Molecular Bases of Drug Resistance in Hepatocellular Carcinoma

 , , ,
, , , 
Abstract
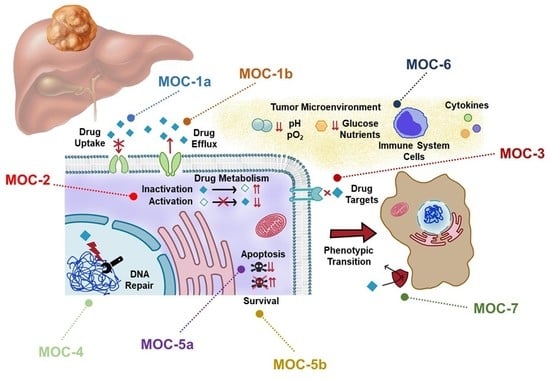
:
1. Introduction
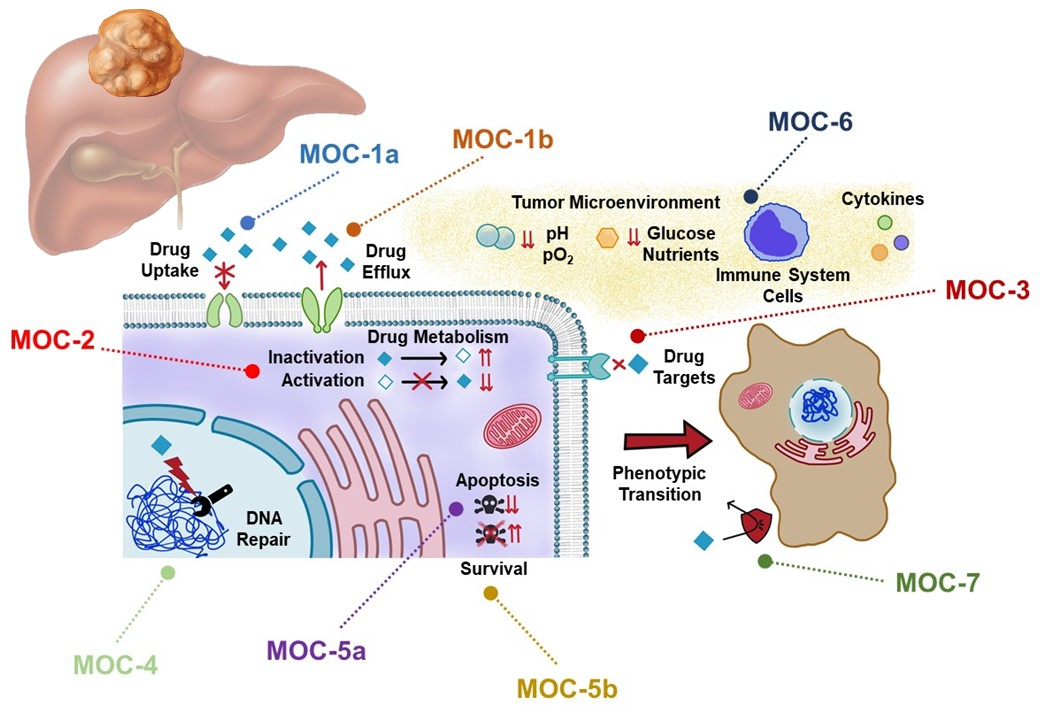
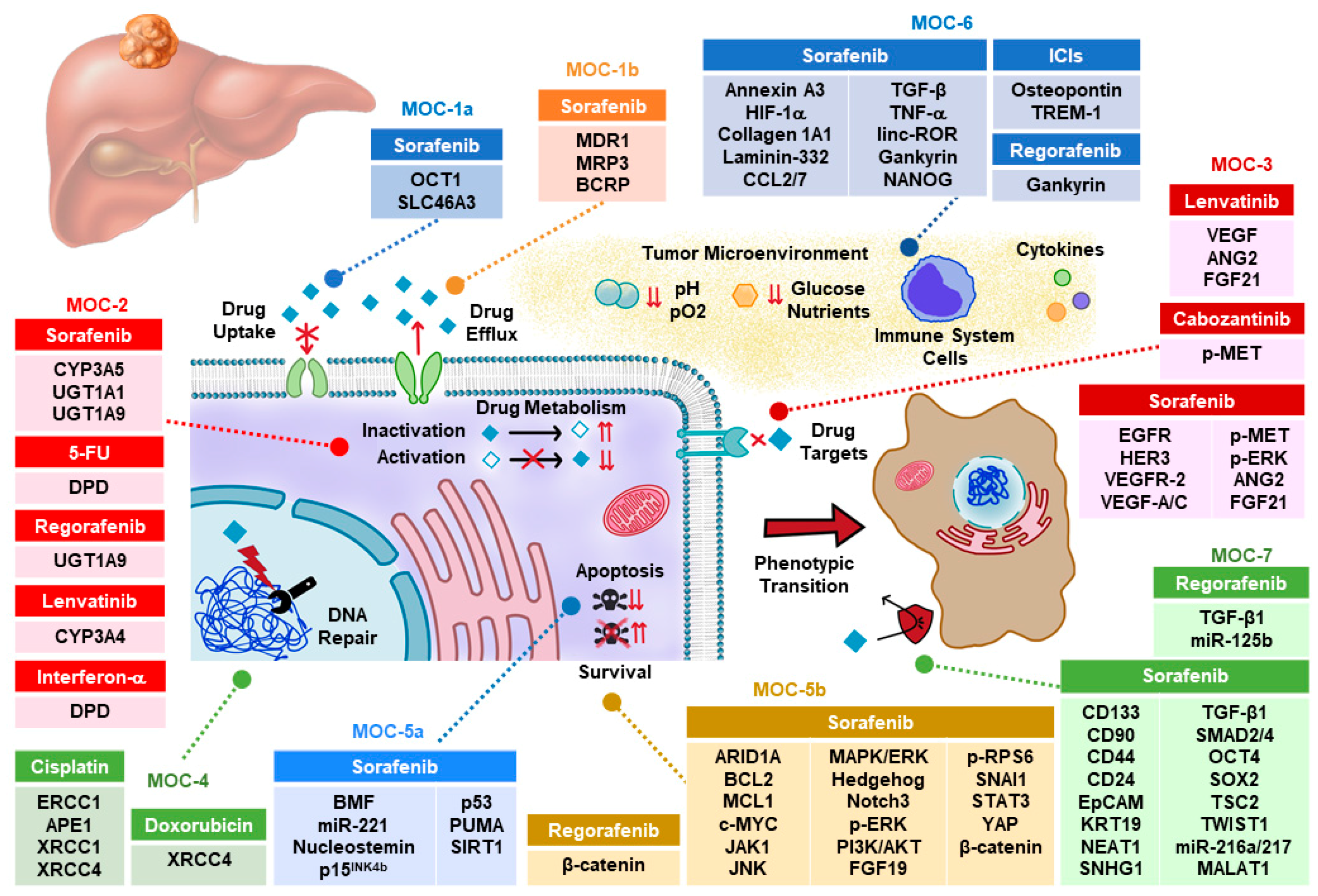
2. Drug Uptake and Export (MOC-1)
2.1. Drug Uptake Carriers (MOC-1a)
2.2. Drug Export Pumps (MOC-1b)
3. Drug Metabolism (MOC-2)
4. Changes in Drug Targets (MOC-3)
5. DNA Repairing (MOC-4)
6. Balance between Pro-Survival and Pro-Apoptotic Factors (MOC-5)
6.1. Pro-Apoptotic Factors (MOC-5a)
6.2. Survival Pathways (MOC-5b)
7. Adaptation to Tumor Microenvironment (MOC-6)
8. Phenotypic Transition (MOC-7)
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, J.J.G.; Briz, O.; Herraez, E.; Lozano, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Osorio-Padilla, L.M.; Santos-Llamas, A.I.; Serrano, M.A.; et al. Molecular bases of the poor response of liver cancer to chemotherapy. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Cervello, M.; Emma, M.R.; Augello, G.; Cusimano, A.; Giannitrapani, L.; Soresi, M.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Gulino, A.; et al. New landscapes and horizons in hepatocellular carcinoma therapy. Aging (Albany NY) 2020, 12, 3053–3094. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [Green Version]
- Vogel, A.; Saborowski, A. Current strategies for the treatment of intermediate and advanced hepatocellular carcinoma. Cancer Treat Rev. 2020, 82, 101946. [Google Scholar] [CrossRef] [Green Version]
- Couri, T.; Pillai, A. Goals and targets for personalized therapy for HCC. Hepatol. Int. 2019, 13, 125–137. [Google Scholar] [CrossRef]
- Marin, J.J.; Monte, M.J.; Blazquez, A.G.; Macias, R.I.; Serrano, M.A.; Briz, O. The role of reduced intracellular concentrations of active drugs in the lack of response to anticancer chemotherapy. Acta Pharmacol. Sin. 2014, 35, 1–10. [Google Scholar] [CrossRef]
- Chen, M.; Neul, C.; Schaeffeler, E.; Frisch, F.; Winter, S.; Schwab, M.; Koepsell, H.; Hu, S.; Laufer, S.; Baker, S.D.; et al. Sorafenib Activity and Disposition in Liver Cancer Does Not Depend on Organic Cation Transporter 1. Clin. Pharmacol. Ther. 2020, 107, 227–237. [Google Scholar] [CrossRef]
- Herraez, E.; Lozano, E.; Macias, R.I.; Vaquero, J.; Bujanda, L.; Banales, J.M.; Marin, J.J.; Briz, O. Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 2013, 58, 1065–1073. [Google Scholar] [CrossRef]
- Al-Abdulla, R.; Lozano, E.; Macias, R.I.R.; Monte, M.J.; Briz, O.; O’Rourke, C.J.; Serrano, M.A.; Banales, J.M.; Avila, M.A.; Martinez-Chantar, M.L.; et al. Epigenetic events involved in organic cation transporter 1-dependent impaired response of hepatocellular carcinoma to sorafenib. Br. J. Pharmacol. 2019, 176, 787–800. [Google Scholar] [CrossRef]
- Geier, A.; Macias, R.I.; Bettinger, D.; Weiss, J.; Bantel, H.; Jahn, D.; Al-Abdulla, R.; Marin, J.J. The lack of the organic cation transporter OCT1 at the plasma membrane of tumor cells precludes a positive response to sorafenib in patients with hepatocellular carcinoma. Oncotarget 2017, 8, 15846–15857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, S.; Hsu, B.; Miles, D.; Aftab, D.; Wang, R.; Nguyen, L. Metabolism and Disposition of Cabozantinib in Healthy Male Volunteers and Pharmacologic Characterization of Its Major Metabolites. Drug Metab. Dispos. Biol. Fate Chem. 2015, 43, 1190–1207. [Google Scholar] [CrossRef] [PubMed]
- Ohya, H.; Shibayama, Y.; Ogura, J.; Narumi, K.; Kobayashi, M.; Iseki, K. Regorafenib is transported by the organic anion transporter 1B1 and the multidrug resistance protein 2. Biol. Pharm. Bull. 2015, 38, 582–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, H.; Tsujimoto, M.; Takeuchi, A.; Tanaka, M.; Ikegami, Y.; Tagami, M.; Abe, S.; Hashimoto, M.; Minegaki, T.; Nishiguchi, K. Substrate-dependent effects of molecular-targeted anticancer agents on activity of organic anion transporting polypeptide 1B1. Xenobiotica Fate Foreign Compd. Biol. Syst. 2018, 48, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.I.; Hu, S.; Roberts, J.L.; Gibson, A.A.; Orwick, S.J.; Li, L.; Sparreboom, A.; Baker, S.D. Contribution of OATP1B1 and OATP1B3 to the disposition of sorafenib and sorafenib-glucuronide. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1458–1466. [Google Scholar] [CrossRef] [Green Version]
- Durmus, S.; van Hoppe, S.; Schinkel, A.H. The impact of Organic Anion-Transporting Polypeptides (OATPs) on disposition and toxicity of antitumor drugs: Insights from knockout and humanized mice. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 27, 72–88. [Google Scholar] [CrossRef]
- Hu, D.G.; Marri, S.; McKinnon, R.A.; Mackenzie, P.I.; Meech, R. Deregulation of the Genes that Are Involved in Drug Absorption, Distribution, Metabolism, and Excretion in Hepatocellular Carcinoma. J. Pharmacol. Exp. Ther. 2019, 368, 363–381. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zheng, B.; Meng, S.; Xu, Y.; Guo, J.; Chen, L.J.; Xiao, J.; Zhang, W.; Tan, Z.R.; Tang, J.; et al. Increased expression of SLC46A3 to oppose the progression of hepatocellular carcinoma and its effect on sorafenib therapy. Biomed. Pharmacother. = Biomed. Pharmacother. 2019, 114, 108864. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, P.M.; Lin, P.Y.; Hsiau, Y.T.; Chu, P.Y. ABCG2 Overexpression Confers Poor Outcomes in Hepatocellular Carcinoma of Elderly Patients. Anticancer Res. 2016, 36, 2983–2988. [Google Scholar]
- Gao, B.; Yang, F.M.; Yu, Z.T.; Li, R.; Xie, F.; Chen, J.; Luo, H.J.; Zhang, J.C. Relationship between the expression of MDR1 in hepatocellular cancer and its biological behaviors. Int. J. Clin. Exp. Pathol. 2015, 8, 6995–7001. [Google Scholar] [PubMed]
- Tandia, M.; Mhiri, A.; Paule, B.; Saffroy, R.; Cailliez, V.; Noe, G.; Farinotti, R.; Bonhomme-Faivre, L. Correlation between clinical response to sorafenib in hepatocellular carcinoma treatment and polymorphisms of P-glycoprotein (ABCB1) and of breast cancer resistance protein (ABCG2): Monocentric study. Cancer Chemother. Pharmacol. 2017, 79, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Tomonari, T.; Takeishi, S.; Taniguchi, T.; Tanaka, T.; Tanaka, H.; Fujimoto, S.; Kimura, T.; Okamoto, K.; Miyamoto, H.; Muguruma, N.; et al. MRP3 as a novel resistance factor for sorafenib in hepatocellular carcinoma. Oncotarget 2016, 7, 7207–7215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozeki, T.; Nagahama, M.; Fujita, K.; Suzuki, A.; Sugino, K.; Ito, K.; Miura, M. Influence of CYP3A4/5 and ABC transporter polymorphisms on lenvatinib plasma trough concentrations in Japanese patients with thyroid cancer. Sci. Rep. 2019, 9, 5404. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.G.; Wang, Z.H.; Dong, W.; He, X.D.; Liu, F.C.; Liu, H. Specific CYP450 Genotypes in the Chinese Population Affect Sorafenib Toxicity in HBV/HCV-associated Hepatocellular Carcinoma Patients. Biomed. Environ. Sci. BES 2018, 31, 586–595. [Google Scholar] [CrossRef]
