosensitizers. Trends Pharmacol. Sci. 2018, 39, 24–48. [Google Scholar] [CrossRef]
- Wardman, P. Chemical radiosensitizers for use in radiotherapy. Clin. Oncol. (R. Coll. Radiol.) 2007, 196, 397–417. [Google Scholar] [CrossRef]
- Hodgkiss, R.; Middleton, R. Enhancement of Misonidazole Radiosensitization by an Inhibitor of Glutathione Biosynthesis. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1983, 43, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Seefeldt, T.; Chen, W.; Carlson, L.; Stoebner, A.; Hanson, S.; Foll, R.; Matthees, D.P.; Palakurthi, S.; Guan, X. Increase in thiol oxidative stress via glutathione reductase inhibition as a novel approach to enhance cancer sensitivity to X-ray irradiation. Free. Radic. Biol. Med. 2009, 47, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bache, M.; Zschornak, M.P.; Passin, S.; Keßler, J.; Wichmann, H.; Kappler, M.; Paschke, R.; Kaluđerović, G.N.; Kommera, H.; Taubert, H.; et al. Increased betulinic acid induced cytotoxicity and radiosensitivity in glioma cells under hypoxic conditions. Radiat. Oncol. 2011, 6, 111. [Google Scholar] [CrossRef] [Green Version]
- Raleigh, D.R.; Haas-Kogan, D.A. Molecular targets and mechanisms of radiosensitization using DNA damage response pathways. Futur. Oncol. 2013, 9, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Adams, G.E. Chemical radiosensitization of hypoxic cells. Br. Med Bull. 1973, 29, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.F.; Adams, E.G.; Denekamp, J. Radiosensitizers of hypoxic cells in solid tumors. Cancer Treat. Rev. 1976, 3, 227–256. [Google Scholar] [CrossRef]
- Goel, S.; Ni, D.; Cai, W. Harnessing the Power of Nanotechnology for Enhanced Radiation Therapy. ACS Nano 2017, 11, 5233–5237. [Google Scholar] [CrossRef]
- Kunz-Schughart, L.A.; Dubrovska, A.; Peitzsch, C.; Ewe, A.; Aigner, A.; Schellenburg, S.; Muders, M.H.; Hampel, S.; Cirillo, G.; Iemma, F.; et al. Nanoparticles for radiooncology: Mission, vision, challenges. Biomater 2017, 120, 155–184. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T.S.; Blackstock, A.; McGinn, C. The mechanism of action of radiosensitization of conventional chemotherapeutic agents. Semin. Radiat. Oncol. 2003, 13, 13–21. [Google Scholar] [CrossRef]
- Wilson, G.D.; Bentzen, S.M.; Harari, P. Biologic Basis for Combining Drugs With Radiation. Semin. Radiat. Oncol. 2006, 16, 2–9. [Google Scholar] [CrossRef]
- Shewach, D.S.; Lawrence, T.S. Radiosensitization of human solid tumor cell lines with gemcitabine. Semin. Oncol. 1996, 23, 65–71. [Google Scholar] [PubMed]
