FGF2 Induces Resistance to Nilotinib through MAPK Pathway Activation in KIT Mutated Melanoma

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
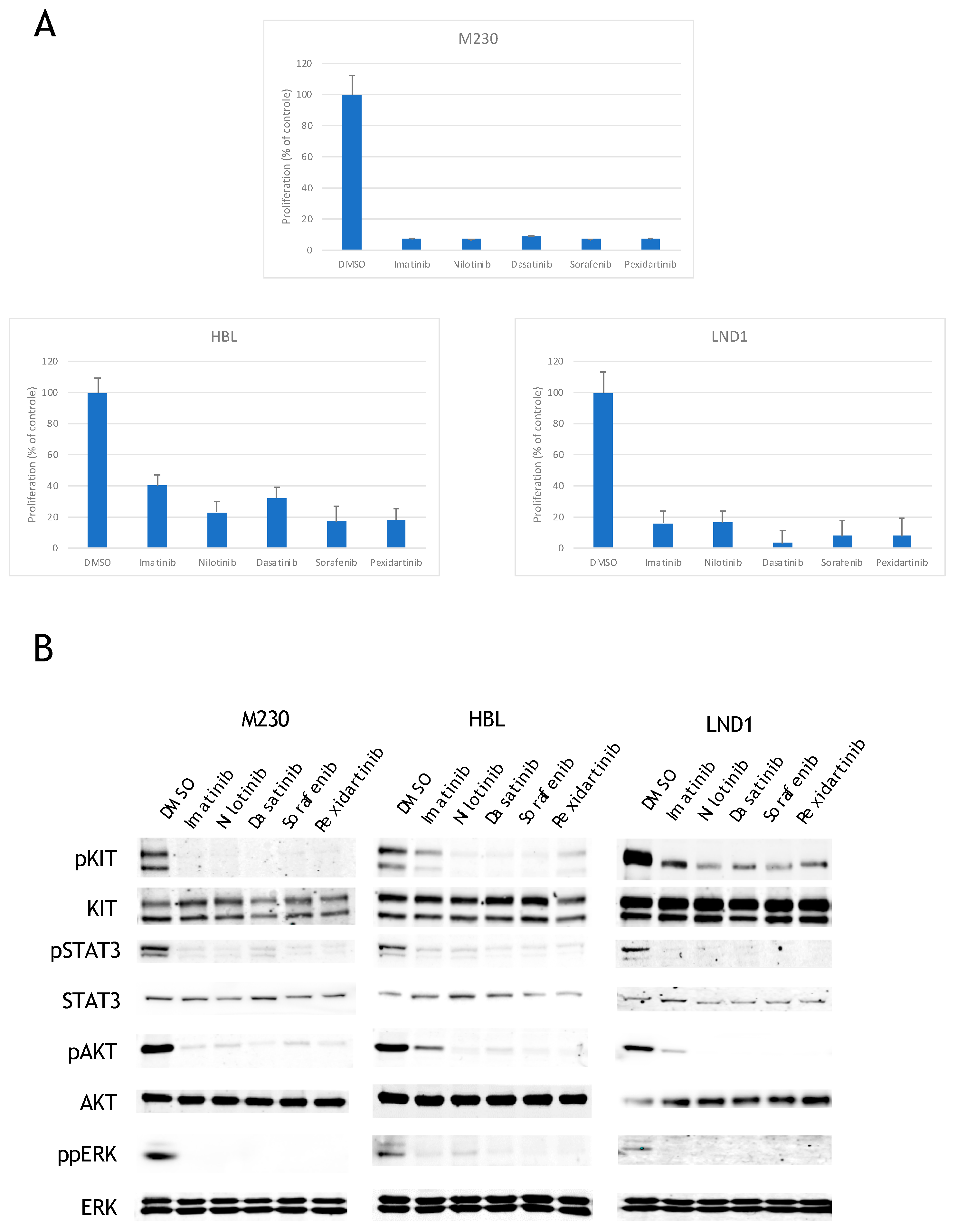
2.1. KIT Mutated Melanoma Are Sensitive to All KIT Inhibitors
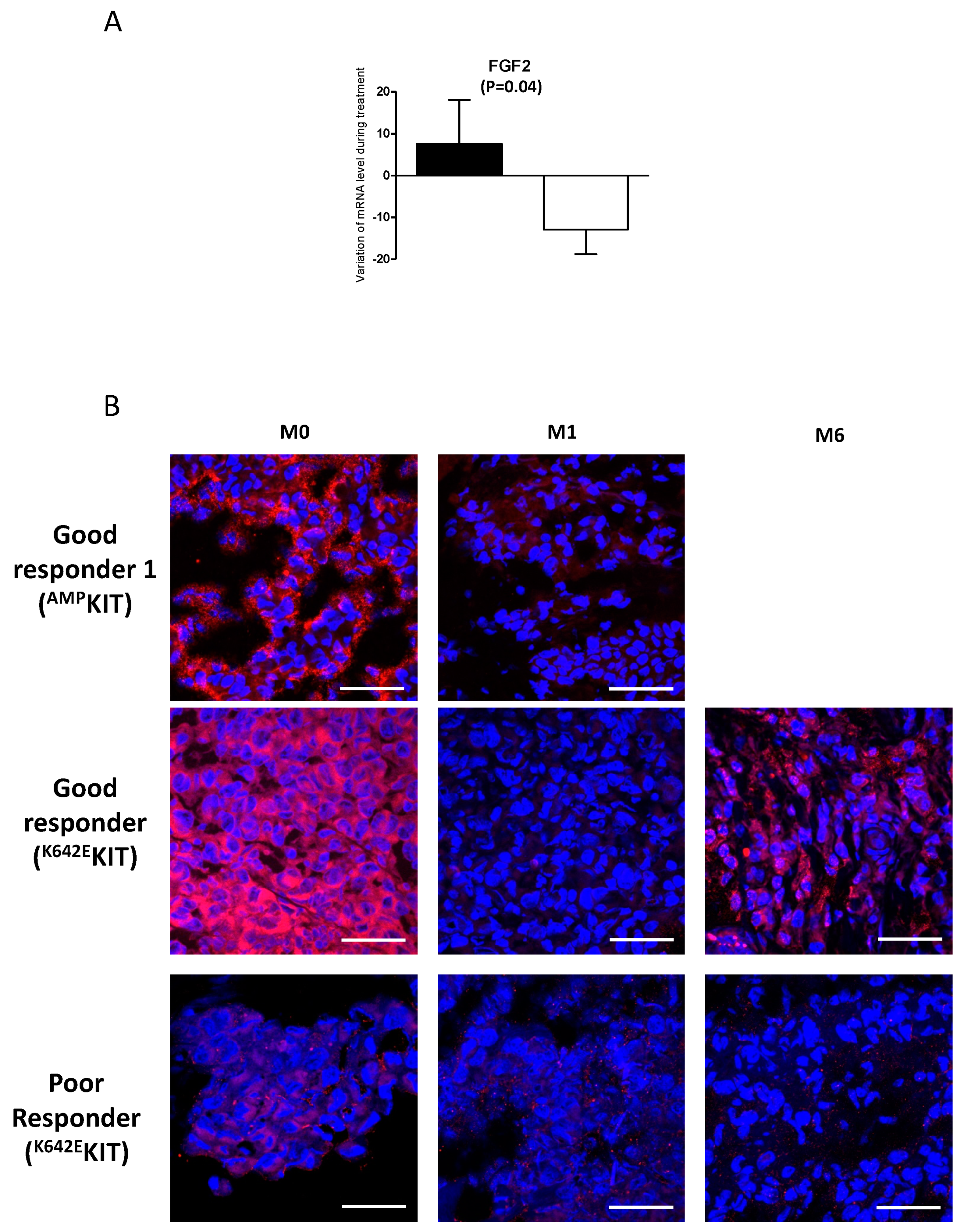
2.2. A Link Between FGF2 and Resistance
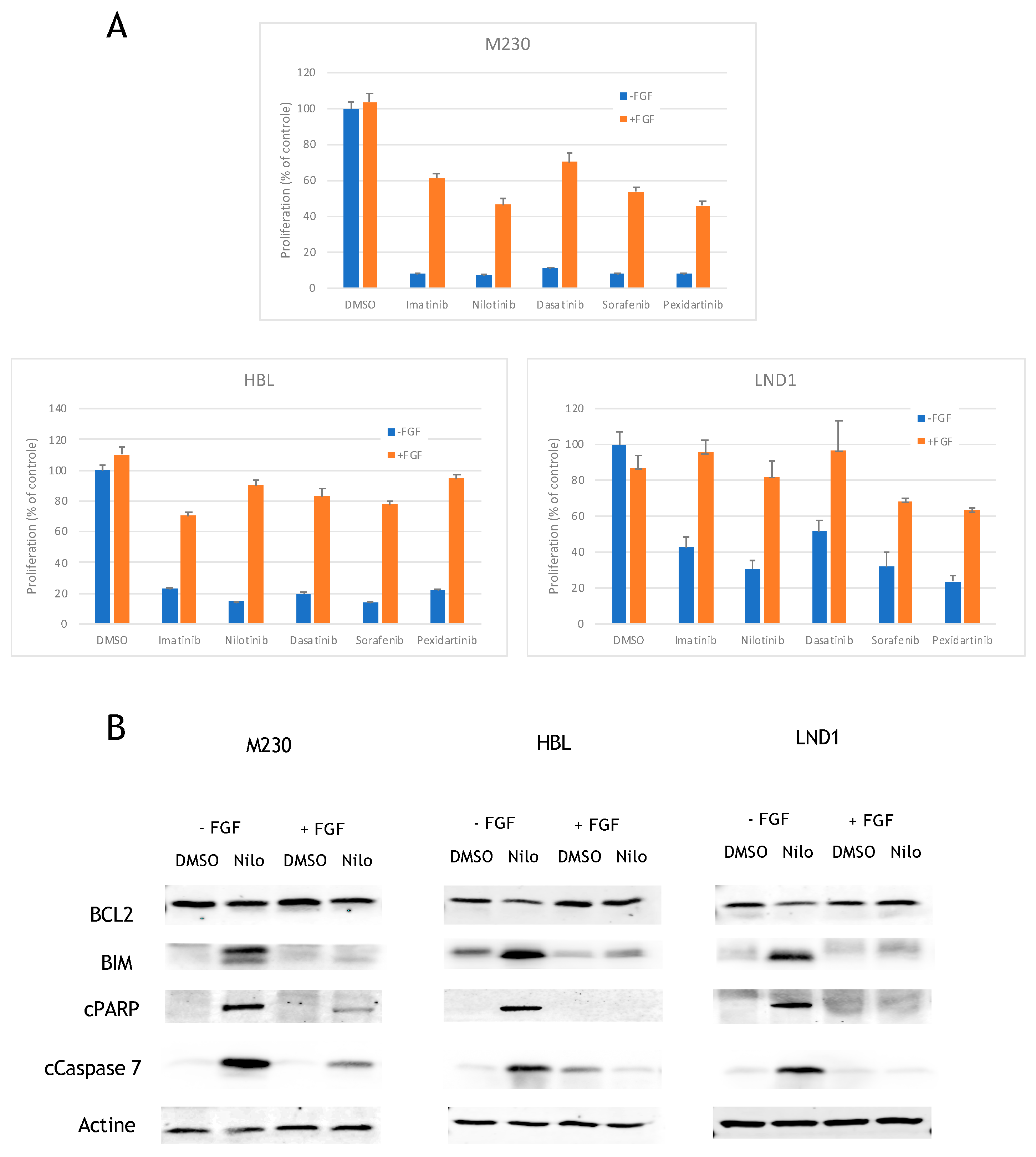
2.3. FGF2 Prevents BCL2 Reduction and BIM Induction to Inhibit Apoptosis
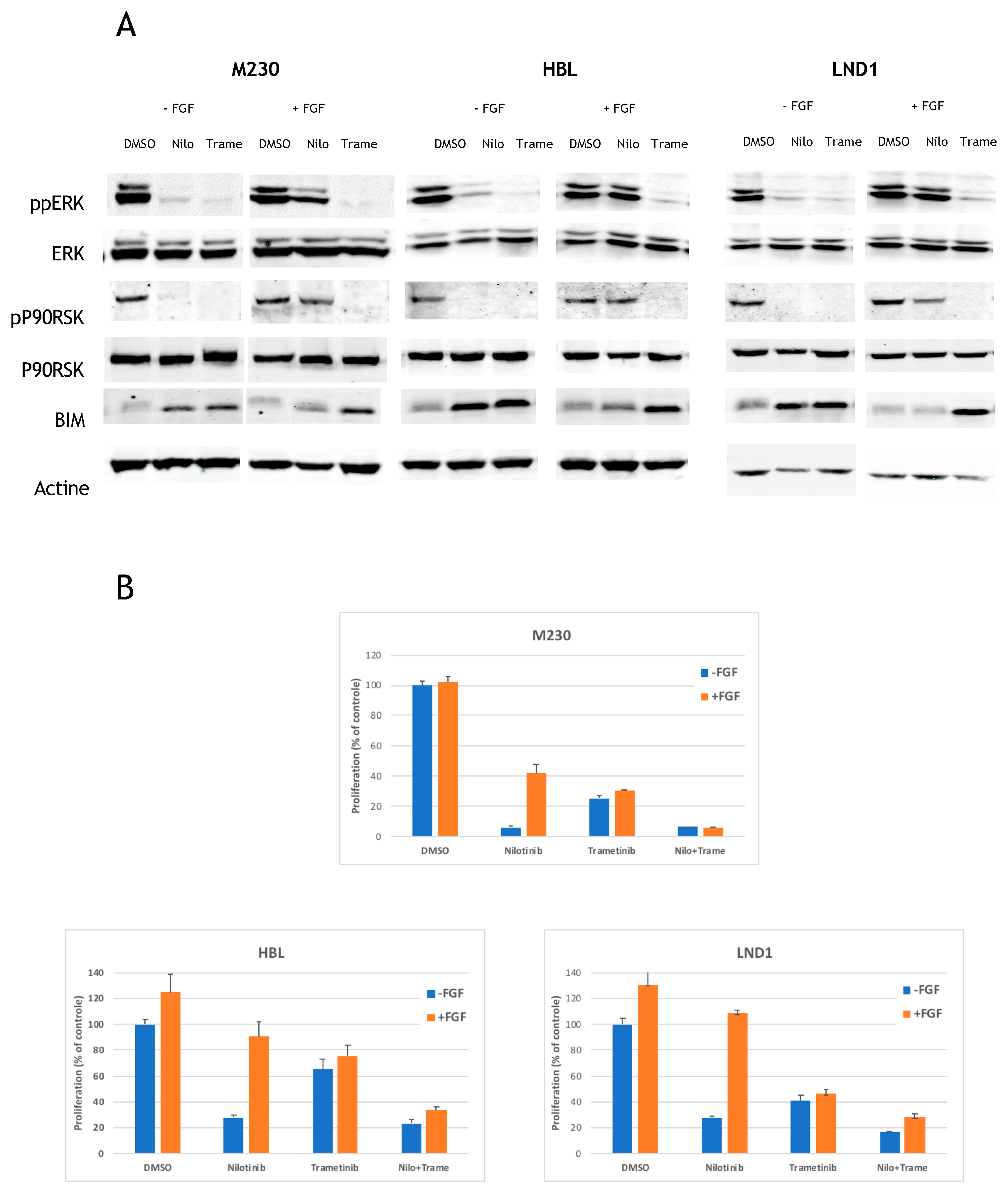
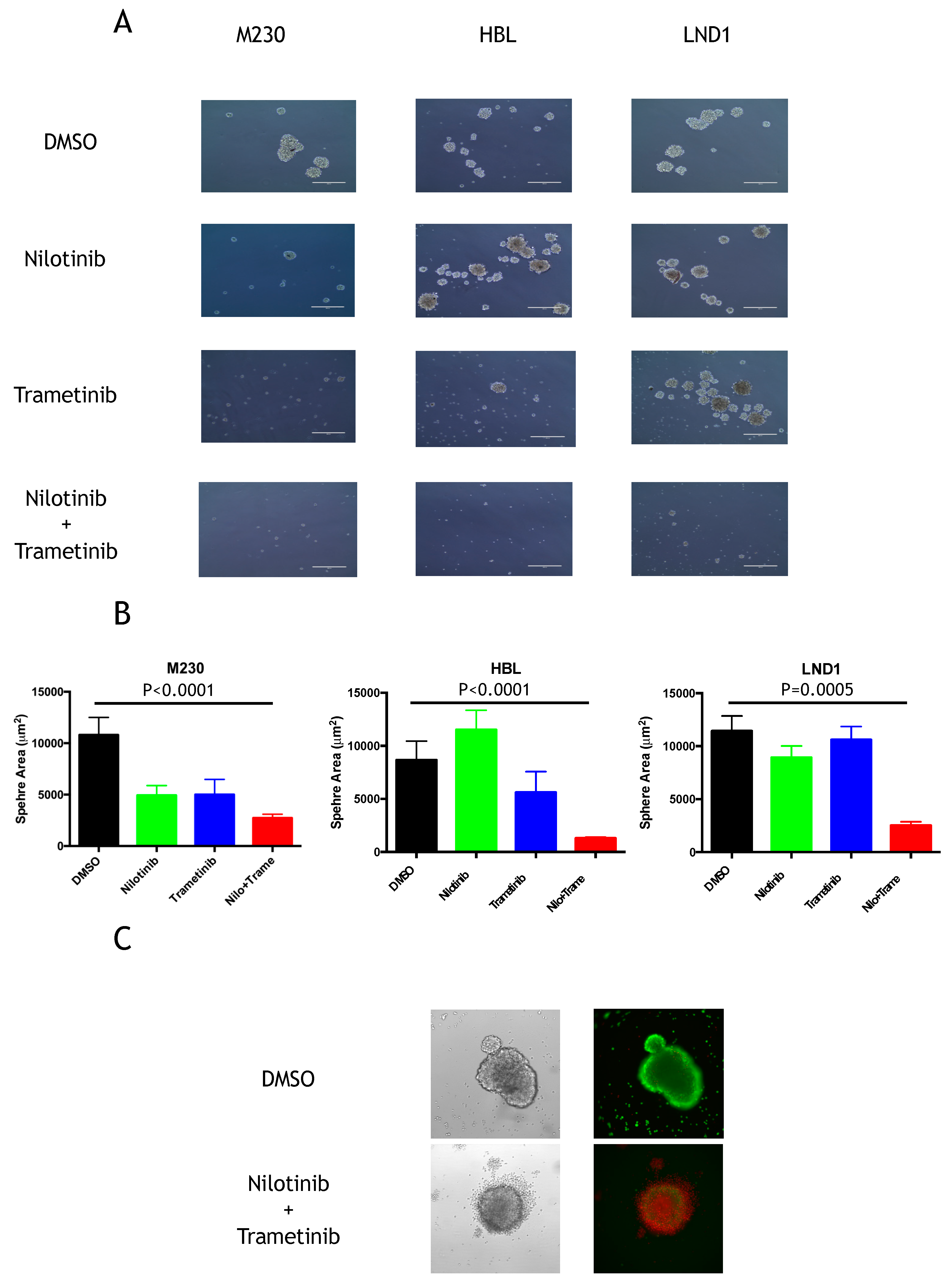
2.4. MEK Inhibition Reverses the FGF2-Mediated Resistance to KIT Inhibition
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Three-Dimensional Spheroid Growth
4.3. Proliferation Assay
4.4. Western Blotting
4.5. Immunofluorescence Staining
4.6. Flow Cytometry
4.7. Pharmacodynamic Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wehrle-Haller, B. The role of Kit-ligand in melanocyte development and epidermal homeostasis. Pigment. Cell Res. 2003, 16, 287–296. [Google Scholar] [CrossRef]
- Lennartsson, J.; Jelacic, T.; Linnekin, D.; Shivakrupa, R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells 2005, 23, 16–43. [Google Scholar] [CrossRef]
