The ATF6-EGF Pathway Mediates the Awakening of Slow-Cycling Chemoresistant Cells and Tumor Recurrence by Stimulating Tumor Angiogenesis

 , and
, and
Abstract
:1. Introduction
2. Results
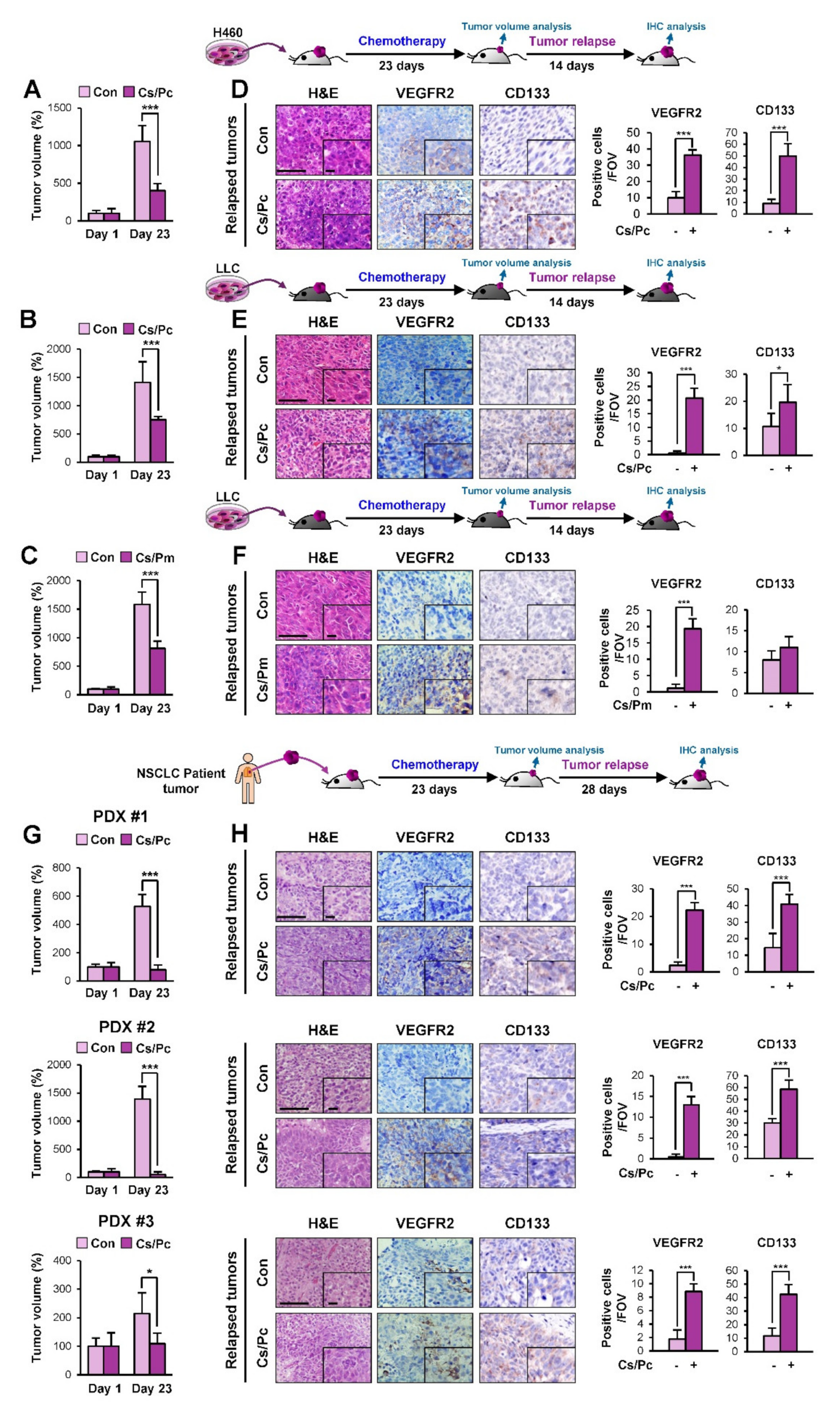
2.1. Increased Vasculature in NSCLC Cell Line- and Patient-Derived Xenograft Tumors that Relapsed Upon Completion of Chemotherapy
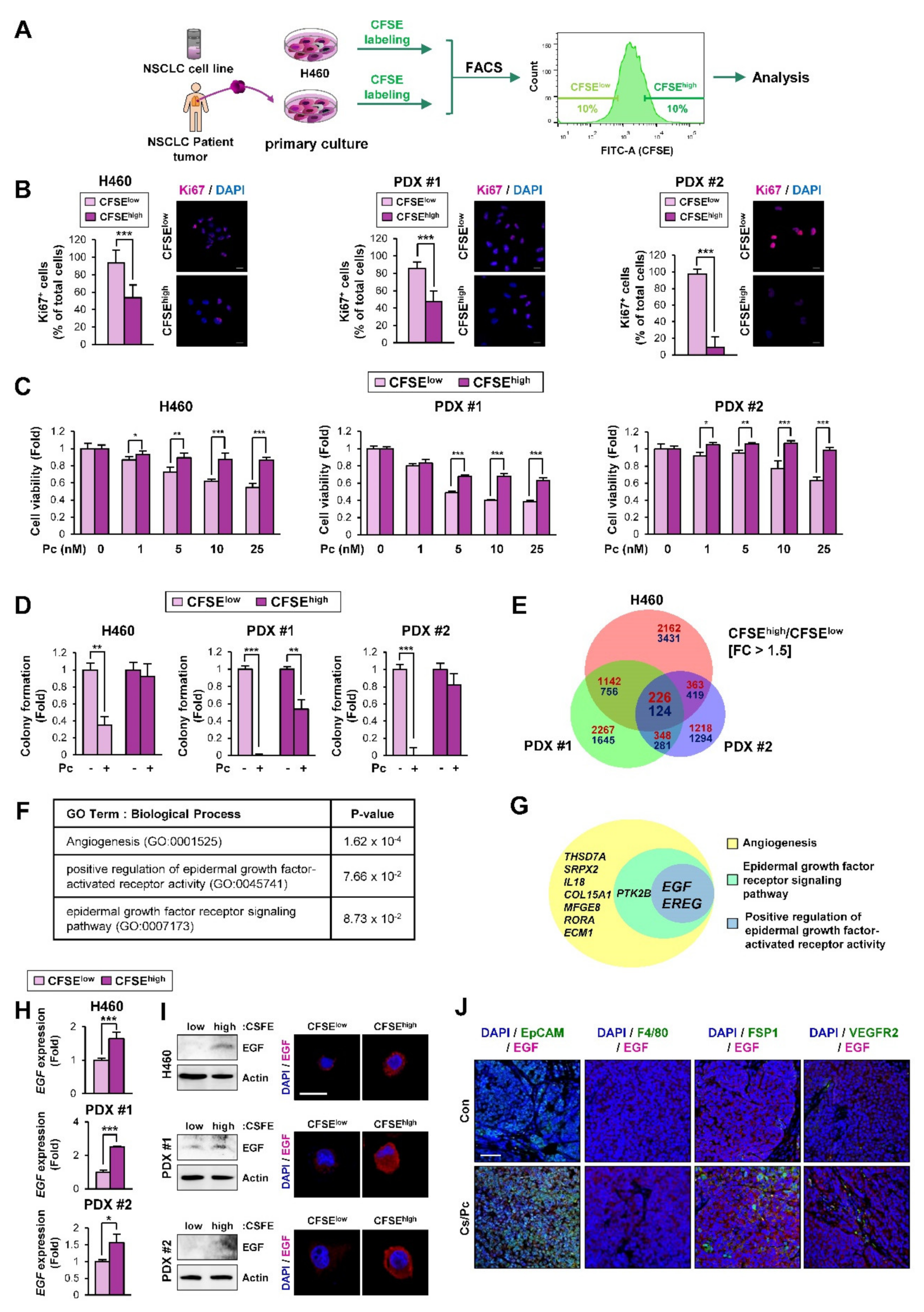
2.2. EGF-Related Gene Sets Are Significantly Enriched in NSCLC Cells with Quiescence-Like Phenotypes Derived from the NSCLC Cell Line and PDX Tumors
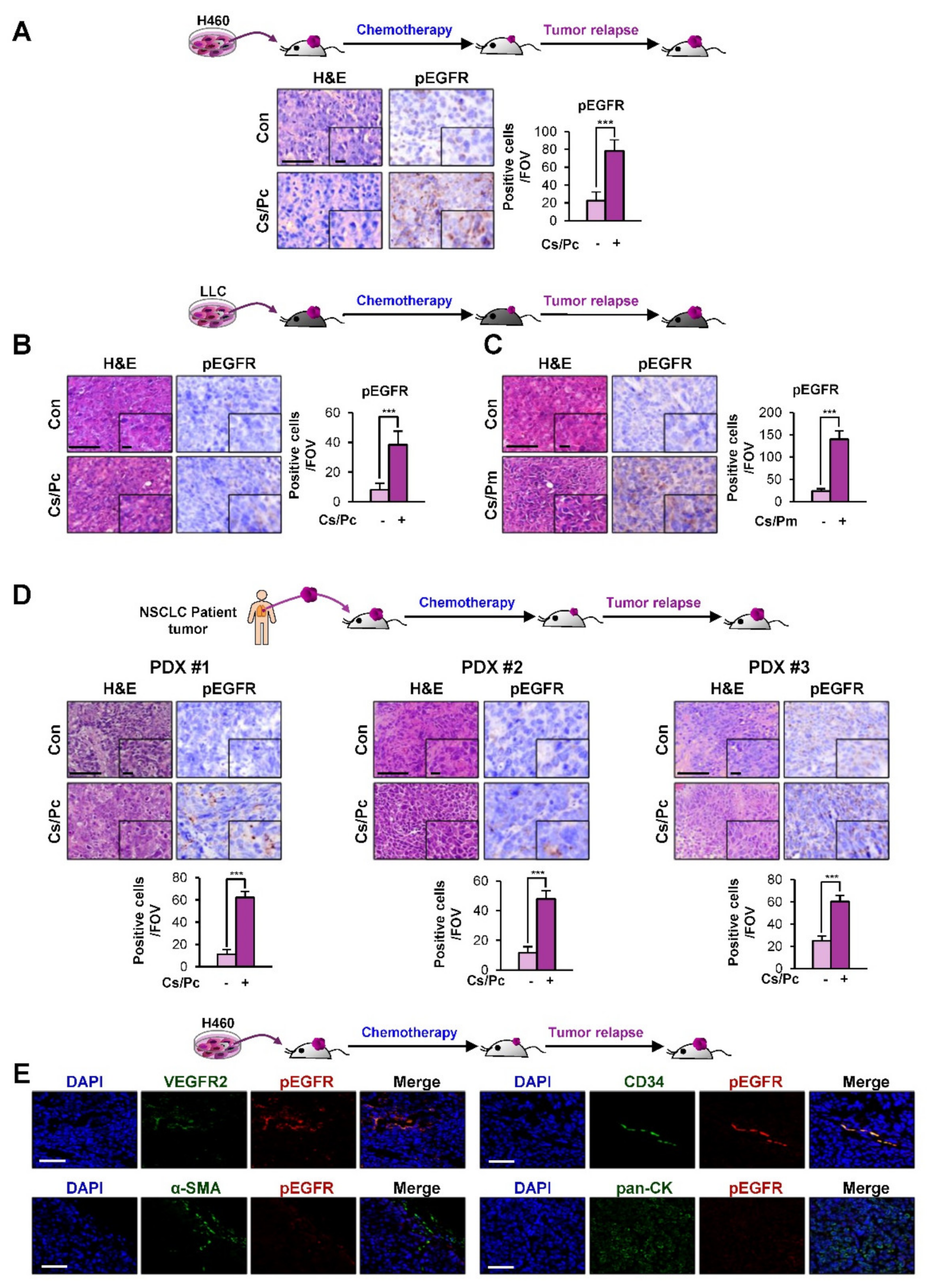
2.3. Activation of EGFR in Residual NSCLC Cell Line-Derived and PDX Tumors after Chemotherapy
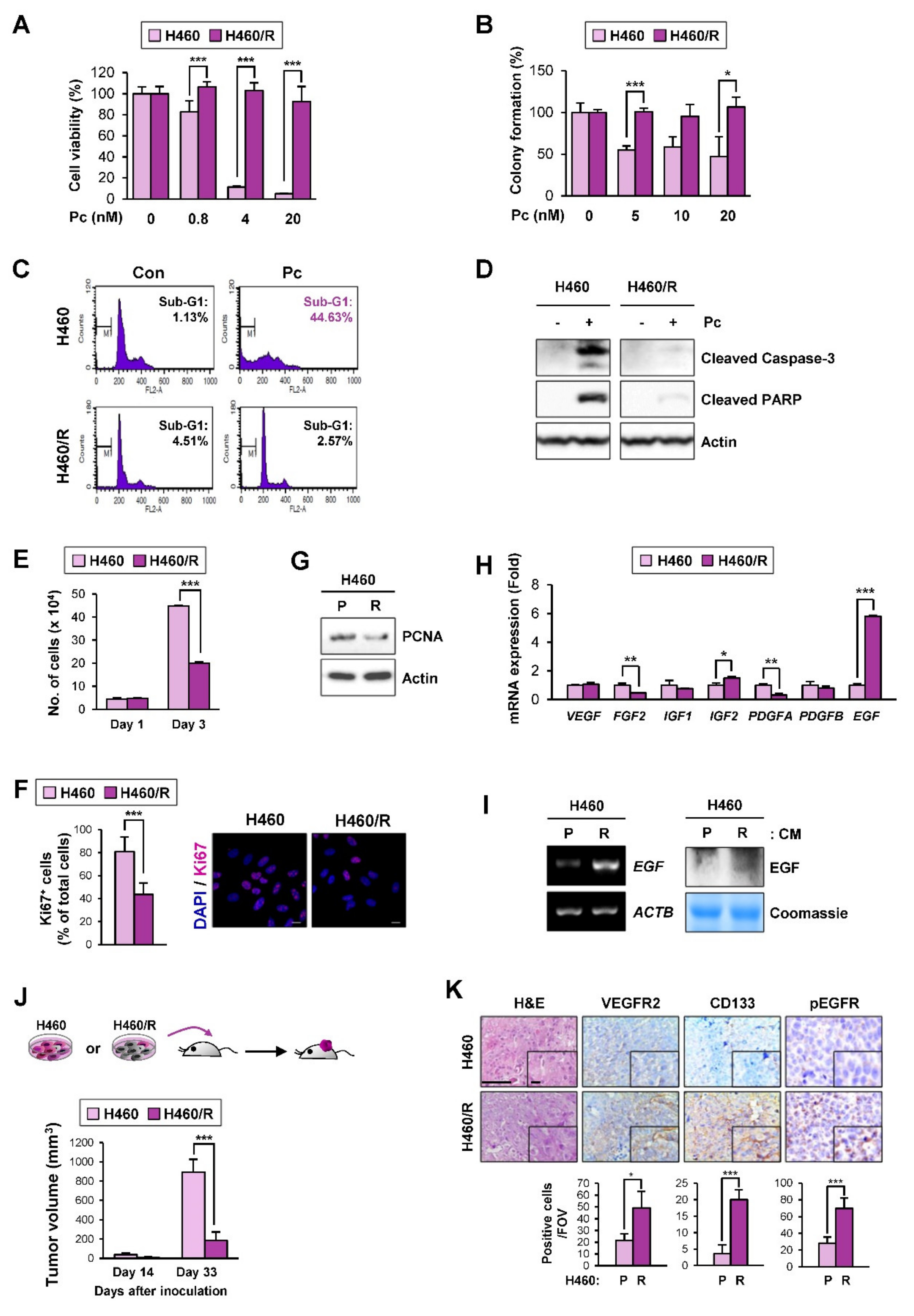
2.4. EGF Expression Was Significantly Upregulated in the H460 NSCLC Cell Subpopulation Carrying Quiescence-Like Phenotypes
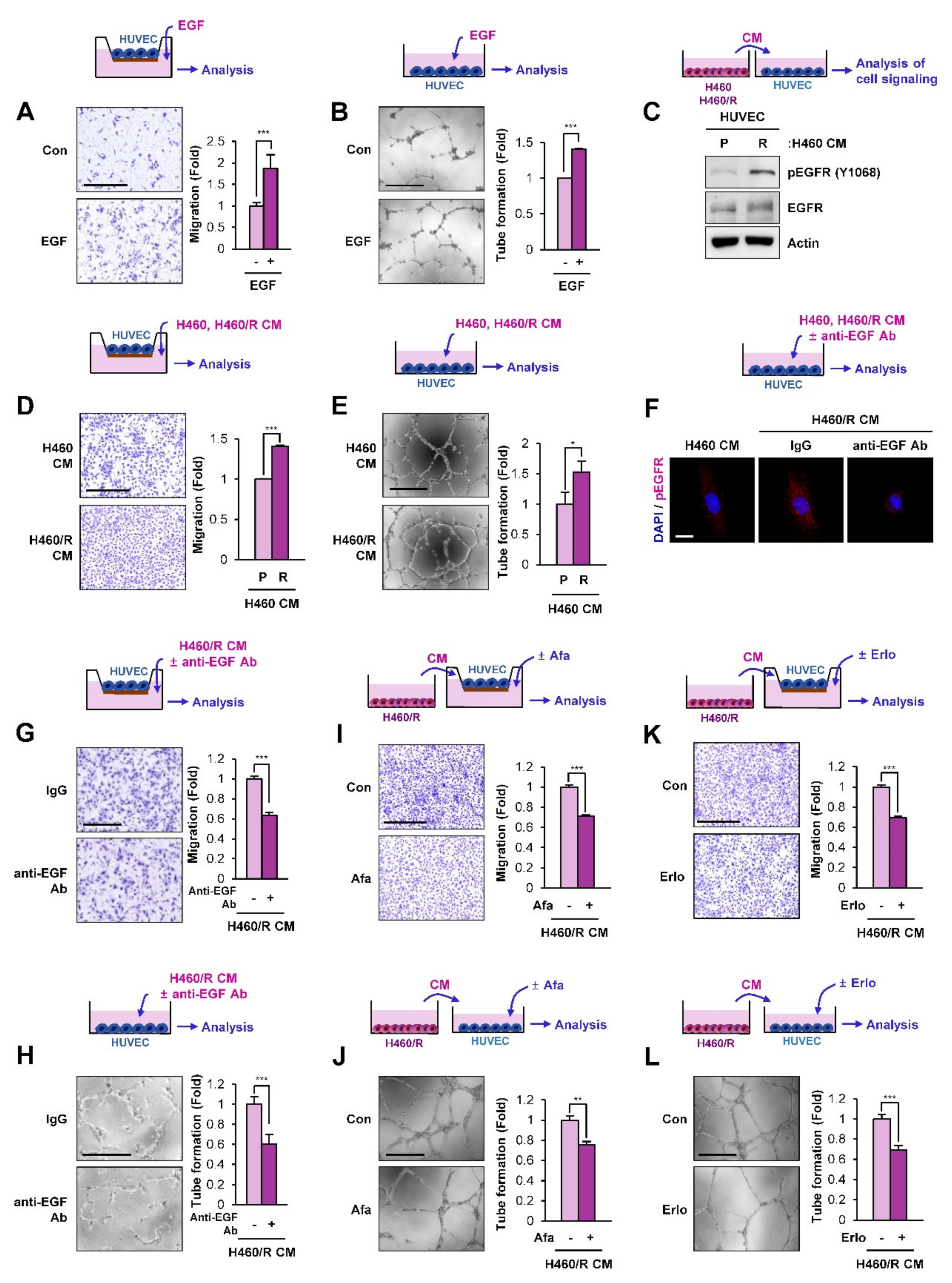
2.5. SCC-Derived EGF Stimulates Migration and Vascular Endothelial Cell Tube Formation