- Lawrence, T.S.; Eisbruch, A.; McGinn, C.J.; Fields, M.T.; Shewach, D.S. Radiosensitization by gemcitabine. Oncol. (Williston Park) 1999, 13, 55–60. [Google Scholar]
- Azria, D.; Jacot, W.; Prost, P.; Culine, S.; Ychou, M.; Lemanski, C.; Dubois, J.-B. Gemcitabine et radiations ionisantes: radiosensibilisation ou association radiochimiothérapique [Gemcitabine and ionizing radiations: radiosensitization or radio-chemotherapy combination]. Bull. Cancer 2002, 89, 369–379. [Google Scholar] [PubMed]
- Cuneo, K.C.; Mehta, R.K.; Kurapati, H.; Thomas, D.G.; Lawrence, T.S.; Nyati, M.K. Enhancing the Radiation Response in KRAS Mutant Colorectal Cancers Using the c-Met Inhibitor Crizotinib. Transl. Oncol. 2018, 12, 209–216. [Google Scholar] [CrossRef]
- Nolte, E.M.; Joubert, A.; Lakier, R.; Van Rensburg, A.; Mercier, A.E. Exposure of Breast and Lung Cancer Cells to a Novel Estrone Analog Prior to Radiation Enhances Bcl-2-Mediated Cell Death. Int. J. Mol. Sci. 2018, 19, 2887. [Google Scholar] [CrossRef] [Green Version]
- Rey, S.; Schito, L.; Koritzinsky, M.; Wouters, B. Molecular targeting of hypoxia in radiotherapy. Adv. Drug Deliv. Rev. 2017, 109, 45–62. [Google Scholar] [CrossRef]
- Horsman, M.R.; Overgaard, J. The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat. Res. 2016, 57, i90–i98. [Google Scholar] [CrossRef] [Green Version]
- Bousquet, P.A.; Meltzer, S.; Sønstevold, L.; Esbensen, Y.; Dueland, S.; Flatmark, K.; Sitter, B.; Bathen, T.F.; Seierstad, T.; Redalen, K.R.; et al. Markers of Mitochondrial Metabolism in Tumor Hypoxia, Systemic Inflammation, and Adverse Outcome of Rectal Cancer. Transl. Oncol. 2019, 12, 76–83. [Google Scholar] [CrossRef]
- Chapman, J.D.; Webb, R.G.; Borsa, J. Radiosensitization of mammalian cells by p-nitroacetophenone. I. Characterization in asynchronous and synchronous populations. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1971, 19, 561–573. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.E.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Overgaard, J.; Hansen, H.S.; Andersen, A.; Hjelm-Hansen, M.; Jørgensen, K.; Sandberg, E.; Berthelsen, A.; Hammer, R.; Pedersen, M. Misonidazole combined with split-course radiotherapy in the treatment of invasive carcinoma of larynx and pharynx: Report from the DAHANCA 2 study. Int. J. Radiat. Oncol. 1989, 16, 1065–1068. [Google Scholar] [CrossRef]