- Hemesath, T.J.; Price, E.R.; Takemoto, C.; Badalian, T.; Fisher, D.E. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature 1998, 391, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.; Sondak, V.K.; Weber, J.S. c-KIT signaling as the driving oncogenic event in sub-groups of melanomas. Histol. Histopathol. 2009, 24, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef]
- Bastian, B.C.; Esteve-Puig, R. Targeting activated KIT signaling for melanoma therapy. J. Clin. Oncol. 2013, 31, 3288–3290. [Google Scholar] [CrossRef]
- Gong, H.Z.; Zheng, H.Y.; Li, J. The clinical significance of KIT mutations in melanoma: A meta-analysis. Melanoma Res. 2018, 28, 259–270. [Google Scholar] [CrossRef]
- Dumaz, N.; Andre, J.; Sadoux, A.; Laugier, F.; Podgorniak, M.P.; Mourah, S.; Lebbe, C. Driver KIT mutations in melanoma cluster in four hotspots. Melanoma Res. 2015, 25, 88–90. [Google Scholar] [CrossRef]
- Jiang, X.; Zhou, J.; Yuen, N.K.; Corless, C.L.; Heinrich, M.C.; Fletcher, J.A.; Demetri, G.D.; Widlund, H.R.; Fisher, D.E.; Hodi, F.S. Imatinib targeting of KIT-mutant oncoprotein in melanoma. Clin. Cancer Res. 2008, 14, 7726–7732. [Google Scholar] [CrossRef] [Green Version]