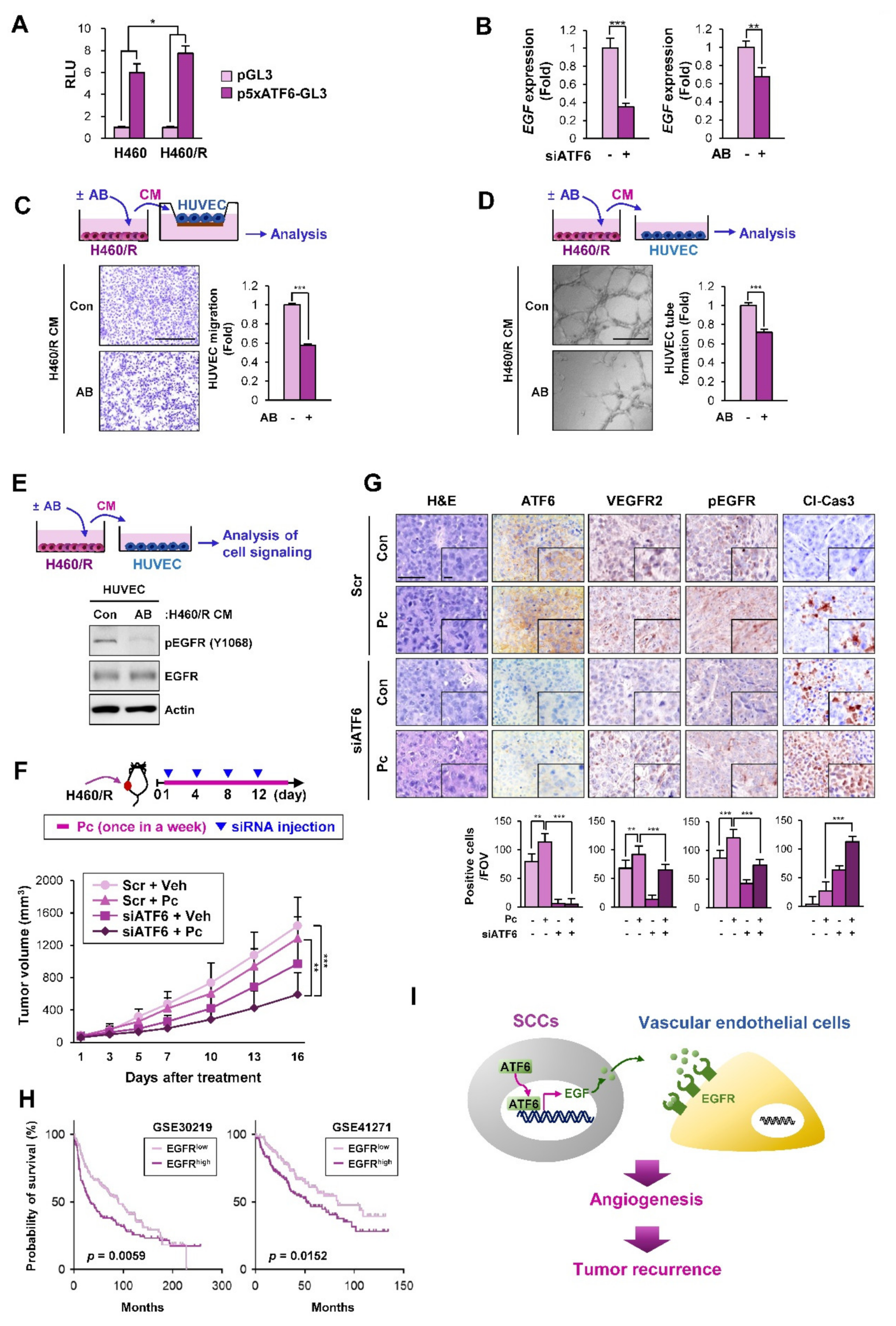
2.6. ATF6 Mediates the Angiogenesis-Stimulating Activities of SCCs by the Transcriptional Upregulation of EGF
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Cell Viability Assay
4.4. Anchorage-Dependent Colony Formation Assay
4.5. Anchorage-Independent Colony Formation Assay
4.6. Cell Cycle Analysis
4.7. Migration Assay
4.8. Tube Formation Assay
4.9. Transfection
4.10. Western Blot Analysis
4.11. RT-PCR and Real-Time PCR
4.12. Luciferase Reporter Assay
4.13. Animal Studies
4.14. IHC Analysis
4.15. Immunofluorescence
4.16. RNA-Sequencing Analysis
4.17. In Silico Analysis
4.18. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruebo, M.; Vilaboa, N.; Saez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; Gonzalez-Fernandez, A. Assessment of the evolution of cancer treatment therapies. Cancers (Basel) 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, J.