Integrative Radiogenomics Approach for Risk Assessment of Post-Operative Metastasis in Pathological T1 Renal Cell Carcinoma: A Pilot Retrospective Cohort Study

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
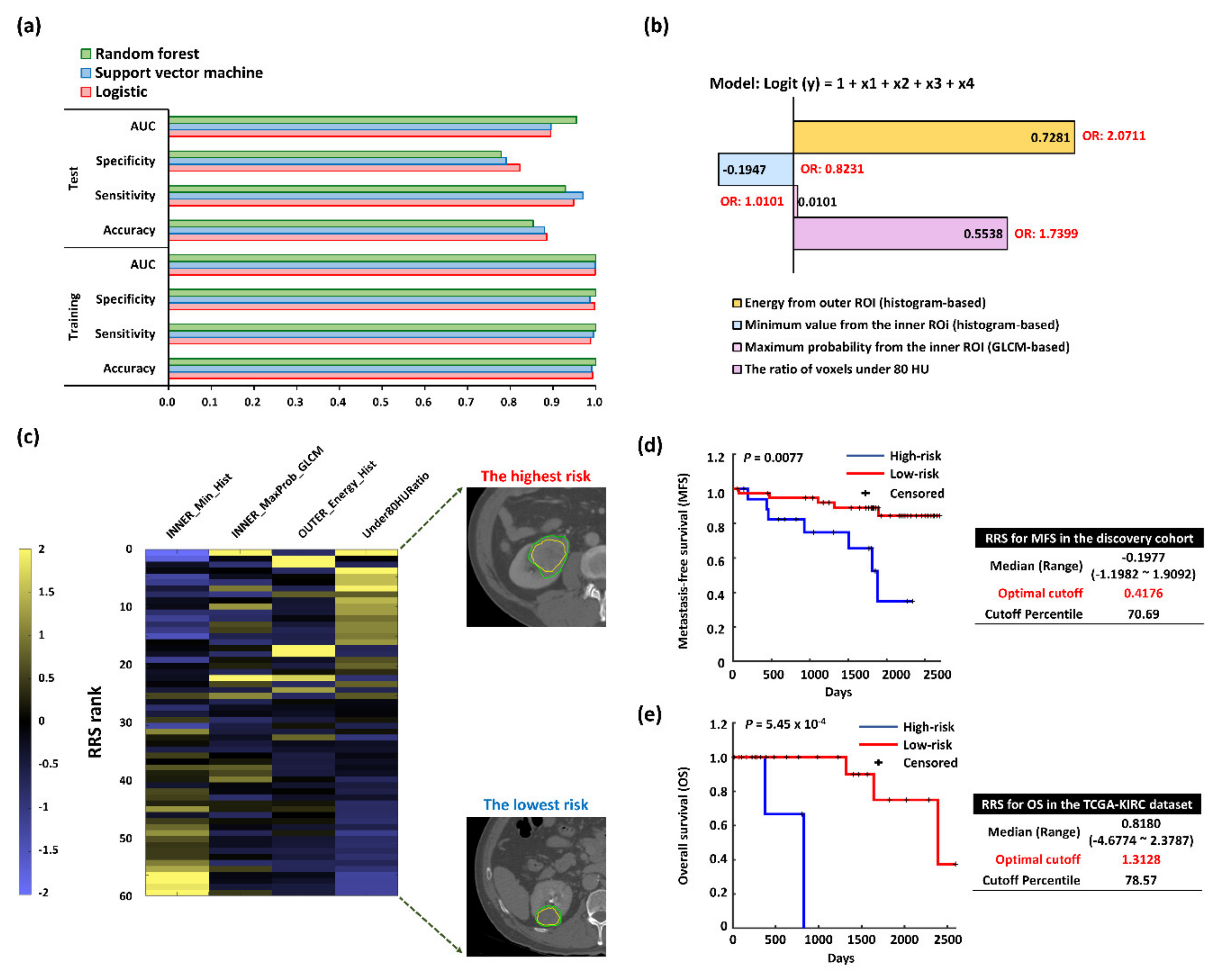
2.1. Selected Prognostic Radiomics Features from the Discovery Cohort
2.2. Radiomics Risk Score (RRS) Predicting Postsurgical Metastasis
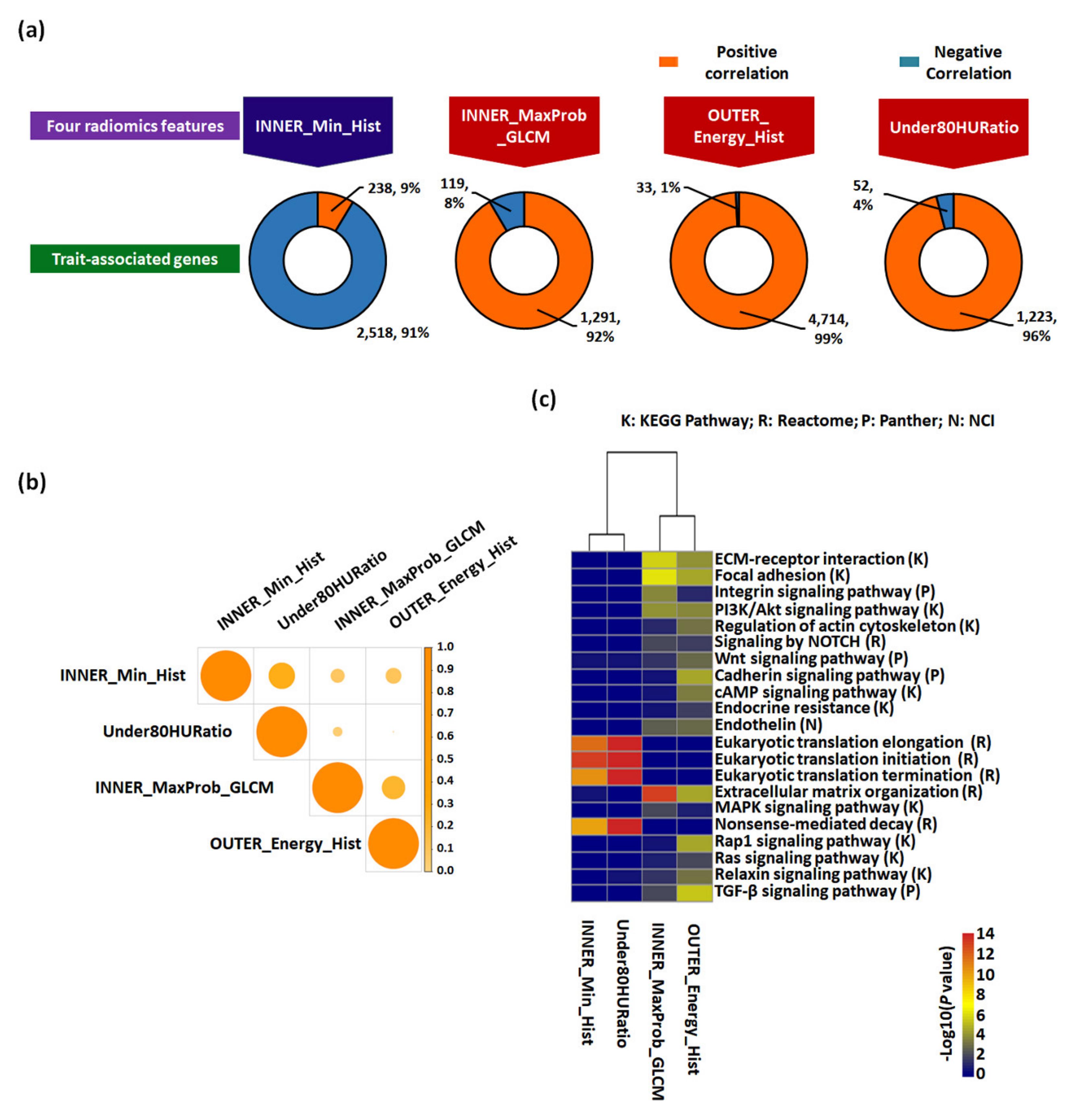
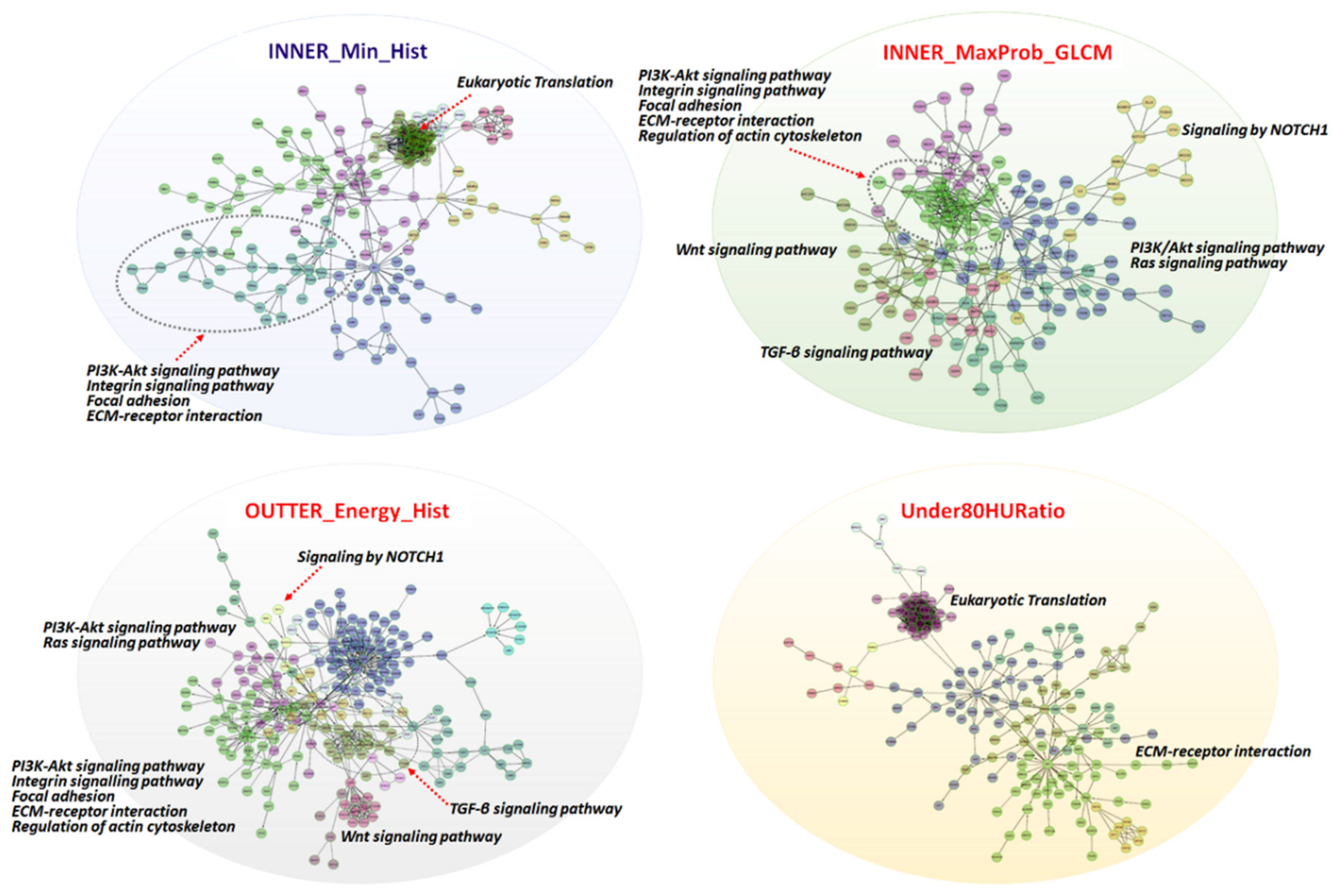
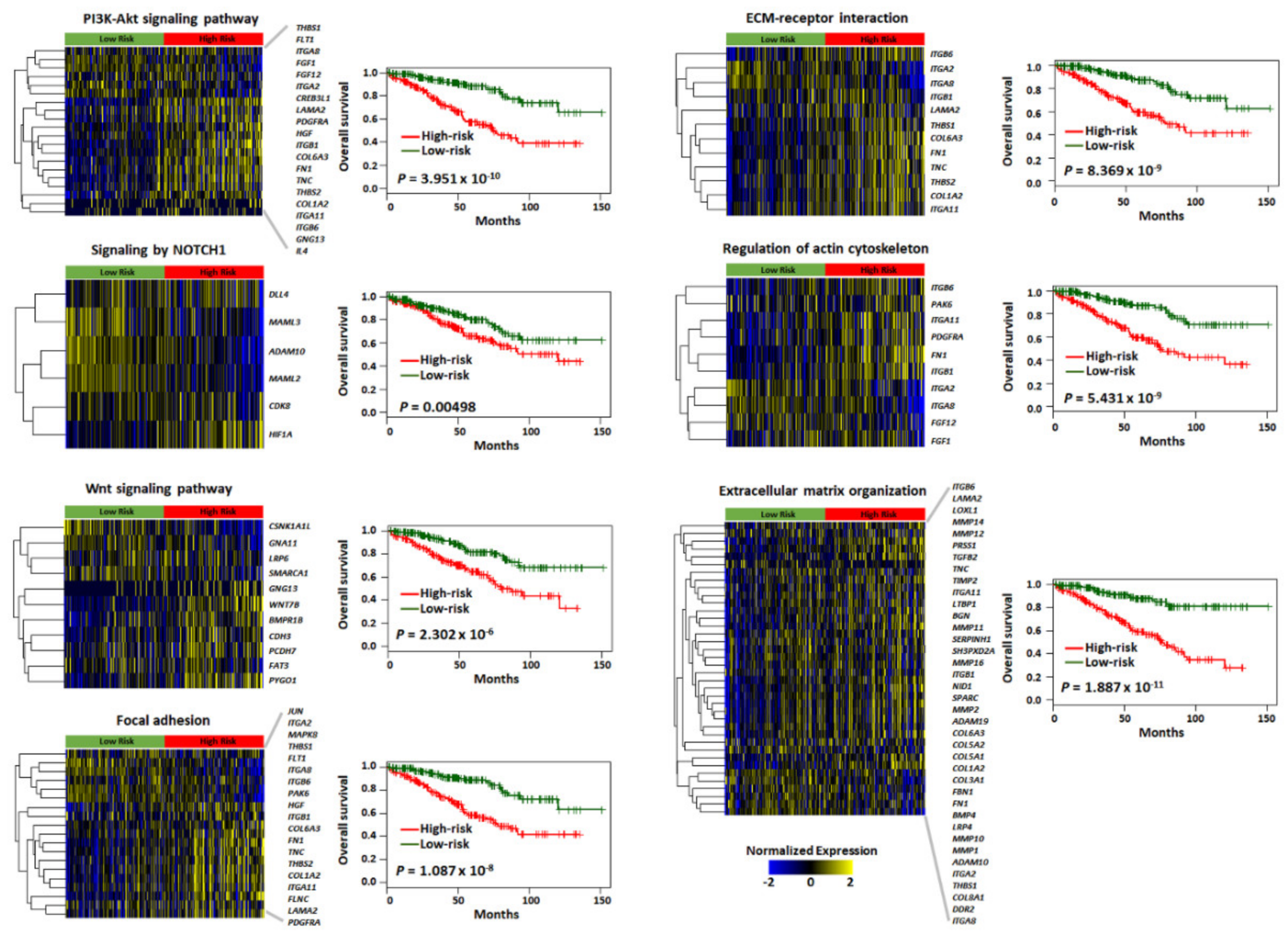
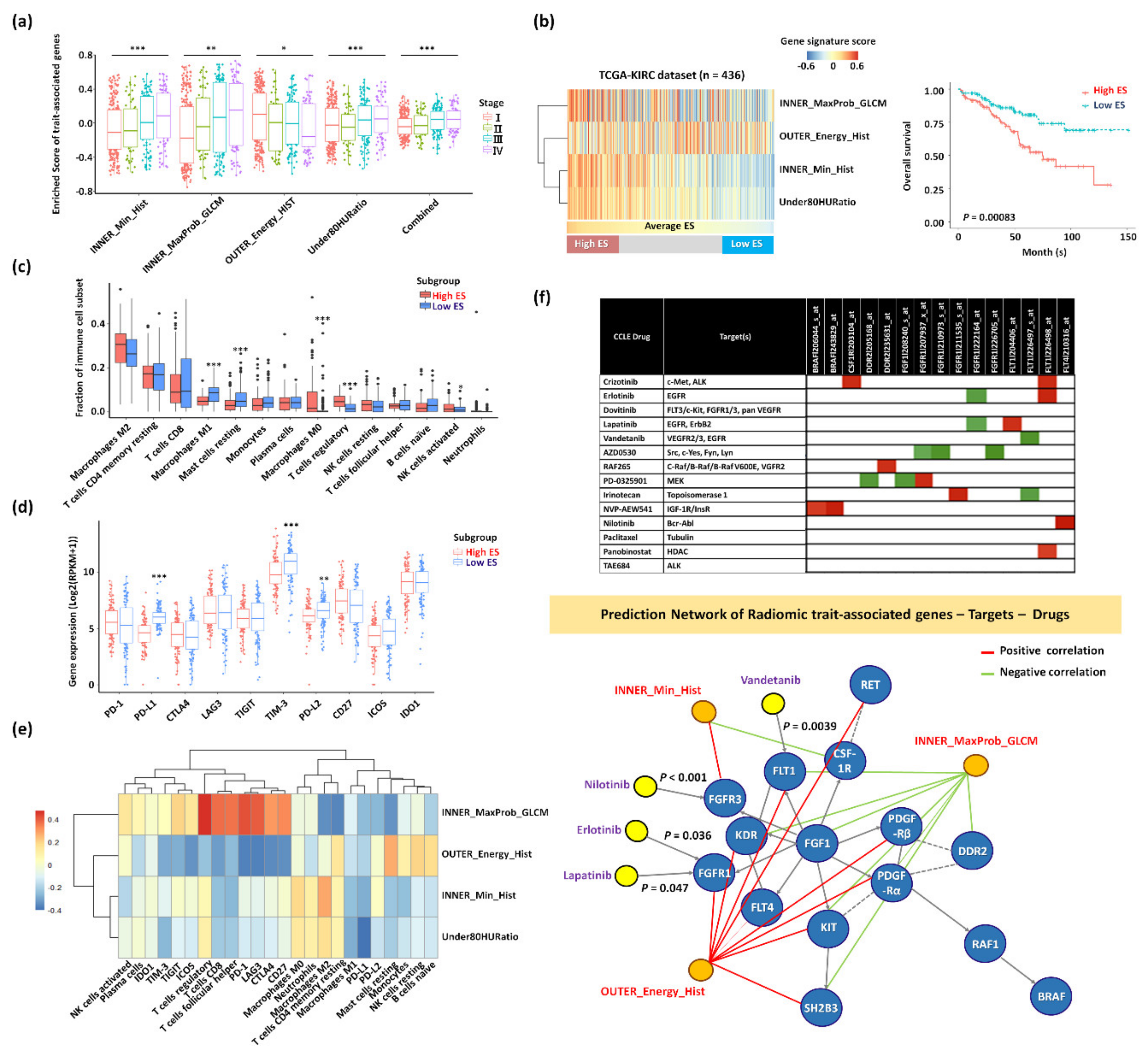
2.3. Functional Enrichment and Prognostic Assessment of Trait-Associated Gene Sets
2.4. Trait-Associated Genes Reflective of the Tumor Immune Microenvironment and the Predicted Drug Sensitivity
3. Discussion
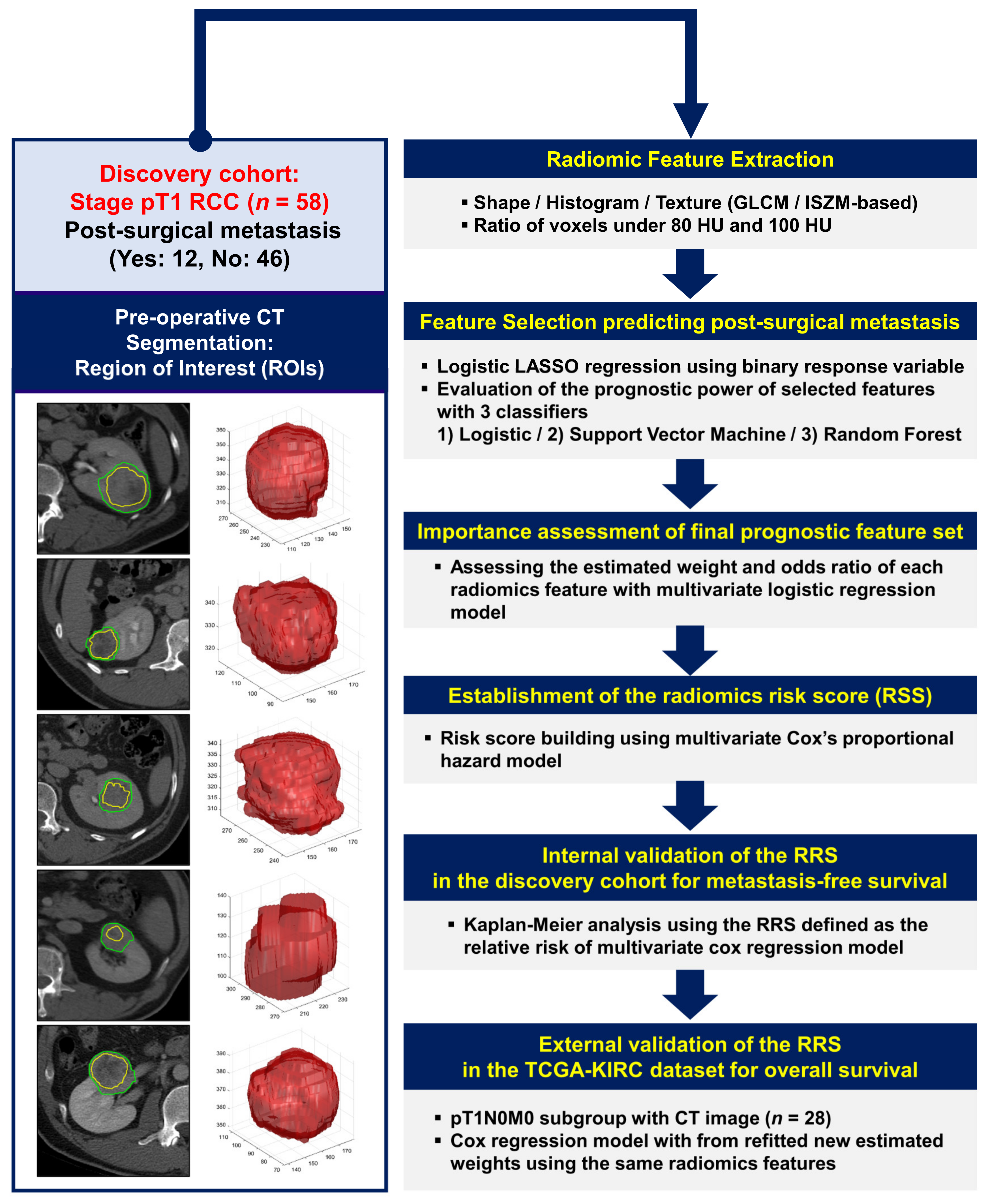
4. Materials and Methods
4.1. Patient Cohorts and Image Data Collection
4.2. CT Imaging and Specification of the ROI
4.3. Image Analysis and Texture Feature Extraction
4.4. Radiomics Model Building and Survival Analysis
4.5. Determination of Radiomics-Feature-Associated Genes
4.6. Functional Analysis of Pathways and Networks, Tumor Immune Microenvironments, and the Predicted Drug Response of Trait-Associated Genes
4.7. Statistical Analysis
4.8. Data Access
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hancock, S.B.; Georgiades, C.S. Kidney Cancer. Cancer J. 2016, 22, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Myszczyszyn, A.; Czarnecka, A.M.; Matak, D.; Szymanski, L.; Lian, F.; Kornakiewicz, A.; Bartnik, E.; Kukwa, W.; Kieda, C.; Szczylik, C. The Role of Hypoxia and Cancer Stem Cells in Renal Cell Carcinoma Pathogenesis. Stem Cell Rev. 2015, 11, 919–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, S.R.; Taneja, K.; Cheng, L. Renal cell carcinoma staging: Pitfalls, challenges, and updates. Histopathology 2019, 74, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.C.; Novick, A.C.; Belldegrun, A.; Blute, M.L.; Chow, G.K.; Derweesh, I.H.; Faraday, M.M.; Kaouk, J.H.; Leveillee, R.J.; Matin, S.F.; et al. Guideline for management of the clinical T1 renal mass. J. Urol. 2009, 182, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.W.; Van