- Zhao, H.; Zhao, Y.; Guo, Y.; Huang, Y.; Lin, S.; Xue, C.; Xu, F.; Zhang, Y.; Zhao, L.; Hu, Z.; et al. Clinical significance of the thymidylate synthase, dihydropyrimidine dehydrogenase, and thymidine phosphorylase mRNA expressions in hepatocellular carcinoma patients receiving 5-fluorouracil-based transarterial chemoembolization treatment. OncoTargets Ther. 2013, 6, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, Y.; Kuramochi, H.; Nakajima, G.; Katagiri, S.; Yamamoto, M. Elevated levels of mRNAs encoding dihydropyrimidine dehydrogenase and thymidylate synthase are associated with improved survival of patients with hepatocellular carcinoma treated with S-1. Oncol. Lett. 2017, 14, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.P.; Liu, Z.Y.; Zhao, Y.M.; He, X.G.; Pan, Q.; Zhang, N.; Zhou, J.M.; Wang, L.R.; Wang, M.; Zhan, D.H.; et al. Dihydropyrimidine dehydrogenase predicts survival and response to interferon-alpha in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Bins, S.; Lenting, A.; El Bouazzaoui, S.; van Doorn, L.; Oomen-de Hoop, E.; Eskens, F.A.; van Schaik, R.H.; Mathijssen, R.H. Polymorphisms in SLCO1B1 and UGT1A1 are associated with sorafenib-induced toxicity. Pharmacogenomics 2016, 17, 1483–1490. [Google Scholar] [CrossRef]
- Ge, Y.; Chen, S.; Mu, W.; Ba, Q.; Li, J.; Chen, P.; Wang, X.; Wang, H. Epigenetic regulation of UDP-Glucuronosyltransferase by microRNA-200a/-183: Implications for responses to sorafenib treatment in patients with hepatocellular carcinoma. Cancer Lett. 2019, 454, 14–25. [Google Scholar] [CrossRef]
- Ba, H.L.; Mbatchi, L.; Gattacceca, F.; Evrard, A.; Lacarelle, B.; Blanchet, B.; Ciccolini, J.; Salas, S. Pharmacogenetics and pharmacokinetics modeling of unexpected and extremely severe toxicities after sorafenib intake. Pharmacogenomics 2020, 21, 173–179. [Google Scholar] [CrossRef]
- Boudou-Rouquette, P.; Narjoz, C.; Golmard, J.L.; Thomas-Schoemann, A.; Mir, O.; Taieb, F.; Durand, J.P.; Coriat, R.; Dauphin, A.; Vidal, M.; et al. Early sorafenib-induced toxicity is associated with drug exposure and UGTIA9 genetic polymorphism in patients with solid tumors: A preliminary study. PLoS ONE 2012, 7, e42875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacre, A.; Lanthier, N.; Dano, H.; Aydin, S.; Leggenhager, D.; Weber, A.; Dekairelle, A.F.; De Cuyper, A.; Gala, J.L.; Humblet, Y.; et al. Regorafenib induced severe toxic hepatitis: Characterization and discussion. Liver Int. Off. J. Int. Assoc. Study Liver 2016, 36, 1590–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.Q.; Ongkeko, W.M.; Chen, L.; Yang, Z.F.; Lu, P.; Chen, K.K.; Lopez, J.P.; Poon, R.T.; Fan, S.T. Octamer 4 (Oct4) mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology 2010, 52, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Hao, X.; Yan, M.; Yao, M.; Ge, C.; Gu, J.; Li, J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int. J. Cancer 2010, 126, 2067–2078. [Google Scholar] [CrossRef]
- Hu, S.; Chen, Z.; Franke, R.; Orwick, S.; Zhao, M.; Rudek, M.A.; Sparreboom, A.; Baker, S.D. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6062–6069. [Google Scholar] [CrossRef] [Green Version]
- Kort, A.; Durmus, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Brain and Testis Accumulation of Regorafenib is Restricted by Breast Cancer Resistance Protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1). Pharm. Res. 2015, 32, 2205–2216. [Google Scholar] [CrossRef] [Green Version]
- Shumaker, R.C.; Aluri, J.; Fan, J.; Martinez, G.; Thompson, G.A.; Ren, M. Effect of rifampicin on the pharmacokinetics of lenvatinib in healthy adults. Clin. Drug Investig. 2014, 34, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Nies, A.T.; Konig, J.; Pfannschmidt, M.; Klar, E.; Hofmann, W.J.; Keppler, D. Expression of the multidrug resistance proteins MRP2 and MRP3 in human hepatocellular carcinoma. Int. J. Cancer 2001, 94, 492–499. [Google Scholar] [CrossRef]
- Shibayama, Y.; Nakano, K.; Maeda, H.; Taguchi, M.; Ikeda, R.; Sugawara, M.; Iseki, K.; Takeda, Y.; Yamada, K. Multidrug resistance protein 2 implicates anticancer drug-resistance to sorafenib. Biol. Pharm. Bull. 2011, 34, 433–435. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Sane, R.; Ohlfest, J.R.; Elmquist, W.F. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J. Pharmacol. Exp. Ther. 2011, 336, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Hsieh, Y.L.; Hung, C.M.; Chien, P.H.; Chien, Y.F.; Chen, L.C.; Tu, C.Y.; Chen, C.H.; Hsu, S.C.; Lin, Y.M.; et al. BCRP/ABCG2 inhibition sensitizes hepatocellular carcinoma cells to sorafenib. PLoS ONE 2013, 8, e83627. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Franz, C.; Xiao, Z.; Mohr, E.; Serba, S.; Buchler, M.W.; Schemmer, P. Sorafenib modulates the gene expression of multi-drug resistance mediating ATP-binding cassette proteins in experimental hepatocellular carcinoma. Anticancer Res. 2010, 30, 4503–4508. [Google Scholar]
- Xiang, Q.F.; Zhang, D.M.; Wang, J.N.; Zhang, H.W.; Zheng, Z.Y.; Yu, D.C.; Li, Y.J.; Xu, J.; Chen, Y.J.; Shang, C.Z. Cabozantinib reverses multidrug resistance of human hepatoma HepG2/adr cells by modulating the function of P-glycoprotein. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Caudle, K.E.; Thorn, C.F.; Klein, T.E.; Swen, J.J.; McLeod, H.L.; Diasio, R.B.; Schwab, M. Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clin. Pharmacol. Ther. 2013, 94, 640–645. [Google Scholar] [CrossRef] [Green Version]
- Lathia, C.; Lettieri, J.; Cihon, F.; Gallentine, M.; Radtke, M.; Sundaresan, P. Lack of effect of ketoconazole-mediated CYP3A inhibition on sorafenib clinical pharmacokinetics. Cancer Chemother. Pharmacol. 2006, 57, 685–692. [Google Scholar] [CrossRef]
- Ye, L.; Yang, X.; Guo, E.; Chen, W.; Lu, L.; Wang, Y.; Peng, X.; Yan, T.; Zhou, F.; Liu, Z. Sorafenib metabolism is significantly altered in the liver tumor tissue of hepatocellular carcinoma patient. PLoS ONE 2014, 9, e96664. [Google Scholar] [CrossRef] [Green Version]
- Peer, C.J.; Sissung, T.M.; Kim, A.; Jain, L.; Woo, S.; Gardner, E.R.; Kirkland, C.T.; Troutman, S.M.; English, B.C.; Richardson, E.D.; et al. Sorafenib is an inhibitor of UGT1A1 but is metabolized by UGT1A9: Implications of genetic variants on pharmacokinetics and hyperbilirubinemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2099–2107. [Google Scholar] [CrossRef] [Green Version]