-C.; Yang, H.; Zhang, X.; Liu, L.-L.; Li, Y.; Zeng, T.-T.; Zhu, Y.-H.; Li, X.-D.; Li, Y.; et al. Expansion of cancer stem cell pool initiates lung cancer recurrence before angiogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E8948–E8957. [Google Scholar] [CrossRef] [Green Version]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artal Cortes, A.; Calera Urquizu, L.; Hernando Cubero, J. Adjuvant chemotherapy in non-small cell lung cancer: State-of-the-art. Transl. Lung Cancer Res. 2015, 4, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Suzuki, A.; Tatematsu, T.; Shitara, M.; Hikosaka, Y.; Okuda, K.; Moriyama, S.; Yano, M.; Fujii, Y. Prognosis of recurrent non-small cell lung cancer following complete resection. Oncol. Lett. 2014, 7, 1300–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subotic, D.; Van Schil, P.; Grigoriu, B. Optimising treatment for post-operative lung cancer recurrence. Eur. Respir. J. 2016, 47, 374–378. [Google Scholar] [CrossRef]
- Yeh, A.C.; Ramaswamy, S. Mechanisms of Cancer Cell Dormancy—Another Hallmark of Cancer? Cancer Res. 2015, 75, 5014–5022. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.L.; Zhang, M.; Tang, Y.L.; Liang, X.H. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. Onco Targets Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.C.; Rajbhandari, N.; Liu, C.; Sakamoto, K.; Zhang, Q.; Triplett, A.A.; Batra, S.K.; Opavsky, R.; Felsher, D.W.; DiMaio, D.J.; et al. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res. 2013, 73, 1821–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 is a promising molecular target in the diagnosis of cancer (review). Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, A.C.; Adam, A.P.; Zhang, L.; Aguirre-Ghiso, J.A. Tumor cell dormancy induced by p38SAPK and ER-stress signaling: An adaptive advantage for metastatic cells? Cancer Biol. Ther. 2006, 5, 729–735. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef] [PubMed]