- Urtasun, R.C.; Chapman, J.D.; Feldstein, M.L.; Band, R.P.; Rabin, H.R.; Wilson, A.F.; Marynowski, B.; Starreveld, E.; Shnitka, T. Peripheral neuropathy related to misonidazole: Incidence and pathology. Br. J. Cancer Suppl. 1978, 3, 271–275. [Google Scholar] [PubMed]
- Takaoka, T.; Shibamoto, Y.; Matsuo, M.; Sugie, C.; Murai, T.; Ogawa, Y.; Miyakawa, A.; Manabe, Y.; Kondo, T.; Nakajima, K.; et al. Biological effects of hydrogen peroxide administered intratumorally with or without irradiation in murine tumors. Cancer Sci. 2017, 108, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.J.; Oprysko, P.R.; Shuman, A.L.; Jenkins, W.T.; Brandt, G.; Evans, S. Radiosensitization of hypoxic tumor cells by dodecafluoropentane: a gas-phase perfluorochemical emulsion. Cancer Res. 2002, 62, 3626–3629. [Google Scholar]
- Harrison, D.K.; Vaupel, P. Heterogeneity in Tissue Oxygenation: From Physiological Variability in Normal Tissues to Pathophysiological Chaos in Malignant Tumours. Adv. Exp. Med. Biol. 2014, 812, 25–31. [Google Scholar]
- Gallez, B.; Neveu, M.-A.; Danhier, P.; Jordan, B.F. Manipulation of tumor oxygenation and radiosensitivity through modification of cell respiration. A critical review of approaches and imaging biomarkers for therapeutic guidance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 700–711. [Google Scholar] [CrossRef]
- Benej, M.; Hong, X.; Vibhute, S.; Scott, S.; Wu, J.; Graves, E.; Le, Q.-T.; Koong, A.C.; Giaccia, A.J.; Yu, B.; et al. Papaverine and its derivatives radiosensitize solid tumors by inhibiting mitochondrial metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 10756–10761. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.E.; Khuntia, D.; Robins, H.I.; Mehta, M.P. Radiotherapy and radiosensitizers in the treatment of glioblastoma multiforme. Clin. Adv. Hematol. Oncol. 2007, 5, 894–915. [Google Scholar]
- Bayin, N.S.; Ma, L.; Placantonakis, D.G.; Barcellos-Hoff, M.H. Evaluation of Radioresponse and Radiosensitizers in Glioblastoma Organotypic Cultures. In Glioblastoma. Methods in Molecular Biology; Placantonakis, D., Ed.; Humana Press: New York, NY, USA, 2018; Volume 1741, pp. 171–182. [Google Scholar]
- Sigmond, J.; Honeywell, R.J.; Postma, T.J.; Dirven, C.M.F.; De Lange, S.M.; Van Der Born, K.; Laan, A.C.; Baayen, J.C.A.; Van Groeningen, C.J.; Bergman, A.M.; et al. Gemcitabine uptake in glioblastoma multiforme: potential as a radiosensitizer. Ann. Oncol. 2009, 20, 182–187. [Google Scholar] [CrossRef]