Amstel, P.; Martens, R.M.; Kooi, I.E.; Wesseling, P.; De Langen, A.J.; Menke-Van der Houven van Oordt, C.W.; Jansen, B.H.E.; Moll, A.C.; Dorsman, J.C.; et al. Non-invasive tumor genotyping using radiogenomic biomarkers, a systematic review and oncology-wide pathway analysis. Oncotarget 2018, 9, 20134–20155. [Google Scholar] [CrossRef]
- Mazurowski, M.A. Radiogenomics: What it is and why it is important. J. Am. Coll. Radiol. 2015, 12, 862–866. [Google Scholar] [CrossRef]
- Bai, H.X.; Lee, A.M.; Yang, L.; Zhang, P.; Davatzikos, C.; Maris, J.M.; Diskin, S.J. Imaging genomics in cancer research: Limitations and promises. Br. J. Radiol. 2016, 89, 20151030. [Google Scholar] [CrossRef] [Green Version]
- Davatzikos, C.; Rathore, S.; Bakas, S.; Pati, S.; Bergman, M.; Kalarot, R.; Sridharan, P.; Gastounioti, A.; Jahani, N.; Cohen, E.; et al. Cancer imaging phenomics toolkit: Quantitative imaging analytics for precision diagnostics and predictive modeling of clinical outcome. J Med. Imaging (Bellingham) 2018, 5, 011018. [Google Scholar] [CrossRef]
- Acharya, U.R.; Hagiwara, Y.; Sudarshan, V.K.; Chan, W.Y.; Ng, K.H. Towards precision medicine: From quantitative imaging to radiomics. J. Zhejiang Univ. Sci. B 2018, 19, 6–24. [Google Scholar] [CrossRef] [Green Version]
- Sala, E.; Mema, E.; Himoto, Y.; Veeraraghavan, H.; Brenton, J.D.; Snyder, A.; Weigelt, B.; Vargas, H.A. Unravelling tumour heterogeneity using next-generation imaging: Radiomics, radiogenomics, and habitat imaging. Clin. Radiol. 2017, 72, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Alessandrino, F.; Shinagare, A.B.; Bosse, D.; Choueiri, T.K.; Krajewski, K.M. Radiogenomics in renal cell carcinoma. Abdom. Radiol. (N. Y.) 2019, 44, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Alessandrino, F.; Krajewski, K.M.; Shinagare, A.B. Update on Radiogenomics of Clear Cell Renal Cell Carcinoma. Eur. Urol. Focus 2016, 2, 572–573. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, N.; Jonasch, E.; Zapala, M.; Korn, R.L.; Aganovic, L.; Zhao, H.; Tumkur Sitaram, R.; Tibshirani, R.J.; Banerjee, S.; Brooks, J.D.; et al. The Radiogenomic Risk Score: Construction of a Prognostic Quantitative, Noninvasive Image-based Molecular Assay for Renal Cell Carcinoma. Radiology 2015, 277, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Karlo, C.A.; Di Paolo, P.L.; Chaim, J.; Hakimi, A.A.; Ostrovnaya, I.; Russo, P.; Hricak, H.; Motzer, R.; Hsieh, J.J.; Akin, O. Radiogenomics of clear cell renal cell carcinoma: Associations between CT imaging features and mutations. Radiology 2014, 270, 464–471. [Google Scholar] [CrossRef]
- Shinagare, A.B.; Vikram, R.; Jaffe, C.; Akin, O.; Kirby, J.; Huang, E.; Freymann, J.; Sainani, N.I.; Sadow, C.A.; Bathala, T.K.; et al. Radiogenomics of clear cell renal cell carcinoma: Preliminary findings of The Cancer Genome Atlas-Renal Cell Carcinoma (TCGA-RCC) Imaging Research Group. Abdom. Imaging 2015, 40, 1684–1692. [Google Scholar] [CrossRef]
- Manley, B.J.; Reznik, E.; Ghanaat, M.; Kashan, M.; Becerra, M.F.; Casuscelli, J.; Tennenbaum, D.; Redzematovic, A.; Carlo, M.I.; Sato, Y.; et al. Characterizing recurrent and lethal small renal masses in clear cell renal cell carcinoma using recurrent somatic mutations. Urol. Oncol. 2019, 37, 12–17. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, H.J.; Cho, N.H.; Kim, J.; Jang, W.S.; Heo, J.E.; Ham, W.S. Risk Prediction Tool for Aggressive Tumors in Clinical T1 Stage Clear Cell Renal Cell Carcinoma Using Molecular Biomarkers. Comput. Struct. Biotechnol. J. 2019, 17, 371–377. [Google Scholar] [CrossRef]
- Incoronato, M.; Aiello, M.; Infante, T.; Cavaliere, C.; Grimaldi, A.M.; Mirabelli, P.; Monti, S.; Salvatore, M. Radiogenomic Analysis of Oncological Data: A Technical Survey. Int. J. Mol. Sci. 2017, 18, 805. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.; Vendt, B.; Smith, K.; Freymann, J.; Kirby, J.; Koppel, P.; Moore, S.; Phillips, S.; Maffitt, D.; Pringle, M.; et al. The Cancer Imaging Archive (TCIA): Maintaining and operating a public information repository. J. Digit. Imaging 2013, 26, 1045–1057. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, B.; Panayiotou, E.; Burnand, K.; Dizdarevic, S.; Miles, K. Tumour heterogeneity in non-small cell lung carcinoma assessed by CT texture analysis: A potential marker of survival. Eur. Radiol. 