- Obacz, J.; Avril, T.; Le Reste, P.J.; Urra, H.; Quillien, V.; Hetz, C.; Chevet, E. Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Indraccolo, S.; Stievano, L.; Minuzzo, S.; Tosello, V.; Esposito, G.; Piovan, E.; Zamarchi, R.; Chieco-Bianchi, L.; Amadori, A. Interruption of tumor dormancy by a transient angiogenic burst within the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2006, 103, 4216–4221. [Google Scholar] [CrossRef] [Green Version]
- Erez, N. Angiogenic awakening. Nature 2013, 500, 37. [Google Scholar] [CrossRef] [PubMed]
- Shaked, Y.; McAllister, S.; Fainaru, O.; Almog, N. Tumor dormancy and the angiogenic switch: Possible implications of bone marrow- derived cells. Curr. Pharm. Des. 2014, 20, 4920–4933. [Google Scholar] [CrossRef]
- Almog, N.; Ma, L.; Raychowdhury, R.; Schwager, C.; Erber, R.; Short, S.; Hlatky, L.; Vajkoczy, P.; Huber, P.E.; Folkman, J.; et al. Transcriptional Switch of Dormant Tumors to Fast-Growing Angiogenic Phenotype. Cancer Res. 2009, 69, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, N.; Houghton, J.; Lyle, S. Slow-cycling therapy-resistant cancer cells. Stem Cells Dev. 2012, 21, 1822–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebinger, S.; Ozdemir, E.Z.; Ziegenhain, C.; Tiedt, S.; Castro Alves, C.; Grunert, M.; Dworzak, M.; Lutz, C.; Turati, V.A.; Enver, T.; et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell 2016, 30, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Van Cruijsen, H.; Giaccone, G.; Hoekman, K. Epidermal growth factor receptor and angiogenesis: Opportunities for combined anticancer strategies. Int. J. Cancer 2005, 117, 883–888. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://www.biovenn.nl/index.php (accessed on 20 December 2019).