- Van Nifterik, K.A.; Berg, J.V.D.; Stalpers, L.J.; LaFleur, M.V.M.; Leenstra, S.; Slotman, B.J.; Hulsebos, T.J.; Sminia, P. Differential Radiosensitizing Potential of Temozolomide in MGMT Promoter Methylated Glioblastoma Multiforme Cell Lines. Int. J. Radiat. Oncol. 2007, 69, 1246–1253. [Google Scholar] [CrossRef]
- Metro, G.; Fabi, A.; Mirri, M.A.; Vidiri, A.; Pace, A.; Carosi, M.; Russillo, M.; Maschio, M.; Giannarelli, D.; Pellegrini, D.; et al. Phase II study of fixed dose rate gemcitabine as radiosensitizer for newly diagnosed glioblastoma multiforme. Cancer Chemother. Pharmacol. 2009, 65, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Setua, S.; Ouberai, M.; Piccirillo, S.G.M.; Watts, C.; Welland, M. Cisplatin-tethered gold nanospheres for multimodal chemo-radiotherapy of glioblastoma. Nanoscale 2014, 6, 10865–10873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz van Haperen, V.W.; Veerman, G.; Vermorken, J.B.; Peters, G.J. 2′,2′-Difluoro-deoxycytidine (gemcitabine) incorporation into RNA and DNA of tumour cell lines. Biochem. Pharmacol. 1993, 46, 762–766. [Google Scholar] [CrossRef]
- Ouédraogo, Z.G.; Müller-Barthélémy, M.; Kemeny, J.-L.; Dedieu, V.; Biau, J.; Khalil, T.; Raoelfils, L.I.; Granzotto, A.; Pereira, B.; Beaudoin, C.; et al. STAT3 Serine 727 Phosphorylation: A Relevant Target to Radiosensitize Human Glioblastoma. Brain Pathol. 2015, 26, 18–30. [Google Scholar] [CrossRef]
- Lesueur, P.; Chevalier, F.; El-Habr, E.; Junier, M.-P.; Chneiweiss, H.; Castéra, L.; Muller, E.; Stefan, D.; Saintigny, Y. Radiosensitization Effect of Talazoparib, a Parp Inhibitor, on Glioblastoma Stem Cells Exposed to Low and High Linear Energy Transfer Radiation. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Narayan, R.S.; Gasol, A.; Slangen, P.L.; Cornelissen, F.M.; Lagerweij, T.; Veldman, H.Y.; Dik, R.; Berg, J.V.D.; Slotman, B.J.; Wurdinger, T.; et al. Identification of MEK162 as a Radiosensitizer for the Treatment of Glioblastoma. Mol. Cancer Ther. 2017, 17, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Prados, M.D.; Chang, S.M.; Butowski, N.; DeBoer, R.; Parvataneni, R.; Carliner, H.; Kabuubi, P.; Ayers-Ringler, J.; Rabbitt, J.; Page, M.; et al. Phase II Study of Erlotinib Plus Temozolomide During and After Radiation Therapy in Patients With Newly Diagnosed Glioblastoma Multiforme or Gliosarcoma. J. Clin. Oncol. 2009, 27, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Yu, H.-H.M.; Stieber, V.; Malone, S.C.; et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: results of NRG Oncology RTOG 0913. Neuro Oncol. 2017, 20, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. 