2012, 22, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, B.; Skogen, K.; Pressney, I.; Coutroubis, D.; Miles, K. Tumour heterogeneity in oesophageal cancer assessed by CT texture analysis: Preliminary evidence of an association with tumour metabolism, stage, and survival. Clin. Radiol. 2012, 67, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Coroller, T.P.; Grossmann, P.; Hou, Y.; Rios Velazquez, E.; Leijenaar, R.T.; Hermann, G.; Lambin, P.; Haibe-Kains, B.; Mak, R.H.; Aerts, H.J. CT-based radiomic signature predicts distant metastasis in lung adenocarcinoma. Radiother. Oncol. 2015, 114, 345–350. [Google Scholar] [CrossRef]
- Goh, V.; Ganeshan, B.; Nathan, P.; Juttla, J.K.; Vinayan, A.; Miles, K.A. Assessment of response to tyrosine kinase inhibitors in metastatic renal cell cancer: CT texture as a predictive biomarker. Radiology 2011, 261, 165–171. [Google Scholar] [CrossRef]
- Prasanna, P.; Patel, J.; Partovi, S.; Madabhushi, A.; Tiwari, P. Radiomic features from the peritumoral brain parenchyma on treatment-naive multi-parametric MR imaging predict long versus short-term survival in glioblastoma multiforme: Preliminary findings. Eur. Radiol. 2017, 27, 4188–4197. [Google Scholar] [CrossRef]
- Braman, N.M.; Etesami, M.; Prasanna, P.; Dubchuk, C.; Gilmore, H.; Tiwari, P.; Plecha, D.; Madabhushi, A. Intratumoral and peritumoral radiomics for the pretreatment prediction of pathological complete response to neoadjuvant chemotherapy based on breast DCE-MRI. Breast Cancer Res. 2017, 19, 57. [Google Scholar] [CrossRef]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef]
- Pooler, B.D.; Pickhardt, P.J.; O’Connor, S.D.; Bruce, R.J.; Patel, S.R.; Nakada, S.Y. Renal cell carcinoma: Attenuation values on unenhanced CT. AJR Am. J. Roentgenol. 2012, 198, 1115–1120. [Google Scholar] [CrossRef] [Green Version]
- Ganeshan, B.; Goh, V.; Mandeville, H.C.; Ng, Q.S.; Hoskin, P.J.; Miles, K.A. Non-small cell lung cancer: Histopathologic correlates for texture parameters at CT. Radiology 2013, 266, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Hopper, K.D.; Diehl, L.F.; Cole, B.A.; Lynch, J.C.; Meilstrup, J.W.; McCauslin, M.A. The significance of necrotic mediastinal lymph nodes on CT in patients with newly diagnosed Hodgkin disease. AJR Am. J. Roentgenol. 