- Hurst, R.E.; Hauser, P.J.; You, Y.; Bailey-Downs, L.C.; Bastian, A.; Matthews, S.M.; Thorpe, J.; Earle, C.; Bourguignon, L.Y.; Ihnat, M.A. Identification of novel drugs to target dormant micrometastases. BMC Cancer 2015, 15, 404. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.; Belani, C.P. Paclitaxel for non-small cell lung cancer. Expert Opin. Pharmacother. 2004, 5, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Kubben, F.J.; Peeters-Haesevoets, A.; Engels, L.G.; Baeten, C.G.; Schutte, B.; Arends, J.W.; Stockbrugger, R.W.; Blijham, G.H. Proliferating cell nuclear antigen (PCNA): A new marker to study human colonic cell proliferation. Gut 1994, 35, 530–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minkovsky, N.; Berezov, A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 1336–1346. [Google Scholar]
- Schettino, C.; Bareschino, M.A.; Ricci, V.; Ciardiello, F. Erlotinib: An EGF receptor tyrosine kinase inhibitor in non-small-cell lung cancer treatment. Expert Rev. Respir. Med. 2008, 2, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Lee, H.J.; Hwang, S.J.; Min, H.Y.; Kang, H.N.; Park, A.Y.; Hyun, S.Y.; Sim, J.Y.; Lee, H.J.; Jang, H.J.; et al. The Interplay between Slow-Cycling, Chemoresistant Cancer Cells and Fibroblasts Creates a Proinflammatory Niche for Tumor Progression. Cancer Res. 2020, 80, 2257–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shen, J.; Arenzana, N.; Tirasophon, W.; Kaufman, R.J.; Prywes, R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 2000, 275, 27013–27020. [Google Scholar] [CrossRef]
- Barr, S.C.; Warner, K. Characterization of a serine protease activity in Sarcocystis neurona merozoites. J. Parasitol. 2003, 89, 385–388. [Google Scholar] [CrossRef]
- Wang, F.T.; Sun, W.; Zhang, J.T.; Fan, Y.Z. Cancer-associated fibroblast regulation of tumor neo-angiogenesis as a therapeutic target in cancer. Oncol. Lett. 2019, 17, 3055–3065. [Google Scholar] [CrossRef] [Green Version]
- Laitala, A.; Erler, J.T. Hypoxic Signalling in Tumour Stroma. Front. Oncol. 2018, 8, 189. [Google Scholar] [CrossRef]
- Yeldag, G.; Rice, A.; Del Rio Hernandez, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers (Basel) 2018, 10, 471. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.; Carlin, S.; Burke, S.A.; Wen, B.; Yang, K.M.; Ling, C.C. Immunohistochemical detection of changes in tumor hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero Vazquez, A.; Garcia Flores, P.; Romero Ortiz, A.; Del Moral, R.G.; Alcazar-Navarrete, B. Changes in non-small cell lung cancer diagnosis, molecular testing and prognosis 2011–2016. J. Thorac. Dis. 2018, 10, 5468–5475. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Rajbhandari, N.; Wagner, K.U. Cancer cell dormancy in novel mouse models for reversible pancreatic cancer: A lingering challenge in the development of targeted therapies. Cancer Res. 2014, 74, 2138–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valcourt, J.R.; Lemons, J.M.; Haley, E.M.; Kojima, M.; Demuren, O.O.; Coller, H.A. Staying alive: Metabolic adaptations to quiescence. Cell Cycle 2012, 11, 1680–1696. [Google Scholar] [CrossRef] [Green Version]
- Coller, H.A.; Sang, L.; Roberts, J.M. A new description of cellular quiescence. PLoS Biol. 2006, 4, e83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Akagi, M.; Kawaguchi, M.; Liu, W.; McCarty, M.F.; Takeda, A.; Fan, F.; Stoeltzing, O.; Parikh, A.A.; Jung, Y.D.; Bucana, C.D.; et al. Induction of neuropilin-1 and vascular endothelial growth factor by epidermal growth factor in human gastric cancer cells. Br. J. Cancer 2003, 88, 796–802. [Google Scholar] [CrossRef] [Green Version]