- Monsel, G.; Ortonne, N.; Bagot, M.; Bensussan, A.; Dumaz, N. c-Kit mutants require hypoxia-inducible factor 1alpha to transform melanocytes. Oncogene 2010, 29, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalley, K.S.; Contractor, R.; Nguyen, T.K.; Xiao, M.; Edwards, R.; Muthusamy, V.; King, A.J.; Flaherty, K.T.; Bosenberg, M.; Herlyn, M.; et al. Identification of a novel subgroup of melanomas with KIT/cyclin-dependent kinase-4 overexpression. Cancer Res. 2008, 68, 5743–5752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, D.; Carvajal, R.D. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am. J. Clin. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Demetri, G.D.; von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.; Roberts, P.J.; Heinz, D.; et al. Long-term results from a randomized phase II trial of standard-versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef]
- Cho, J.H.; Kim, K.M.; Kwon, M.; Kim, J.H.; Lee, J. Nilotinib in patients with metastatic melanoma harboring KIT gene aberration. Investig. New Drugs 2012, 30, 2008–2014. [Google Scholar] [CrossRef]
- Delyon, J.; Chevret, S.; Jouary, T.; Dalac, S.; Dalle, S.; Guillot, B.; Arnault, J.P.; Avril, M.F.; Bedane, C.; Bens, G.; et al. STAT3 Mediates Nilotinib Response in KIT-Altered Melanoma: A Phase II Multicenter Trial of the