- Thillai, K.; Srikandarajah, K.; Ross, P. Regorafenib as treatment for patients with advanced hepatocellular cancer. Future Oncol. 2017, 13, 2223–2232. [Google Scholar] [CrossRef]
- Gerisch, M.; Hafner, F.T.; Lang, D.; Radtke, M.; Diefenbach, K.; Cleton, A.; Lettieri, J. Mass balance, metabolic disposition, and pharmacokinetics of a single oral dose of regorafenib in healthy human subjects. Cancer Chemother. Pharmacol. 2018, 81, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Miners, J.O.; Chau, N.; Rowland, A.; Burns, K.; McKinnon, R.A.; Mackenzie, P.I.; Tucker, G.T.; Knights, K.M.; Kichenadasse, G. Inhibition of human UDP-glucuronosyltransferase enzymes by lapatinib, pazopanib, regorafenib and sorafenib: Implications for hyperbilirubinemia. Biochem. Pharmacol. 2017, 129, 85–95. [Google Scholar] [CrossRef] [PubMed]
- De Mattia, E.; Cecchin, E.; Guardascione, M.; Foltran, L.; Di Raimo, T.; Angelini, F.; D’Andrea, M.; Toffoli, G. Pharmacogenetics of the systemic treatment in advanced hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 3870–3896. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Xu, L.; Yuan, C.; Li, X.; Zhang, X.; Wang, W.; Guo, X.; Ouyang, Y.; Qiao, L.; Wang, Z.; et al. Activation of EGFR-KLF4 positive feedback loop results in acquired resistance to sorafenib in hepatocellular carcinoma. Mol. Carcinogenesis 2019, 58, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Blivet-Van Eggelpoel, M.J.; Chettouh, H.; Fartoux, L.; Aoudjehane, L.; Barbu, V.; Rey, C.; Priam, S.; Housset, C.; Rosmorduc, O.; Desbois-Mouthon, C. Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J. Hepatol. 2012, 57, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Negri, F.V.; Dal Bello, B.; Porta, C.; Campanini, N.; Rossi, S.; Tinelli, C.; Poggi, G.; Missale, G.; Fanello, S.; Salvagni, S.; et al. Expression of pERK and VEGFR-2 in advanced hepatocellular carcinoma and resistance to sorafenib treatment. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 2001–2008. [Google Scholar] [CrossRef]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef] [Green Version]
- Caruso, S.; Calatayud, A.L.; Pilet, J.; La Bella, T.; Rekik, S.; Imbeaud, S.; Letouze, E.; Meunier, L.; Bayard, Q.; Rohr-Udilova, N.; et al. Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology 2019, 157, 760–776. [Google Scholar] [CrossRef]
- Finn, R.S.; Kudo, M.; Cheng, A.L.; Wyrwicz, L.; Ngan, R.; Blanc, J.F.; Baron, A.D.; Vogel, A.; Ikeda, M.; Piscaglia, F.; et al. Final analysis of serum biomarkers in patients (pts) from the phase III study of lenvatinib (LEN) vs sorafenib (SOR) in unresectable hepatocellular carcinoma (uHCC) [REFLECT]. Ann. Oncol. 2018, 29, viii17–viii18. [Google Scholar] [CrossRef]
- Scartozzi, M.; Faloppi, L.; Svegliati Baroni, G.; Loretelli, C.; Piscaglia, F.; Iavarone, M.; Toniutto, P.; Fava, G.; De Minicis, S.; Mandolesi, A.; et al. VEGF and VEGFR genotyping in the prediction of clinical outcome for HCC patients receiving sorafenib: The ALICE-1 study. Int. J. Cancer 2014, 135, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Faloppi, L.; Puzzoni, M.; Casadei Gardini, A.; Silvestris, N.; Masi, G.; Marisi, G.; Vivaldi, C.; Gadaleta, C.D.; Ziranu, P.; Bianconi, M.; et al. Angiogenesis Genotyping and Clinical Outcomes in Patients with Advanced Hepatocellular Carcinoma Receiving Sorafenib: The ALICE-2 Study. Target. Oncol. 2020, 15, 115–126. [Google Scholar] [CrossRef]
- Zheng, Y.B.; Zhan, M.X.; Zhao, W.; Liu, B.; Huang, J.W.; He, X.; Fu, S.R.; Zhao, Y.; Li, Y.; Hu, B.S.; et al. The relationship of kinase insert domain receptor gene polymorphisms and clinical outcome in advanced hepatocellular carcinoma patients treated with sorafenib. Med. Oncol. 2014, 31, 209. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Shirabe, K.; Morita, K.; Umeda, K.; Kayashima, H.; Uchiyama, H.; Soejima, Y.; Taketomi, A.; Maehara, Y. Evaluation of ERCC1 expression for cisplatin sensitivity in human hepatocellular carcinoma. Ann. Surg. Oncol. 2011, 18, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.Q. XLF-mediated NHEJ activity in hepatocellular carcinoma therapy resistance. BMC Cancer 2017, 17, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.C.; Wang, F.; Quan, Q.Q. Roles of XRCC1/XPD/ERCC1 Polymorphisms in Predicting Prognosis of Hepatocellular Carcinoma in Patients Receiving Transcatheter Arterial Chemoembolization. Genet. Test. Mol. Biomark. 2016, 20, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhao, J. Effect of APE1 and XRCC1 gene polymorphism on susceptibility to hepatocellular carcinoma and sensitivity to cisplatin. Int. J. Clin. Exp. Med. 2015, 8, 9931–9936. [Google Scholar] [PubMed]
- Lu, J.; Wang, X.Z.; Zhang, T.Q.; Huang, X.Y.; Yao, J.G.; Wang, C.; Wei, Z.H.; Ma, Y.; Wu, X.M.; Luo, C.Y.; et al. Prognostic significance of XRCC4 expression in hepatocellular carcinoma. Oncotarget 2017, 8, 87955–87970. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, E.; Stein, I.; Andreozzi, M.; Nemeth, J.; Shoham, A.; Pappo, O.; Schweitzer, N.; Tornillo, L.; Kanarek, N.; Quagliata, L.; et al. Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to sorafenib treatment. Cancer Discov. 2014, 4, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M. Immune Checkpoint Blockade in Hepatocellular Carcinoma: 2017 Update. Liver Cancer 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Daud, A.I.; Wolchok, J.D.; Robert, C.; Hwu, W.J.; Weber, J.S.; Ribas, A.; Hodi, F.S.; Joshua, A.M.; Kefford, R.; Hersey, P.; et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 4102–4109. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Turhal, N.S.; Bas, E.; Er, O.; Aliustaoglu, M.; Seber, S.; Dane, F.; Korkmaz, T.; Soyuer, I.; Ozkara, S.; Celikel, C. ERCC1 is not expressed in hepatocellular cancer: A turkish oncology group, gastrointestinal oncology subgroup study. Off. J. Balkan Union Oncol. 2010, 15, 794–796. [Google Scholar]
- Zheng, X.; Chen, K.; Liu, X.; Jiang, G.; Liu, H. High expression of ERCC5 predicts a poor prognosis in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 3664–3670. [Google Scholar]
- Fautrel, A.; Andrieux, L.; Musso, O.; Boudjema, K.; Guillouzo, A.; Langouet, S. Overexpression of the two nucleotide excision repair genes ERCC1 and XPC in human hepatocellular carcinoma. J. Hepatol. 2005, 43, 288–293. [Google Scholar] [CrossRef]