1990, 155, 267–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotker, A.M.; Karlo, C.A.; Zheng, J.; Moskowitz, C.S.; Russo, P.; Hricak, H.; Akin, O. Clear Cell Renal Cell Carcinoma: Associations Between CT Features and Patient Survival. AJR Am. J. Roentgenol. 2016, 206, 1023–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.D.; Kim, C.K.; Park, B.K.; Kim, B. Utility of iodine overlay technique and virtual unenhanced images for the characterization of renal masses by dual-energy CT. AJR Am. J. Roentgenol. 2011, 197, W1076–W1082. [Google Scholar] [CrossRef]
- Ahmed, F.S.; Akin, O.; Shaish, H.; Luk, L.; Guo, X.; Yang, H.; Zabor, E.; Ostrovnaya, I.; Hakimi, A.A.; Zhao, B.; et al. Nonenhancing Component of Clear Cell Renal Cell Carcinoma on Computed Tomography Correlates with Tumor Necrosis and Stage and Serves as a Size-Independent Prognostic Biomarker. J. Comput. Assist. Tomogr. 2019, 43, 628–633. [Google Scholar] [CrossRef]
- Klatte, T.; Rossi, S.H.; Stewart, G.D. Prognostic factors and prognostic models for renal cell carcinoma: A literature review. World J. Urol. 2018, 36, 1943–1952. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Massague, J. Genetic determinants of cancer metastasis. Nat. Rev. Genet. 2007, 8, 341–352. [Google Scholar] [CrossRef]
- O’Mahony, F.C.; Faratian, D.; Varley, J.; Nanda, J.; Theodoulou, M.; Riddick, A.C.; Harrison, D.J.; Stewart, G.D. The use of automated quantitative analysis to evaluate epithelial-to-mesenchymal transition associated proteins in clear cell renal cell carcinoma. PLoS ONE 2012, 7, e31557. [Google Scholar] [CrossRef] [Green Version]
- Guillen-Ahlers, H. Wnt signaling in renal cancer. Curr. Drug Targets 2008, 9, 591–600. [Google Scholar] [CrossRef]
- Liu, Z.; Fu, Q.; Fu, H.; Wang, Z.; Xu, L.; An, H.; Li, Y.; Xu, J. A three-molecule score based on Notch pathway predicts poor prognosis in non-metastasis clear cell renal cell carcinoma. Oncotarget 2016, 7, 68559–68570. [Google Scholar] [CrossRef] [Green Version]