French Skin Cancer Network. J. Investig. Derm. 2018, 138, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for Melanomas Harboring Mutationally Activated or Amplified KIT Arising on Mucosal, Acral, and Chronically Sun-Damaged Skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef] [Green Version]
- Minor, D.R.; Kashani-Sabet, M.; Garrido, M.; O’Day, S.J.; Hamid, O.; Bastian, B.C. Sunitinib therapy for melanoma patients with KIT mutations. Clin. Cancer Res. 2012, 18, 1457–1463. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, Y.; Choi, J.W.; Kim, Y.S. Correlation of imatinib resistance with the mutational status of KIT and PDGFRA genes in gastrointestinal stromal tumors: A meta-analysis. J. Gastrointest. Liver Dis. 2013, 22, 413–418. [Google Scholar]
- Si, L.; Xu, X.; Kong, Y.; Flaherty, K.T.; Chi, Z.; Cui, C.; Sheng, X.; Li, S.; Dai, J.; Yu, W.; et al. Major response to everolimus in melanoma with acquired imatinib resistance. J. Clin. Oncol. 2012, 30, e37–e40. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Kim, S.Y.; Kim, Y.J.; Sim, M.H.; Kim, S.T.; Kim, N.K.D.; Kim, K.; Park, W.; Kim, J.H.; Jang, K.T.; et al. Emergence of CTNNB1 mutation at acquired resistance to KIT inhibitor in metastatic melanoma. Clin. Transl. Oncol. 