2015, 92, 986–992. [Google Scholar] [CrossRef] [Green Version]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: results of Alliance N0874/ABTC 02. Neuro Oncol. 2018, 20, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Q.; Kaley, T.J.; Duda, D.G.; Schiff, D.; Lassman, A.B.; Wong, E.T.; Mikkelsen, T.; Purow, B.W.; Muzikansky, A.; Ancukiewicz, M.; et al. A Multicenter, Phase II, Randomized, Noncomparative Clinical Trial of Radiation and Temozolomide with or without Vandetanib in Newly Diagnosed Glioblastoma Patients. Clin. Cancer Res. 2015, 21, 3610–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butowski, N.; Chang, S.M.; Lamborn, K.R.; Polley, M.; Pieper, R.; Costello, J.F.; Vandenberg, S.; Parvataneni, R.; Nicole, A.; Sneed, P.K.; et al. Phase II and pharmacogenomics study of enzastaurin plus temozolomide during and following radiation therapy in patients with newly diagnosed glioblastoma multiforme and gliosarcoma. Neuro Oncol. 2011, 13, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Ye, X.; Chamberlain, M.; Mikkelsen, T.; Batchelor, T.; Desideri, S.; Piantadosi, S.; Fisher, J.; Fine, H.A. Talampanel With Standard Radiation and Temozolomide in Patients With Newly Diagnosed Glioblastoma: A Multicenter Phase II Trial. J. Clin. Oncol. 2009, 27, 4155–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, B.L.; Grogan, P.T.; Mladek, A.C.; Schroeder, M.A.; Kitange, G.J.; Decker, P.A.; Giannini, C.; Wu, W.; Ballman, K.V.; James, C.D.; et al. Radiosensitizing Effects of Temozolomide Observed in vivo only in a Subset of O6-Methylguanine-DNA Methyltransferase Methylated Glioblastoma Multiforme Xenografts. Int. J. Radiat. Oncol. 2009, 75, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.-T.; Nguyen, N.T.; Choi, Y.J.; Zhang, G.; Nguyen, H.N.; Filka, E.; Green, S.; Yong, W.H.; Liau, L.M.; Green, R.M.; et al. Phase 2 Study of Bortezomib Combined With Temozolomide and Regional Radiation Therapy for Upfront Treatment of Patients With Newly Diagnosed Glioblastoma Multiforme: Safety and Efficacy Assessment. Int. J. Radiat. Oncol. 2018, 100, 1195–1203. [Google Scholar] [CrossRef]
- Jue, T.R.; Nozue, K.; Lester, A.; Joshi, S.; Schroder, L.B.W.; Whittaker, S.P.; Nixdorf, S.; Rapkins, R.W.; Khasraw, M.; McDonald, K.L. Veliparib in combination with radiotherapy for the treatment of MGMT unmethylated glioblastoma. J. Transl. Med. 2017, 15, 61. [Google Scholar] [CrossRef] [Green Version]