- Rousseau, B.; Menard, L.; Haurie, V.; Taras, D.; Blanc, J.F.; Moreau-Gaudry, F.; Metzler, P.; Hugues, M.; Boyault, S.; Lemiere, S.; et al. Overexpression and role of the ATPase and putative DNA helicase RuvB-like 2 in human hepatocellular carcinoma. Hepatology 2007, 46, 1108–1118. [Google Scholar] [CrossRef]
- Mao, Y.Q.; Houry, W.A. The Role of Pontin and Reptin in Cellular Physiology and Cancer Etiology. Front. Mol. Biosci. 2017, 4, 58. [Google Scholar] [CrossRef] [Green Version]
- Raymond, A.A.; Benhamouche, S.; Neaud, V.; Di Martino, J.; Javary, J.; Rosenbaum, J. Reptin regulates DNA double strand breaks repair in human hepatocellular carcinoma. PLoS ONE 2015, 10, e0123333. [Google Scholar] [CrossRef]
- Mello, T.; Materozzi, M.; Zanieri, F.; Simeone, I.; Ceni, E.; Bereshchenko, O.; Polvani, S.; Tarocchi, M.; Marroncini, G.; Nerlov, C.; et al. Liver haploinsufficiency of RuvBL1 causes hepatic insulin resistance and enhances hepatocellular carcinoma progression. Int. J. Cancer 2020, 146, 3410–3422. [Google Scholar] [CrossRef]
- Yan, T.; Liu, F.; Gao, J.; Lu, H.; Cai, J.; Zhao, X.; Sun, Y. Multilevel regulation of RUVBL2 expression predicts poor prognosis in hepatocellular carcinoma. Cancer Cell Int. 2019, 19, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santonocito, C.; Scapaticci, M.; Nedovic, B.; Annicchiarico, E.B.; Guarino, D.; Leoncini, E.; Boccia, S.; Gasbarrini, A.; Capoluongo, E. XRCC1 Arg399Gln gene polymorphism and hepatocellular carcinoma risk in the Italian population. Int. J. Biol. Mark. 2017, 32, e190–e194. [Google Scholar] [CrossRef]
- Guo, L.Y.; Jin, X.P.; Niu, W.; Li, X.F.; Liu, B.H.; Wang, Y.L. Association of XPD and XRCC1 genetic polymorphisms with hepatocellular carcinoma risk. Asian Pac. J. Cancer Prev. APJCP 2012, 13, 4423–4426. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, Q.; Ye, J.; Pan, S.; Ge, L. Associations between three XRCC1 polymorphisms and hepatocellular carcinoma risk: A meta-analysis of case-control studies. PLoS ONE 2018, 13, e0206853. [Google Scholar] [CrossRef] [PubMed]
- Cun, Y.; Dai, N.; Xiong, C.; Li, M.; Sui, J.; Qian, C.; Li, Z.; Wang, D. Silencing of APE1 enhances sensitivity of human hepatocellular carcinoma cells to radiotherapy in vitro and in a xenograft model. PLoS ONE 2013, 8, e55313. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Yang, J.; Li, Y. Association of hOGG1 Ser326Cys polymorphism with susceptibility to hepatocellular carcinoma. Int. J. Clin. Exp. Med. 2015, 8, 8977–8985. [Google Scholar] [PubMed]
- Zhang, H.; Mizumachi, T.; Carcel-Trullols, J.; Li, L.; Naito, A.; Spencer, H.J.; Spring, P.M.; Smoller, B.R.; Watson, A.J.; Margison, G.P.; et al. Targeting human 8-oxoguanine DNA glycosylase (hOGG1) to mitochondria enhances cisplatin cytotoxicity in hepatoma cells. Carcinogenesis 2007, 28, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, E.J.; Brissett, N.C.; Plocinski, P.; Carlberg, T.; Doherty, A.J. Molecular basis for DNA strand displacement by NHEJ repair polymerases. Nucleic Acids Res. 2016, 44, 2173–2186. [Google Scholar] [CrossRef] [Green Version]
- Fujimaki, S.; Matsuda, Y.; Wakai, T.; Sanpei, A.; Kubota, M.; Takamura, M.; Yamagiwa, S.; Yano, M.; Ohkoshi, S.; Aoyagi, Y. Blockade of ataxia telangiectasia mutated sensitizes hepatoma cell lines to sorafenib by interfering with Akt signaling. Cancer Lett. 2012, 319, 98–108. [Google Scholar] [CrossRef]
- Lin, X.; Kim, H.K.; Howell, S.B. The role of DNA mismatch repair in cisplatin mutagenicity. J. Inorg. Biochem. 1999, 77, 89–93. [Google Scholar] [CrossRef]
- Wang, L.; Bani-Hani, A.; Montoya, D.P.; Roche, P.C.; Thibodeau, S.N.; Burgart, L.J.; Roberts, L.R. hMLH1 and hMSH2 expression in human hepatocellular carcinoma. Int. J. Oncol. 2001, 19, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.F.; Chang, C.W.; Wei, R.J.; Shiue, Y.L.; Wang, S.N.; Yeh, Y.T. Involvement of DNA damage response pathways in hepatocellular carcinoma. BioMed Res. Int. 2014, 2014, 153867. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Hernandez-Gea, V. Hepatocellular carcinoma: Reasons for phase III failure and novel perspectives on trial design. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2072–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.G.; Merchant, J.L.; Lai, P.B.; Ho, R.L.; Hu, X.; Okada, M.; Huang, S.F.; Chui, A.K.; Law, D.J.; Li, Y.G.; et al. Mutation of p53 in recurrent hepatocellular carcinoma and its association with the expression of ZBP-89. Am. J. Pathol. 2003, 162, 1823–1829. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.G.; Wang, X.W.; Budhu, A.; Kim, Y.H.; Kwon, S.M.; Tang, Z.Y.; Sun, Z.; Harris, C.C.; Thorgeirsson, S.S. Association of TP53 mutations with stem cell-like gene expression and survival of patients with hepatocellular carcinoma. Gastroenterology 2011, 140, 1063–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.; Nault, J.C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef]
- Garten, A.; Grohmann, T.; Kluckova, K.; Lavery, G.G.; Kiess, W.; Penke, M. Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. Int. J. Mol. Sci. 2019, 20, 4048. [Google Scholar] [CrossRef] [Green Version]
- Hua, L.; Hu, B.; Yan, D.; Liu, J.; Shen, Y.; Zhao, F.; Shen, C.; Chen, B.; Cui, X. Upregulated expression of Nucleostemin/GNL3 is associated with poor prognosis and Sorafenib Resistance in Hepatocellular Carcinoma. Pathol. Res. Pract. 2017, 213, 688–697. [Google Scholar] [CrossRef]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. CR 2018, 37, 30. [Google Scholar] [CrossRef] [Green Version]
- Weng, X.; Zeng, L.; Yan, F.; He, M.; Wu, X.; Zheng, D. Cyclin-dependent kinase inhibitor 2B gene is associated with the sensitivity of hepatoma cells to Sorafenib. OncoTargets Ther. 2019, 12, 5025–5036. [Google Scholar] [CrossRef] [Green Version]
- Gramantieri, L.; Fornari, F.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Croce, C.M.; Bolondi, L.; Negrini, M. MicroRNA-221 targets Bmf in hepatocellular carcinoma and correlates with tumor multifocality. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5073–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornari, F.; Pollutri, D.; Patrizi, C.; La Bella, T.; Marinelli, S.; Casadei Gardini, A.; Marisi, G.; Baron Toaldo, M.; Baglioni, M.; Salvatore, V.; et al. In Hepatocellular Carcinoma miR-221 Modulates Sorafenib Resistance through Inhibition of Caspase-3-Mediated Apoptosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3953–3965. [Google Scholar] [CrossRef] [Green Version]