2017, 19, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Oba, J.; Kim, S.H.; Wang, W.L.; Macedo, M.P.; Carapeto, F.; McKean, M.A.; Van Arnam, J.; Eterovic, A.K.; Sen, S.; Kale, C.R.; et al. Targeting the HGF/MET Axis Counters Primary Resistance to KIT Inhibition in KIT-Mutant Melanoma. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Chraybi, M.; Abd Alsamad, I.; Copie-Bergman, C.; Baia, M.; André, J.; Dumaz, N.; Ortonne, N. Oncogene abnormalities in a series of primary melanomas of the sinonasal tract: NRAS mutations and CCND1 amplification are more frequent than KIT or BRAF mutations. Human Pathol. 2013, 44, 1902–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Carvajal, R.D.; Dummer, R.; Hauschild, A.; Daud, A.; Bastian, B.C.; Markovic, S.N.; Queirolo, P.; Arance, A.; Berking, C.; et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann. Oncol. 2017, 28, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Shoushtari, A.N.; Munhoz, R.R.; Kuk, D.; Ott, P.A.; Johnson, D.B.; Tsai, K.K.; Rapisuwon, S.; Eroglu, Z.; Sullivan, R.J.; Luke, J.J.; et al. The efficacy of anti-PD-1 agents in acral and mucosal melanoma. Cancer 2016, 122, 3354–3362. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Larkin, J.; Sosman, J.A.; Lebbe, C.; Brady, B.; Neyns, B.; Schmidt, H.; Hassel, J.C.; Hodi, F.S.; Lorigan, P.; et al. Efficacy and Safety of Nivolumab Alone or in Combination With Ipilimumab in Patients With Mucosal Melanoma: A Pooled Analysis. J. Clin. Oncol. 2017, 35, 226–235. [Google Scholar] [CrossRef]
- Han, Y.; Gu, Z.; Wu, J.; Huang, X.; Zhou, R.; Shi, C.; Tao, W.; Wang, L.; Wang, Y.; Zhou, G.; et al. Repurposing Ponatinib as a Potent Agent against KIT Mutant Melanomas. Theranostics 2019, 9, 1952–1964. [Google Scholar] [CrossRef]
- Li, F.; Huynh, H.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. FGFR-Mediated Reactivation of MAPK Signaling Attenuates Antitumor Effects of Imatinib in Gastrointestinal Stromal Tumors. Cancer Discov. 2015, 5, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 Promotes Gastrointestinal Stromal Tumor Cell Growth and Drug Resistance. Cancer Res. 2015, 75, 880–891. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartlidge, R.A.; Thomas, G.R.; Cagnol, S.; Jong, K.A.; Molton, S.A.; Finch, A.J.; McMahon, M. Oncogenic BRAF(V600E) inhibits BIM expression to promote melanoma cell survival. Pigment. Cell Melanoma Res. 2008, 21, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belloc, F.; Moreau-Gaudry, F.; Uhalde, M.; Cazalis, L.; Jeanneteau, M.; Lacombe, F.; Praloran, V.; Mahon, F.X. Imatinib and nilotinib induce apoptosis of chronic myeloid leukemia cells through a Bim-dependant pathway modulated by cytokines. Cancer Biol. 2007, 6, 912–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tétu, P.; Delyon, J.; André, J.; Reger de Moura, C.; Sabbah, M.; Ghanem, G.E.; Battistella, M.; Mourah, S.; Lebbé, C.; Dumaz, N. FGF2 Induces Resistance to Nilotinib through MAPK Pathway Activation in KIT Mutated Melanoma. Cancers 2020, 12, 1062. https://doi.org/10.3390/cancers12051062
Tétu P, Delyon J, André J, Reger de Moura C, Sabbah M, Ghanem GE, Battistella M, Mourah S, Lebbé C, Dumaz N. FGF2 Induces Resistance to Nilotinib through MAPK Pathway Activation in KIT Mutated Melanoma. Cancers. 2020; 12(5):1062. https://doi.org/10.3390/cancers12051062
Chicago/Turabian StyleTétu, Pauline, Julie Delyon, Jocelyne André, Coralie Reger de Moura, Malak Sabbah, Ghanem E Ghanem, Maxime Battistella, Samia Mourah, Céleste Lebbé, and Nicolas Dumaz. 2020. "FGF2 Induces Resistance to Nilotinib through MAPK Pathway Activation in KIT Mutated Melanoma" Cancers 12, no. 5: 1062. https://doi.org/10.3390/cancers12051062