- Ichiyanagi, O.; Naito, S.; Ito, H.; Kabasawa, T.; Narisawa, T.; Kanno, H.; Kurota, Y.; Kurokawa, M.; Fukuhara, H.; Sakurai, T.; et al. Levels of 4EBP1/eIF4E Activation in Renal Cell Carcinoma Could Differentially Predict Its Early and Late Recurrence. Clin. Genitourin. Cancer 2018, 16, e1029–e1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.H.; Serie, D.J.; Parasramka, M.; Cheville, J.C.; Bot, B.M.; Tan, W.; Wang, L.; Joseph, R.W.; Hilton, T.; Leibovich, B.C.; et al. Differential gene expression profiling of matched primary renal cell carcinoma and metastases reveals upregulation of extracellular matrix genes. Ann. Oncol. 2017, 28, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Mikami, S.; Oya, M.; Mizuno, R.; Kosaka, T.; Katsube, K.; Okada, Y. Invasion and metastasis of renal cell carcinoma. Med. Mol. Morphol. 2014, 47, 63–67. [Google Scholar] [CrossRef]
- Chuanyu, S.; Yuqing, Z.; Chong, X.; Guowei, X.; Xiaojun, Z. Periostin promotes migration and invasion of renal cell carcinoma through the integrin/focal adhesion kinase/c-Jun N-terminal kinase pathway. Tumour. Biol. 2017, 39, 1010428317694549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, X.; Li, H.; Ma, X.; Li, X.; Gao, Y.; Ni, D.; Shen, D.; Gu, L.; Wang, B.; Zhang, Y.; et al. High-level S100A6 promotes metastasis and predicts the outcome of T1-T2 stage in clear cell renal cell carcinoma. Cell Biochem. Biophys. 2015, 71, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Keely, P.J.; Westwick, J.K.; Whitehead, I.P.; Der, C.J.; Parise, L.V. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature 1997, 390, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Takagi, K.I.; Suzuki, J.; Imaizumi, A.; Kimura, T.; Mason, R.M.; Kamimura, T.; Zhang, Z. RhoGTPase activation is a key step in renal epithelial mesenchymal transdifferentiation. J. Am. Soc. Nephrol. 2005, 16, 1977–1984. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol. Ther. 2006, 5, 1059–1064. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Gordoa, T.; Garcia-Bermejo, M.L.; Grande, E.; Garrido, P.; Carrato, A.; Molina-Cerrillo, J. Targeting Tyrosine kinases in Renal Cell Carcinoma: “New Bullets against Old Guys”. Int. J. Mol. Sci. 2019, 20, 1901. [Google Scholar] [CrossRef] [Green Version]