- Fernando, J.; Sancho, P.; Fernandez-Rodriguez, C.M.; Lledo, J.L.; Caja, L.; Campbell, J.S.; Fausto, N.; Fabregat, I. Sorafenib sensitizes hepatocellular carcinoma cells to physiological apoptotic stimuli. J. Cell. Physiol. 2012, 227, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Bouattour, M.; Mehta, N.; He, A.R.; Cohen, E.I.; Nault, J.C. Systemic Treatment for Advanced Hepatocellular Carcinoma. Liver Cancer 2019, 8, 341–358. [Google Scholar] [CrossRef]
- Teufel, M.; Seidel, H.; Kochert, K.; Meinhardt, G.; Finn, R.S.; Llovet, J.M.; Bruix, J. Biomarkers Associated With Response to Regorafenib in Patients With Hepatocellular Carcinoma. Gastroenterology 2019, 156, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutusaus, A.; Stefanovic, M.; Boix, L.; Cucarull, B.; Zamora, A.; Blasco, L.; de Frutos, P.G.; Reig, M.; Fernandez-Checa, J.C.; Mari, M.; et al. Antiapoptotic BCL-2 proteins determine sorafenib/regorafenib resistance and BH3-mimetic efficacy in hepatocellular carcinoma. Oncotarget 2018, 9, 16701–16717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Dong, X.; Lv, H.; Xiu, P.; Li, T.; Wang, F.; Xu, Z.; Li, J. Targeting hypoxia-inducible factor-2alpha enhances sorafenib antitumor activity via beta-catenin/C-Myc-dependent pathways in hepatocellular carcinoma. Oncol. Lett. 2015, 10, 778–784. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Wang, X.; Tang, Y.; Huang, S.; Hu, C.A.; Teng, Y. FGF19/FGFR4 signaling contributes to the resistance of hepatocellular carcinoma to sorafenib. J. Exp. Clin. Cancer Res. CR 2017, 36, 8. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yang, X.; Xue, X.; Sun, D.; Cai, P.; Song, Q.; Zhang, B.; Qin, L. HANR Enhances Autophagy-Associated Sorafenib Resistance Through miR-29b/ATG9A Axis in Hepatocellular Carcinoma. OncoTargets Ther. 2020, 13, 2127–2137. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, Y.; Xun, X.; Zhang, C.; Xiang, X.; Cheng, Q.; Hu, S.; Li, Z.; Zhu, J. Hedgehog signaling promotes sorafenib resistance in hepatocellular carcinoma patient-derived organoids. J. Exp. Clin. Cancer Res. CR 2020, 39, 22. [Google Scholar] [CrossRef]
- Ding, X.; He, M.; Chan, A.W.H.; Song, Q.X.; Sze, S.C.; Chen, H.; Man, M.K.H.; Man, K.; Chan, S.L.; Lai, P.B.S.; et al. Genomic and Epigenomic Features of Primary and Recurrent Hepatocellular Carcinomas. Gastroenterology 2019, 157, 1630–1645.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.B.; Lee, M.; Park, S.Y.; Lee, S.; Kim, H.R.; Lee, H.S.; Yoon, J.H.; Kim, Y.J. Sorafenib inhibits cancer side population cells by targeting cJun Nterminal kinase signaling. Mol. Med. Rep. 2015, 12, 8247–8252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.; Huang, X.; Chen, J.; Zhang, K.; Gu, Y.H.; Sun, J.; Cui, S.Y. Long Noncoding RNA MALAT1 Contributes to Sorafenib Resistance by Targeting miR-140-5p/Aurora-A Signaling in Hepatocellular Carcinoma. Mol. Cancer Ther. 2020, 19, 1197–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhou, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Li, H.; Liu, J. LncRNA NEAT1 promotes autophagy via regulating miR-204/ATG3 and enhanced cell resistance to sorafenib in hepatocellular carcinoma. J. Cell. Physiol. 2020, 235, 3402–3413. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, C.; Baglioni, M.; Baron Toaldo, M.; Ventrucci, C.; D’Adamo, S.; Cipone, M.; Chieco, P.; Gramantieri, L.; Bolondi, L. Notch3 inhibition enhances sorafenib cytotoxic efficacy by promoting GSK3b phosphorylation and p21 down-regulation in hepatocellular carcinoma. Oncotarget 2013, 4, 1618–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Chen, J.; Yu, Q.; Ji, T.; Zhang, B.; Xu, J.; Dai, Y.; Xie, Y.; Lin, H.; Liang, X.; et al. Phosphorylated ERK is a potential prognostic biomarker for Sorafenib response in hepatocellular carcinoma. Cancer Med. 2017, 6, 2787–2795. [Google Scholar] [CrossRef]
- Chen, K.F.; Chen, H.L.; Tai, W.T.; Feng, W.C.; Hsu, C.H.; Chen, P.J.; Cheng, A.L. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983.e11. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, L.; Zhu, X.; Wang, P.; Chi, H.; Meng, Z. Activation of phosphatidylinositol 3-kinase/Akt signaling mediates sorafenib-induced invasion and metastasis in hepatocellular carcinoma. Oncol. Rep. 2014, 32, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.F.; Tai, W.T.; Liu, T.H.; Huang, H.P.; Lin, Y.C.; Shiau, C.W.; Li, P.K.; Chen, P.J.; Cheng, A.L. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5189–5199. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Rong, Y.; Huang, Y.; Shi, P.; Wang, X.; Meng, X.; Dong, J.; Wu, C. Cirrhotic stiffness affects the migration of hepatocellular carcinoma cells and induces sorafenib resistance through YAP. J. Cell. Physiol. 2019, 234, 2639–2648. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, R.; Gassler, N.; Bangen, J.M.; Trautwein, C.; Liedtke, C. Pro-apoptotic Sorafenib signaling in murine hepatocytes depends on malignancy and is associated with PUMA expression in vitro and in vivo. Cell Death Dis. 2014, 5, e1030. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, X.; Qu, H.; Qu, B.; Yin, X.; Zhao, H. Cabozantinib induces PUMA-dependent apoptosis in colon cancer cells via AKT/GSK-3beta/NF-kappaB signaling pathway. Cancer Gene Ther. 2019, 27, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Dudgeon, C.; Peng, R.; Wang, P.; Sebastiani, A.; Yu, J.; Zhang, L. Inhibiting oncogenic signaling by sorafenib activates PUMA via GSK3beta and NF-kappaB to suppress tumor cell growth. Oncogene 2012, 31, 4848–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zulehner, G.; Mikula, M.; Schneller, D.; van Zijl, F.; Huber, H.; Sieghart, W.; Grasl-Kraupp, B.; Waldhor, T.; Peck-Radosavljevic, M.; Beug, H.; et al. Nuclear beta-catenin induces an early liver progenitor phenotype in hepatocellular carcinoma and promotes tumor recurrence. Am. J. Pathol. 2010, 176, 472–481. [Google Scholar] [CrossRef] [PubMed]
- van Zijl, F.; Zulehner, G.; Petz, M.; Schneller, D.; Kornauth, C.; Hau, M.; Machat, G.; Grubinger, M.; Huber, H.; Mikulits, W. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009, 5, 1169–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perugorria, M.J.; Olaizola, P.; Labiano, I.; Esparza-Baquer, A.; Marzioni, M.; Marin, J.J.G.; Bujanda, L.; Banales, J.M. Wnt-beta-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 121–136. [Google Scholar] [CrossRef]
- Wong, C.M.; Fan, S.T.; Ng, I.O. beta-Catenin mutation and overexpression in hepatocellular carcinoma: Clinicopathologic and prognostic significance. Cancer 2001, 92, 136–145. [Google Scholar] [CrossRef]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Ang, C.; Miura, J.T.; Gamblin, T.C.; He, R.; Xiu, J.; Millis, S.Z.; Gatalica, Z.; Reddy, S.K.; Yee, N.S.; Abou-Alfa, G.K. Comprehensive multiplatform biomarker analysis of 350 hepatocellular carcinomas identifies potential novel therapeutic options. J. Surg. Oncol. 2016, 113, 55–61. [Google Scholar] [CrossRef]