- Perrotte, P.; Matsumoto, T.; Inoue, K.; Kuniyasu, H.; Eve, B.Y.; Hicklin, D.J.; Radinsky, R.; Dinney, C.P. Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin. Cancer Res. 1999, 5, 257–265. [Google Scholar]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10631–10634. [Google Scholar] [CrossRef] [Green Version]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Zhai, Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am. J. Transplant. 2014, 14, 1552–1561. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, D.J.; Livezey, M.; Yu, L.; Zheng, X.; Andruska, N. Anticipatory UPR Activation: A Protective Pathway and Target in Cancer. Trends Endocrinol. Metab. 2016, 27, 731–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Andruska, N.; Zheng, X.; Shapiro, D.J. Anticipatory activation of the unfolded protein response by epidermal growth factor is required for immediate early gene expression and cell proliferation. Mol. Cell. Endocrinol. 2016, 422, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rousseaux, S.; Debernardi, A.; Jacquiau, B.; Vitte, A.L.; Vesin, A.; Nagy-Mignotte, H.; Moro-Sibilot, D.; Brichon, P.Y.; Lantuejoul, S.; Hainaut, P.; et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci. Transl. Med. 2013, 5, 186ra166. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Larsen, J.E.; Lee, W.; Sun, H.; Shames, D.S.; Dalvi, M.P.; Ramirez, R.D.; Tang, H.; DiMaio, J.M.; Gao, B.; et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol. Cancer Res. 2013, 11, 638–650. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence (5‘-3‘) | Reverse Sequence (5‘-3‘) | Application |
|---|---|---|---|
| EGF | CAGACCCTTGAAGGGGGTGT | ATCTGTCTCCAGGCATTGAG | Real-time PCR |
| EGF | AAGAATGGGGGTCAACCAGT | TGAAGTTGGTTGCATTGACC | Real-time PCR |
| VEGF | CCTGGTGGACATCTTCCAGGAGTACC | GAAGCTCATCTCTCCTATGTGCTGGC | Real-time PCR |
| FGF2 | TGTGCTAACCGTTACCTGGC | CGTTTCAGTGCCACATACCAA | Real-time PCR |
| IGF1 | TGGTGGATGCTCTTCAGTTC | GACAGAGCGAGCTGACTTG | Real-time PCR |
| IGF2 | CCGTGCTTCCGGACAACTT | CTGCTTCCAGGTGTCATATTGC | Real-time PCR |
| PDGFA | TTGGCCACCTTGACGCT | CCTGCCCATTCGGAGGAA | Real-time PCR |
| PDGFB | TTTCTCACCTGGACAGGTCG | GAAGGAGCCTGGGTTCCCT | Real-time PCR |
| EGF | GGGAGCCTGAGCAGAAACTT | CTTCAGTGTATGGGCAA | RT-PCR |
| ATF6 | ATGAAGTTGTGTCAGAGAACC | CTCTTTAGCAGAAAATCCTAG | Real-time PCR |
| HMBS | CATGTCTGGTAACGGCAATG | GTACGAGGCTTTCAATGTTG | Real-time PCR |
| ACTB | TCATTCCAAATATGAGATGCGTTG | TAGAGAGAAGTGGGGTGGCT | Real-time PCR |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, J.; Min, H.-Y.; Pei, H.; Wei, X.; Sim, J.Y.; Park, S.-H.; Hwang, S.J.; Lee, H.-J.; Hong, S.; Shin, Y.K.; et al. The ATF6-EGF Pathway Mediates the Awakening of Slow-Cycling Chemoresistant Cells and Tumor Recurrence by Stimulating Tumor Angiogenesis. Cancers 2020, 12, 1772. https://doi.org/10.3390/cancers12071772
Cho J, Min H-Y, Pei H, Wei X, Sim JY, Park S-H, Hwang SJ, Lee H-J, Hong S, Shin YK, et al. The ATF6-EGF Pathway Mediates the Awakening of Slow-Cycling Chemoresistant Cells and Tumor Recurrence by Stimulating Tumor Angiogenesis. Cancers. 2020; 12(7):1772. https://doi.org/10.3390/cancers12071772
Chicago/Turabian StyleCho, Jaebeom, Hye-Young Min, Honglan Pei, Xuan Wei, Jeong Yeon Sim, Shin-Hyung Park, Su Jung Hwang, Hyo-Jong Lee, Sungyoul Hong, Young Kee Shin, and et al. 2020. "The ATF6-EGF Pathway Mediates the Awakening of Slow-Cycling Chemoresistant Cells and Tumor Recurrence by Stimulating Tumor Angiogenesis" Cancers 12, no. 7: 1772. https://doi.org/10.3390/cancers12071772