- Voon, D.C.; Wang, H.; Koo, J.K.; Chai, J.H.; Hor, Y.T.; Tan, T.Z.; Chu, Y.S.; Mori, S.; Ito, Y. EMT-induced stemness and tumorigenicity are fueled by the EGFR/Ras pathway. PLoS ONE 2013, 8, e70427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Gao, P. Toward Normalization of the Tumor Microenvironment for Cancer Therapy. Integr Cancer Ther. 2019, 18, 1534735419862352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhang, E.; Long, J.; Hu, Z.; Peng, J.; Liu, L.; Tang, F.; Li, L.; Ouyang, Y.; Zeng, Z. Immune infiltration in renal cell carcinoma. Cancer Sci. 2019, 110, 1564–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Hobbs, B.; Amer, A.; Li, X.; Behrens, C.; Canales, J.R.; Cuentas, E.P.; Villalobos, P.; Fried, D.; Chang, J.Y.; et al. Development of an Immune-Pathology Informed Radiomics Model for Non-Small Cell Lung Cancer. Sci. Rep. 2018, 8, 1922. [Google Scholar] [CrossRef] [PubMed]
- Bashir, U.; Siddique, M.M.; McLean, E.; Goh, V.; Cook, G.J. Imaging Heterogeneity in Lung Cancer: Techniques, Applications, and Challenges. AJR Am. J. Roentgenol. 2016, 207, 534–543. [Google Scholar] [CrossRef]
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef] [Green Version]
- Goldsberry, W.N.; Londono, A.; Randall, T.D.; Norian, L.A.; Arend, R.C. A Review of the Role of Wnt in Cancer Immunomodulation. Cancers 2019, 11, 771. [Google Scholar] [CrossRef] [Green Version]
- Baecher-Allan, C.; Anderson, D.E. Immune regulation in tumor-bearing hosts. Curr. Opin. Immunol. 2006, 18, 214–219. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.M.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Klaassen, Z.; Goldberg, H.; Kulkarni, G.S.; Hamilton, R.J.; Fleshner, N.E. Metastatic renal cell carcinoma: Patterns and predictors of metastases-A contemporary population-based series. Urol. Oncol. 2017, 35, 661.e7–661.e14. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Van Griethuysen, J.J.M.; Fedorov, A.; Parmar, C.; Hosny, A.; Aucoin, N.; Narayan, V.; Beets-Tan, R.G.H.; Fillion-Robin, J.C.; Pieper, S.; Aerts, H. Computational Radiomics System to Decode the Radiographic Phenotype. Cancer Res. 2017, 77, e104–e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Bai, Y.; Garcia, E.A.; Li, S. ADASYN: Adaptive Synthetic Sampling Approach for Imbalanced Learning. In Proceedings of the 2008 IEEE International Joint Conference on Neural Networks (IEEE World Congress on Computational Intelligence), Hong Kong, China, 1–8 June 2008; pp. 1322–1328. [Google Scholar]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B (Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Sauerbrei, W.; Royston, P.; Binder, H. Selection of important variables and determination of functional form for continuous predictors in multivariable model building. Stat. Med. 2007, 26, 5512–5528. [Google Scholar] [CrossRef]
- Wu, G.; Dawson, E.; Duong, A.; Haw, R.; Stein, L. ReactomeFIViz: A Cytoscape app for pathway and network-based data analysis. F1000Res 2014, 3, 146. [Google Scholar]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Conley, A.P.; Grimm, E.A.; Roszik, J. A tool for discovering drug sensitivity and gene expression associations in cancer cells. PLoS ONE 2017, 12, e0176763. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.G.; Trevino, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.W.; Cho, H.-h.; Joung, J.-G.; Jeon, H.G.; Jeong, B.C.; Jeon, S.S.; Lee, H.M.; Nam, D.-H.; Park, W.-Y.; Kim, C.K.; et al. Integrative Radiogenomics Approach for Risk Assessment of Post-Operative Metastasis in Pathological T1 Renal Cell Carcinoma: A Pilot Retrospective Cohort Study. Cancers 2020, 12, 866. https://doi.org/10.3390/cancers12040866
Lee HW, Cho H-h, Joung J-G, Jeon HG, Jeong BC, Jeon SS, Lee HM, Nam D-H, Park W-Y, Kim CK, et al. Integrative Radiogenomics Approach for Risk Assessment of Post-Operative Metastasis in Pathological T1 Renal Cell Carcinoma: A Pilot Retrospective Cohort Study. Cancers. 2020; 12(4):866. https://doi.org/10.3390/cancers12040866
Chicago/Turabian StyleLee, Hye Won, Hwan-ho Cho, Je-Gun Joung, Hwang Gyun Jeon, Byong Chang Jeong, Seong Soo Jeon, Hyun Moo Lee, Do-Hyun Nam, Woong-Yang Park, Chan Kyo Kim, and et al. 2020. "Integrative Radiogenomics Approach for Risk Assessment of Post-Operative Metastasis in Pathological T1 Renal Cell Carcinoma: A Pilot Retrospective Cohort Study" Cancers 12, no. 4: 866. https://doi.org/10.3390/cancers12040866