- Boyault, S.; Rickman, D.S.; de Reynies, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Herault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wang, S.C.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.C.; et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 2017, 32, 574–589.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Li, W.; Tian, F.; Jiang, K.; Liu, X.; Cen, J.; He, Q.; Qiu, Z.; Kienast, Y.; Wang, Z.; et al. Arid1a regulates response to anti-angiogenic therapy in advanced hepatocellular carcinoma. J. Hepatol. 2018, 68, 465–475. [Google Scholar] [CrossRef]
- Chen, C.; Lou, T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget 2017, 8, 46691–46703. [Google Scholar] [CrossRef] [Green Version]
- Li, X.P.; Yang, X.Y.; Biskup, E.; Zhou, J.; Li, H.L.; Wu, Y.F.; Chen, M.L.; Xu, F. Co-expression of CXCL8 and HIF-1alpha is associated with metastasis and poor prognosis in hepatocellular carcinoma. Oncotarget 2015, 6, 22880–22889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Blanco, C.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Chen, X.P.; Luo, S.F.; Guan, J.; Zhang, W.G.; Zhang, B.X. Involvement of hypoxia-inducible factor-1-alpha in multidrug resistance induced by hypoxia in HepG2 cells. J. Exp. Clin. Cancer Res. CR 2005, 24, 565–574. [Google Scholar]
- Sermeus, A.; Cosse, J.P.; Crespin, M.; Mainfroid, V.; de Longueville, F.; Ninane, N.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia induces protection against etoposide-induced apoptosis: Molecular profiling of changes in gene expression and transcription factor activity. Mol. Cancer 2008, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Che, N.; Zhou, L.; Luk, S.T.; Kau, P.W.; Chai, S.; Ngan, E.S.; Lo, C.M.; Man, K.; Ding, J.; et al. Efficacy of annexin A3 blockade in sensitizing hepatocellular carcinoma to sorafenib and regorafenib. J. Hepatol. 2018, 69, 826–839. [Google Scholar] [CrossRef]
- Song, Y.; Kim, S.H.; Kim, K.M.; Choi, E.K.; Kim, J.; Seo, H.R. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci. Rep. 2016, 6, 36750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzariti, A.; Mancarella, S.; Porcelli, L.; Quatrale, A.E.; Caligiuri, A.; Lupo, L.; Dituri, F.; Giannelli, G. Hepatic stellate cells induce hepatocellular carcinoma cell resistance to sorafenib through the laminin-332/alpha3 integrin axis recovery of focal adhesion kinase ubiquitination. Hepatology 2016, 64, 2103–2117. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.M.; Zhang, Z.; Hsu, J.L.; Li, C.W.; Lim, S.O.; Sheng, Y.Y.; Zhang, Y.; et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019, 68, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Ungerleider, N.; Han, C.; Zhang, J.; Yao, L.; Wu, T. TGFbeta signaling confers sorafenib resistance via induction of multiple RTKs in hepatocellular carcinoma cells. Mol. Carcinogenesis 2017, 56, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-alpha is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2019, 40, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhou, W.; Yin, S.; Zhou, Y.; Chen, T.; Qian, J.; Su, R.; Hong, L.; Lu, H.; Zhang, F.; et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology 2019, 70, 198–214. [Google Scholar] [CrossRef]
- Takahashi, K.; Yan, I.K.; Kogure, T.; Haga, H.; Patel, T. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio 2014, 4, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Wu, J.; Luo, D.; Jiang, C.; Ding, Y. Exosomes derived from HCC cells induce sorafenib resistance in hepatocellular carcinoma both in vivo and in vitro. J. Exp. Clin. Cancer Res. CR 2016, 35, 159. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Li, Y.; Tian, L.; Shi, H.; Wang, J.; Liang, Y.; Sun, B.; Wang, S.; Zhou, M.; Wu, L.; et al. Gankyrin drives metabolic reprogramming to promote tumorigenesis, metastasis and drug resistance through activating beta-catenin/c-Myc signaling in human hepatocellular carcinoma. Cancer Lett. 2019, 443, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. BioMed Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Kitisin, K.; Jogunoori, W.; Li, C.; Deng, C.X.; Mueller, S.C.; Ressom, H.W.; Rashid, A.; He, A.R.; Mendelson, J.S.; et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2445–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudoureyimu, M.; Zhou, H.; Zhi, Y.; Wang, T.; Feng, B.; Wang, R.; Chu, X. Recent progress in the emerging role of exosome in hepatocellular carcinoma. Cell Prolif. 2019, 52, e12541. [Google Scholar] [CrossRef] [Green Version]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef] [Green Version]
- Mishra, L.; Banker, T.; Murray, J.; Byers, S.; Thenappan, A.; He, A.R.; Shetty, K.; Johnson, L.; Reddy, E.P. Liver stem cells and hepatocellular carcinoma. Hepatology 2009, 49, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Pomyen, Y.; Hernandez, M.O.; Li, C.; Livak, F.; Tang, W.; Dang, H.; Greten, T.F.; Davis, J.L.; Zhao, Y.; et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology 2018, 68, 127–140. [Google Scholar] [CrossRef]
- Xiao, Y.; Lin, M.; Jiang, X.; Ye, J.; Guo, T.; Shi, Y.; Bian, X. The Recent Advances on Liver Cancer Stem Cells: Biomarkers, Separation, and Therapy. Anal. Cell. Pathol. (Amst.) 2017, 2017, 5108653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, S.; Li, M.Y.; Hu, B.G.; Liu, L.P.; Yang, S.L.; Yang, S.; Gong, Z.; Lai, P.B.S.; Chen, G.G. Cancer stem cells in hepatocellular carcinoma: An overview and promising therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816287. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Arao, T.; Furuta, K.; Sakai, K.; Kudo, K.; Kaneda, H.; Tamura, D.; Aomatsu, K.; Kimura, H.; Fujita, Y.; et al. Sorafenib inhibits the hepatocyte growth factor-mediated epithelial mesenchymal transition in hepatocellular carcinoma. Mol. Cancer Ther. 2011, 10, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.F.; Ngai, P.; Ho, D.W.; Yu, W.C.; Ng, M.N.; Lau, C.K.; Li, M.L.; Tam, K.H.; Lam, C.T.; Poon, R.T.; et al. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology 2008, 47, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Park, J.W.; Kim, J.S.; Lee, S.K.; Hong, E.K. Stem Cell Markers Predict the Response to Sorafenib in Patients with Hepatocellular Carcinoma. Gut Liver 2019, 13, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Fernando, J.; Malfettone, A.; Cepeda, E.B.; Vilarrasa-Blasi, R.; Bertran, E.; Raimondi, G.; Fabra, A.; Alvarez-Barrientos, A.; Fernandez-Salguero, P.; Fernandez-Rodriguez, C.M.; et al. A mesenchymal-like phenotype and expression of CD44 predict lack of apoptotic response to sorafenib in liver tumor cells. Int. J. Cancer 2015, 136, E161–E172. [Google Scholar] [CrossRef]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.C.; Chang, Y.S.; Hsu, H.P.; Yen, M.C.; Huang, H.L.; Cho, C.Y.; Wang, C.Y.; Weng, T.Y.; Lai, P.T.; Chen, C.S.; et al. Therapeutics targeting CD90-integrin-AMPK-CD133 signal axis in liver cancer. Oncotarget 2015, 6, 42923–42937. [Google Scholar] [CrossRef]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Antonucci, L.; Nakagawa, H.; Kim, J.Y.; Glitzner, E.; Caruso, S.; Shalapour, S.; Yang, L.; Valasek, M.A.; Lee, S.; et al. Liver Cancer Initiation Requires p53 Inhibition by CD44-Enhanced Growth Factor Signaling. Cancer Cell 2018, 33, 1061–1077.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Yao, Y.; Xu, G.; Zhou, C.; Zhang, Y.; Sun, J.; Jiang, R.; Shao, Q.; Chen, Y. CD24 regulates sorafenib resistance via activating autophagy in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.K.; Ng, L.; Lam, C.S.; Wong, S.K.; Wan, T.M.; Cheng, N.S.; Yau, T.C.; Poon, R.T.; Pang, R.W. The Enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS ONE 2013, 8, e78675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, M.; Yamashita, T.; Okada, H.; Oishi, N.; Nio, K.; Hayashi, T.; Nomura, Y.; Asahina, Y.; Ohwada, M.; Sunagozaka, H.; et al. Sorafenib suppresses extrahepatic metastasis de novo in hepatocellular carcinoma through inhibition of mesenchymal cancer stem cells characterized by the expression of CD90. Sci. Rep. 2017, 7, 11292. [Google Scholar] [CrossRef] [PubMed]
- Govaere, O.; Komuta, M.; Berkers, J.; Spee, B.; Janssen, C.; de Luca, F.; Katoonizadeh, A.; Wouters, J.; van Kempen, L.C.; Durnez, A.; et al. Keratin 19: A key role player in the invasion of human hepatocellular carcinomas. Gut 2014, 63, 674–685. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.N.; Zeng, Q.; Wang, H.Y.; Zhang, B.; Li, S.T.; Nan, X.; Cao, N.; Fu, C.J.; Yan, X.L.; Jia, Y.L.; et al. MicroRNA-125b attenuates epithelial-mesenchymal transitions and targets stem-like liver cancer cells through small mothers against decapentaplegic 2 and 4. Hepatology 2015, 62, 801–815. [Google Scholar] [CrossRef]
- Xiang, D.M.; Sun, W.; Zhou, T.; Zhang, C.; Cheng, Z.; Li, S.C.; Jiang, W.; Wang, R.; Fu, G.; Cui, X.; et al. Oncofetal HLF transactivates c-Jun to promote hepatocellular carcinoma development and sorafenib resistance. Gut 2019, 68, 1858–1871. [Google Scholar] [CrossRef]
- Guan, D.X.; Shi, J.; Zhang, Y.; Zhao, J.S.; Long, L.Y.; Chen, T.W.; Zhang, E.B.; Feng, Y.Y.; Bao, W.D.; Deng, Y.Z.; et al. Sorafenib enriches epithelial cell adhesion molecule-positive tumor initiating cells and exacerbates a subtype of hepatocellular carcinoma through TSC2-AKT cascade. Hepatology 2015, 62, 1791–1803. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Cheng, J.W.; Hu, J.W.; Li, H.; Ma, X.L.; Tang, W.G.; Sun, Y.F.; Guo, W.; Huang, A.; Zhou, K.Q.; et al. KPNA3 Confers Sorafenib Resistance to Advanced Hepatocellular Carcinoma via TWIST Regulated Epithelial-Mesenchymal Transition. J. Cancer 2019, 10, 3914–3925. [Google Scholar] [CrossRef]
- Xia, H.; Ooi, L.L.; Hui, K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, W.; Ye, Y.; Li, H.; Cheng, L.; Zhang, M.; Zheng, S.; Yu, J. LncRNA HOTAIR Contributes to Sorafenib Resistance through Suppressing miR-217 in Hepatic Carcinoma. BioMed Res. Int. 2020, 2020, 9515071. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.X.; et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008, 68, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Yasuchika, K.; Ishii, T.; Katayama, H.; Yoshitoshi, E.Y.; Ogiso, S.; Kita, S.; Yasuda, K.; Fukumitsu, K.; Mizumoto, M.; et al. Keratin 19, a Cancer Stem Cell Marker in Human Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3081–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.H.; Shao, Y.Y.; Chan, S.Y.; Huang, C.Y.; Hsu, C.H.; Cheng, A.L. High Serum Transforming Growth Factor-beta1 Levels Predict Outcome in Hepatocellular Carcinoma Patients Treated with Sorafenib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-beta1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Xu, X.; Tao, Y.; Shan, L.; Chen, R.; Jiang, H.; Qian, Z.; Cai, F.; Ma, L.; Yu, Y. The Role of MicroRNAs in Hepatocellular Carcinoma. J. Cancer 2018, 9, 3557–3569. [Google Scholar] [CrossRef]
- Li, W.; Dong, X.; He, C.; Tan, G.; Li, Z.; Zhai, B.; Feng, J.; Jiang, X.; Liu, C.; Jiang, H.; et al. LncRNA SNHG1 contributes to sorafenib resistance by activating the Akt pathway and is positively regulated by miR-21 in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. CR 2019, 38, 183. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Protein | Change | Drugs Affected | Consequences | Reference |
|---|---|---|---|---|
| Uptake Carriers (MOC-1a) | ||||
| OCT1 | Down-regulation | Sorafenib | Reduced OS | [11] |
| OCT1 | Mutations | Decreased function in vitro | [9] | |
| SLC46A3 | Down-regulation | Reduced OS | [18] | |
| Export Pumps (MOC-1b) | ||||
| BCRP | Up-regulation | Sorafenib | Reduced OS | [19] |
| MDR1 | Up-regulation | Reduced MST | [20] | |
| MDR1 | GV: rs1045642 | Better clinical evolution | [21] | |
| MRP3 | Up-regulation | Decreased cell sensitivity in vitro | [22] | |
| Drug Metabolism (MOC-2) | ||||
| CYP3A4 | GV: rs2242480 | Lenvatinib | Altered plasma levels | [23] |
| CYP3A5 | GV: rs776746 | Sorafenib | Hepatic and renal toxicity | [24] |
| DPD | Up-regulation | 5-Fluorouracil | Higher DPR and lower PFS | [25] |
| DPD | Up-regulation | S-1 | Increased OS | [26] |
| DPD | Up-regulation | Interferon-α | Reduced OS | [27] |
| UGT1A1 | GV: rs8175347 | Sorafenib | Hyperbilirubinemia and toxicity | [28] |
| UGT1A9 | Down-regulation | Reduced OS | [29] | |
| UGT1A9 | GV: rs3832043 | Severe toxicity | [30] | |
| UGT1A9 | GV: rs17868320 | Severe toxicity | [31] | |
| UGT1A9 | GV: rs3832043 | Regorafenib | Severe toxicity | [32] |
| Protein | Change | Drugs Affected | Consequences | Reference |
|---|---|---|---|---|
| Drug Targets (MOC-3) | ||||
| EGFR | Positive feedback EGFR-KLF4 | Sorafenib | Reduced sensitivity (in vitro) | [53] |
| EGFR, HER3 | Increased activity | Reduced sensitivity (in vitro and in vivo) | [54] | |
| p-ERK, VEGFR-2 | Up-regulation | Reduced OS | [55] | |
| p-MET | High levels | Cabozantinib | Increased sensitivity in vitro and in vivo | [56] |