- Sarcar, B.; Kahali, S.; Prabhu, A.H.; Shumway, S.D.; Xu, Y.; DeMuth, T.; Chinnaiyan, P. Targeting radiation-induced G(2) checkpoint activation with the Wee-1 inhibitor MK-1775 in glioblastoma cell lines. Mol. Cancer Ther. 2011, 10, 2405–2414. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.-Q.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.G.; Bodeker, K.L.; Smith, M.C.; Monga, V.; Sandhu, S.; Hohl, R.J.; Carlisle, T.L.; Brown, H.; Hollenbeck, N.J.; Vollstedt, S.; et al. First-in-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined with Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2019, 25, 6590–6597. [Google Scholar] [CrossRef] [Green Version]
- Oronsky, B.; Scicinski, J.; Ning, S.; Peehl, D.; Oronsky, A.; Cabrales, P.; Bednarski, M.; Knox, S. RRx-001, A novel dinitroazetidine radiosensitizer. Investig. New Drugs 2016, 34, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachman, D.G.; Pugh, S.L.; Ashby, L.S.; Thomas, T.A.; Dunbar, E.M.; Narayan, S.; Robins, H.I.; Bovi, J.A.; Rockhill, J.K.; Won, M.; et al. Phase 1/2 trials of Temozolomide, Motexafin Gadolinium, and 60-Gy fractionated radiation for newly diagnosed supratentorial glioblastoma multiforme: final results of RTOG 0513. Int. J. Radiat. Oncol. 2015, 91, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, K.; Unger, E.C. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainer, J.L.; Sheehan, J.P.; Larner, J.M.; Jones, D.R. Trans sodium crocetinate with temozolomide and radiation therapy for glioblastoma multiforme. J. Neurosurg. 2017, 126, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Grimm, S.A.; Marymont, M.; Chandler, J.P.; Muro, K.; Newman, S.B.; Levy, R.M.; Jovanovic, B.; McCarthy, K.; Raizer, J.J. Phase I study of arsenic trioxide and temozolomide in combination with radiation therapy in patients with malignant gliomas. J. Neuro-Oncology 2012, 110, 237–243. [Google Scholar] [CrossRef]
- Kumthekar, P.; Grimm, S.; Chandler, J.; Mehta, M.; Marymont, M.; Levy, R.; Muro, K.; Helenowski, I.; McCarthy, K.; Fountas, L.; et al. A phase II trial of arsenic trioxide and temozolomide in combination with radiation therapy for patients with malignant gliomas. J. Neurooncol. 2017, 133, 589–594. [Google Scholar] [CrossRef]
- Bell, J.B.; Eckerdt, F.; Dhruv, H.D.; Finlay, D.; Peng, S.; Kim, S.; Kroczynska, B.; Beauchamp, E.M.; Alley, K.; Clymer, J.; et al. Differential Response of Glioma Stem Cells to Arsenic Trioxide Therapy Is Regulated by MNK1 and mRNA Translation. Mol. Cancer Res. 2017, 16, 32–46. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, S.; Wada, K.; Nagatani, K.; Otani, N.; Osada, H.; Nawashiro, H. Sulfasalazine and temozolomide with radiation therapy for newly diagnosed glioblastoma. Neurol. India 2014, 62, 42. [Google Scholar] [CrossRef]
- Whittaker, S.; Madani, D.; Joshi, S.; Chung, A.S.; Johns, T.; Day, B.; Khasraw, M.; McDonald, K.L. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov. 2017, 3, 17033. [Google Scholar] [CrossRef]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2014, 9, 192–203. [Google Scholar] [CrossRef]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compter, I.; Eekers, D.; Hoeben, A.; Rouschop, K.; Reymen, B.; Ackermans, L.; Beckervordersantforth, J.; Bauer, N.; Anten, M.; Wesseling, P.; et al. CHLOROBRAIN phase IB trial: The addition of chloroquine, an autophagy inhibitor, to concurrent radiation and temozolomide for newly diagnosed glioblastoma. Ann. Oncol. 2019, 30, v154. [Google Scholar] [CrossRef]




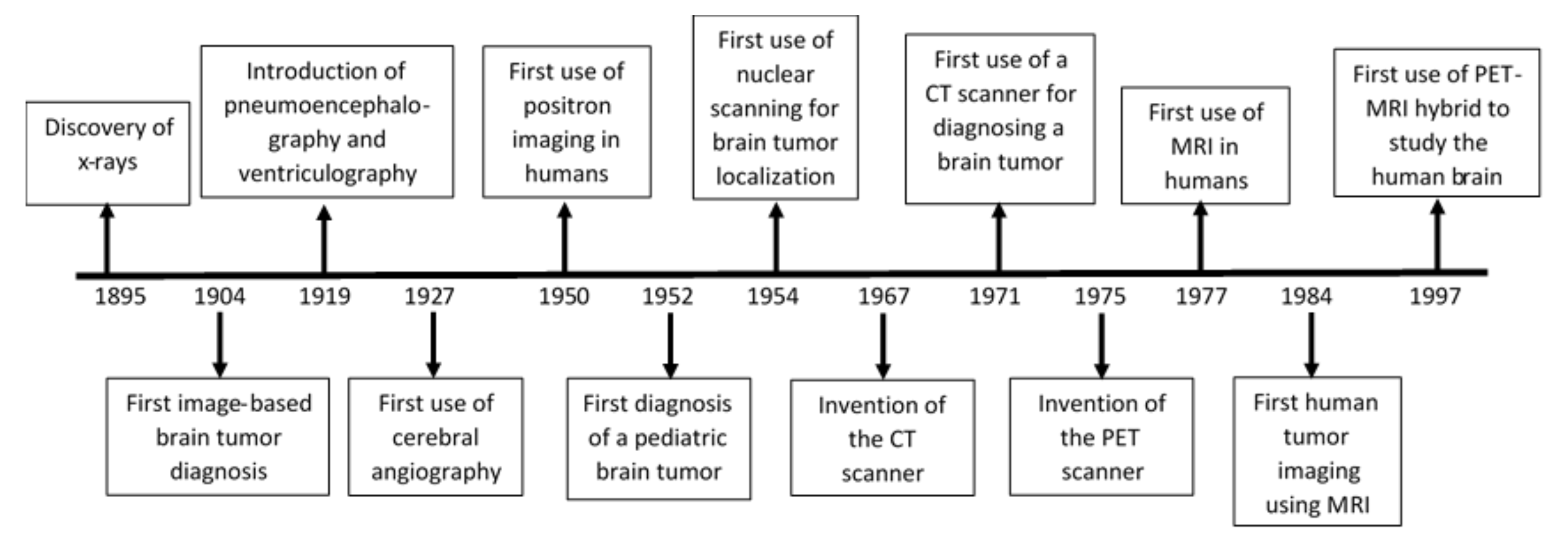{kind=link}
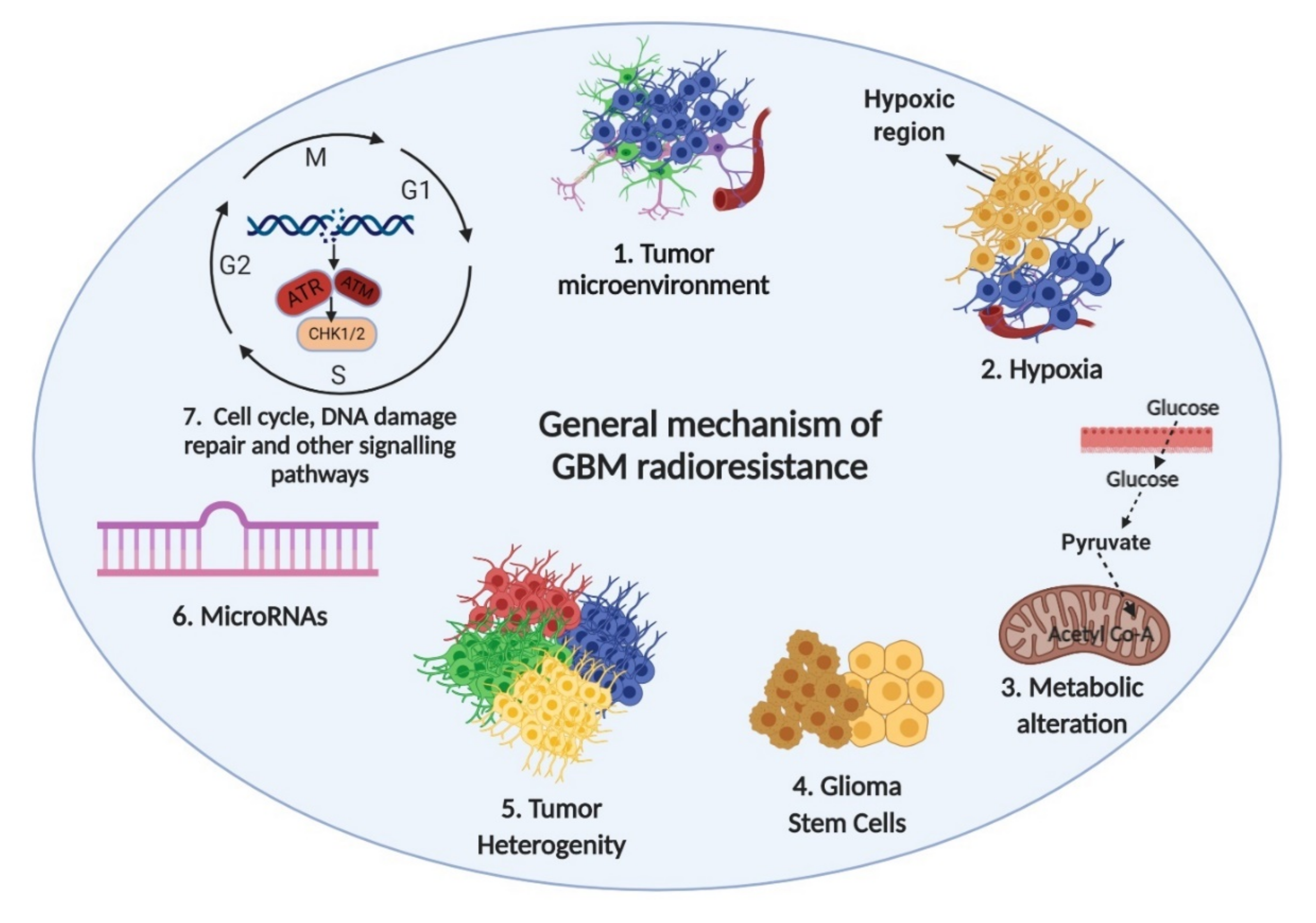{kind=link}
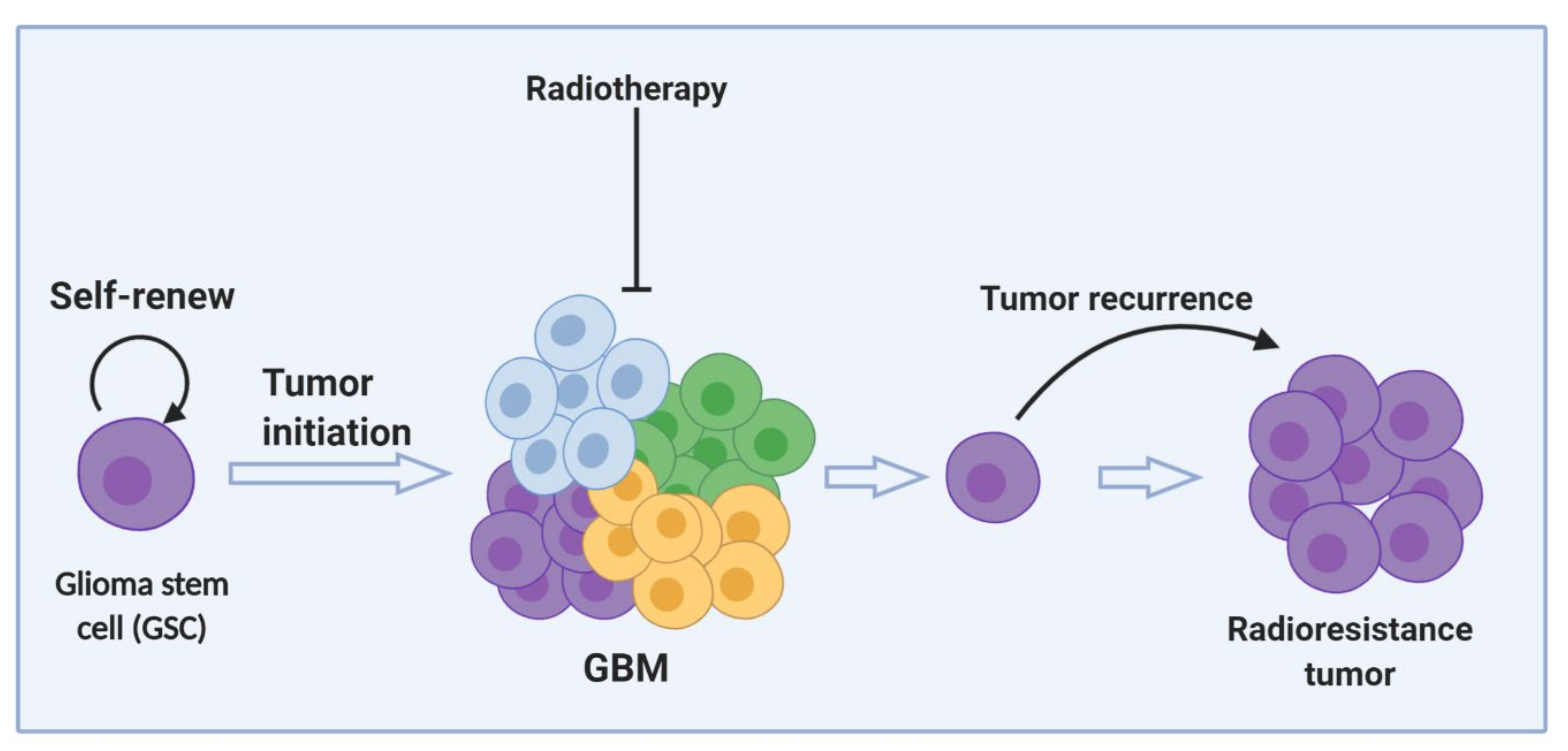{kind=link}
| Name of the Radiosensitizers | Effect | References |
|---|---|---|
| Gemcitabine | Initiates DNA damage by incorporating gemcitabine triphosphate, an active metabolite of gemcitabine, instead of nucleotide deoxycytidine triphosphate (dCTP) | [272,276] |
| Gö6976 | Protein kinase inhibitor | [277] |
| Talazoparib | PARP inhibitor | [278] |
| MEK162 | MAPK inhibitor | [279] |
| Erlotinib | EGFR inhibitor | [280] |
| Everolimus | mTOR inhibitor | [281] |
| Valproate | HDAC | [282] |
| Vorinostat | HDAC inhibitor | [283] |
| Vandetanib | VEGFR2 inhibitor | [284] |
| Enzastaurin | Protein Kinase C (PKC) inhibitor | [285] |
| Talampanel | alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist | [286] |
| TMZ | Alkylates/methylates DNA at N-7 or O-6 positions of guanine residue | [287] |
| Bortezomib | Proteasome inhibitor | [288] |
| Resveratrol | STAT3 inhibitor | [218] |
| Veliparib | PARP inhibitor | [289] |
| Adavosertib | WEE1 inhibitor | [290] |
| Chloroquine | Inhibits autophagy and induces apoptosis | [291,292] |
| Ascorbate | Pro-oxidant | [293] |
| RRx-001 | Macrophage-stimulating agent | [294] |
| Motexafin gadolinium | Inhibits thioredoxin reductase and ribonucleotide reductase | [295] |
| NVX-108 | Carries oxygen to the hypoxic tissue | [296] |
| Trans sodium crocetinate | Enhances oxygen levels in hypoxic tissue | [297] |
| Arsenic trioxide | Activates apoptosis and autophagy | [298,299,300] |