| p-MET | High levels | Sorafenib | Reduced clinical response | [56] |
| p-MET | Gene amplification | Cabozantinib | Increased sensitivity in vitro | [57] |
| VEGF, ANG2, FGF21 | High serum levels | Sorafenib, Lenvatinib | Reduced OS | [58] |
| VEGF-A, VEGF-C | GV: rs2010963, rs4604006 | Sorafenib | Reduced OS and PFS | [59,60] |
| VEGFR-2 | GV: rs2071559, rs1870377 | Reduced OS, PFS and TTP | [61] | |
| DNA Repairing (MOC-4) | ||||
| ERCC1 | Up-regulation | Platinum derivatives | Lower sensitivity in surgically resected tissue | [62] |
| XRCC4 | Up-regulation | Reduced OS and PFS | [63] | |
| XRCC1 | GV: rs25487 | Reduced MST | [64] | |
| XRCC1, APE1 | GV: rs1799782, rs1130409 | Reduced clinical response | [65] | |
| XRCC4 | Down-regulation | Doxorubicin, Cisplatin | Increased OS and PFS | [66] |
| Factor | Change | Drugs Affected | Consequences | Reference |
|---|---|---|---|---|
| Pro-Apoptotic Factors (MOC-5a) | ||||
| BMF | Down-regulation | Sorafenib | Reduced OS and TTP | [101] |
| miR-221 | Up-regulation | Reduced OS and TTP | [101] | |
| miR-221 | High serum levels | Increased DPR | [102] | |
| Nucleostemin | Up-regulation | Reduced sensitivity (in vitro) | [98] | |
| p15INK4b | Down-regulation | Lower survival rate | [100] | |
| p53 | Mutations | Reduced OS | [95] | |
| PUMA | Down-regulation | Reduced sensitivity (in vitro) | [103] | |
| SIRT1 | Up-regulation | Reduced sensitivity (in vitro) | [97] | |
| Survival Pathways (MOC-5b) | ||||
| ARID1A | Mutations | Sorafenib | Reduced OS | [104] |
| β-catenin | GOF mutations | Regorafenib, Sorafenib | Reduced OS and TTP | [105] |
| BCL2, MCL1 | Up-regulation | Sorafenib | Reduced sensitivity (in vitro) | [106] |
| c-MYC | Up-regulation | Reduced sensitivity (in vitro) | [107] | |
| FGF19/FGFR4 | Increased activity | Reduced sensitivity (in vitro) | [108] | |
| HANR | Up-regulation | Reduced sensitivity (in vitro and in vivo) | [109] | |
| Hedgehog pathway | Increased activity | Reduced sensitivity (in HCC patient-derived organoids) | [110] | |
| JAK1 | GOF mutations | Increased DPR | [111] | |
| JNK | Up-regulation | Reduced sensitivity (in vitro) | [112] | |
| MALAT1 | Up-regulation | Reduced OS | [113] | |
| MAPK/ERK pathway | Increased activity | Reduced sensitivity (in vitro) | [57] | |
| NEAT1 | Up-regulation | Reduced OS | [114] | |
| Notch3 | Up-regulation | Reduced sensitivity (in vitro) | [115] | |
| p-ERK | Increased levels | Reduced sensitivity (in vitro and in vivo) | [116] | |
| PI3K/AKT pathway | Increased activity | Reduced sensitivity (in vitro) | [117] | |
| p-RPS6 | Increased levels | Increased recurrence rate | [118] | |
| SNAI1 | Up-regulation | Reduced sensitivity (in vitro) | [119] | |
| STAT3 | Increased activity | Reduced sensitivity (in vitro) | [120] | |
| YAP | Up-regulation | Reduced sensitivity (in vitro) | [121] | |
| Factor | Change | Drugs Affected | Consequences | Reference |
|---|---|---|---|---|
| Hypoxia | ||||
| Annexin A3 | Up-regulation | Sorafenib | Reduced OS | [141] |
| HIF-1α | Up-regulation | Reduced OS and DFS | [136] | |
| HIF-1α | GV: rs12434438 | Reduced OS and TTP | [60] | |
| Fibrosis | ||||
| Collagen 1A1 | Up-regulation | Sorafenib | Reduced sensitivity (in vitro) | [142] |
| Laminin-332 | Up-regulation | Reduced sensitivity (in vitro) | [143] | |
| Immune System and Inflammation | ||||
| CCL2, CCL17 | Up-regulation | Sorafenib | Reduced OS and TTP | [144] |
| Osteopontin | Up-regulation | ICIs | Reduced sensitivity (in vivo) | [145] |
| TGF-β | Up-regulation | Sorafenib | Reduced sensitivity (in vitro) | [146] |
| TNF-α | Up-regulation | Sorafenib | Reduced OS and PFS | [147] |
| TREM-1 | Up-regulation | ICIs | Reduced OS and DFS | [148] |
| Extracellular Microvesicles | ||||
| linc-ROR | Up-regulation | Sorafenib | Reduced sensitivity (in vitro) | [149] |
| HCC-derived Exosomes | High levels | Reduced sensitivity (in vitro and in vivo) | [150] | |
| Metabolic Reprogramming | ||||
| Gankyrin | Up-regulation | Sorafenib, Regorafenib | Reduced sensitivity (in vitro and in vivo) | [151] |
| NANOG | Up-regulation | Sorafenib | Reduced sensitivity (in vitro and in vivo) | [152] |
| Factor | Change | Drugs Affected | Consequences | Ref. |
|---|---|---|---|---|
| Cell Adhesion Proteins | ||||
| CD133, CD90 | Up-regulation | Sorafenib | Reduced PFS | [166] |
| CD133, CD44 | Up-regulation | Reduced sensitivity (in vitro and in vivo) | [174] | |
| CD44 | Up-regulation | Reduced sensitivity (in vitro and in vivo) | [167] | |
| CD24 | Up-regulation | Reduced sensitivity (in vitro) | [173] | |
| EpCAM | Up-regulation | Reduced sensitivity (in vivo) | [175] | |
| Cytokeratins | ||||
| KRT19 | Up-regulation | Sorafenib | Reduced sensitivity (in vitro) | [176] |
| TGF-β Pathway | ||||
| TGF-β1 | Up-regulation | Sorafenib | Reduced sensitivity (in vitro) | [146] |
| TGF-β1 | Up-regulation | Reduced OS and PFS | [105] | |
| SMAD2/4 | Up-regulation | Reduced sensitivity (in vitro) | [177] | |
| Transcription Factors | ||||
| OCT4 | Up-regulation | Sorafenib | Reduced sensitivity (in vitro and in vivo) | [33] |
| OCT4, SOX2 | Up-regulation | Reduced sensitivity (in vitro and in vivo) | [178] | |
| TSC2 | Increased activity | Reduced sensitivity (in vitro and in vivo) | [179] | |
| TWIST1 | Up-regulation | Reduced sensitivity (in vitro and in vivo) | [180] | |
| Non-Coding RNAs | ||||
| miR-216a/217 | Up-regulation | Sorafenib | Reduced DFS | [181] |
| miR-125b | Down-regulation | Regorafenib | Reduced OS | [177] |
| HANR | Up-regulation | Sorafenib | Reduced sensitivity (in vitro and in vivo) | [109] |
| HOTAIR | Up-regulation | Reduced sensitivity (in vitro) | [182] | |
| MALAT1 | Up-regulation | Reduced OS | [113] | |
| NEAT | Up-regulation | Reduced OS | [114] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marin, J.J.G.; Macias, R.I.R.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1663. https://doi.org/10.3390/cancers12061663
Marin JJG, Macias RIR, Monte MJ, Romero MR, Asensio M, Sanchez-Martin A, Cives-Losada C, Temprano AG, Espinosa-Escudero R, Reviejo M, et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers. 2020; 12(6):1663. https://doi.org/10.3390/cancers12061663
Chicago/Turabian StyleMarin, Jose J.G., Rocio I.R. Macias, Maria J. Monte, Marta R. Romero, Maitane Asensio, Anabel Sanchez-Martin, Candela Cives-Losada, Alvaro G. Temprano, Ricardo Espinosa-Escudero, Maria Reviejo, and et al. 2020. "Molecular Bases of Drug Resistance in Hepatocellular Carcinoma" Cancers 12, no. 6: 1663. https://doi.org/10.3390/cancers12061663