| Sulfasalazine | Inhibits cystine uptake | [301] |
| Palbociclib | CDK inhibitor | [302] |
| KU - 55933 | ATM inhibitor | [303] |
| AZD1390 | ATM inhibitor | [304] |
| Study ID | Phase | Diagnosis | Treatment | Outcomes |
|---|---|---|---|---|
| NCT01752491 | I | GBM | Ascorbate, TMZ, and radiotherapy | No dose-limiting toxicities [293] |
| NCT01465347 | I & II | GBM | Trans sodium crocetinate (TSC), TMZ, and radiotherapy | No adverse effects. Suggests radiotherapy and TSC combination is beneficial for GBM treatment. No significant difference in overall survival [297]. |
| NCT00185861 | I | Recurrent malignant glioma | Arsenic trioxide (ATO) and stereotactic radiotherapy | ATO and fractionated stereotactic radiotherapy is well-tolerated [298] |
| NCT04205357 | I | Recurrent GBM | Sulfasalazine and stereotactic radiotherapy | Study ongoing, recruiting patients |
| NCT02871843 | I | GBM, oligodendroglioma, anaplastic oligodendroglioma | RRx-001, TMZ, and radiotherapy | Study ongoing |
| NCT00302159 | II | High-grade gliomas | Valproic acid (VPA), TMZ, and radiotherapy | No adverse effects; VPA in combination with TMZ and radiotherapy can improve outcome [282] |
| NCT00305864 | I & II | GBM | Motexafin gadolinium, TMZ, and radiotherapy | No adverse effects; no significant improvement in overall survival [295] |
| NCT03862430 | II | GBM | NVX-108, TMZ, and radiotherapy | Study ongoing |
| NCT03672721 | I & II | GBM | Carboplatin and radiotherapy | Study ongoing |
| NCT02378532 | I | GBM | Chloroquine, TMZ, and radiotherapy | No adverse effects reported [305] |
| NCT02432417 | II | GBM | Chloroquine, TMZ, and radiotherapy | Study ongoing |
| NCT01849146 | I | Newly diagnosed and recurrent GBM | Adavosertib, TMZ, and radiotherapy | Study ongoing |
| NCT03423628 | I | GBM | AZD1390 and Radiotherapy | Study ongoing |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. https://doi.org/10.3390/cancers12092511
Ali MY, Oliva CR, Noman ASM, Allen BG, Goswami PC, Zakharia Y, Monga V, Spitz DR, Buatti JM, Griguer CE. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers. 2020; 12(9):2511. https://doi.org/10.3390/cancers12092511
Chicago/Turabian StyleAli, Md Yousuf, Claudia R. Oliva, Abu Shadat M. Noman, Bryan G. Allen, Prabhat C. Goswami, Yousef Zakharia, Varun Monga, Douglas R. Spitz, John M. Buatti, and Corinne E. Griguer. 2020. "Radioresistance in Glioblastoma and the Development of Radiosensitizers" Cancers 12, no. 9: 2511. https://doi.org/10.3390/cancers12092511