Clinico-Biological Implications of Modified Levels of Cytokines in Chronic Lymphocytic Leukemia: A Possible Therapeutic Role

 , and
, and
Abstract
:1. Introduction
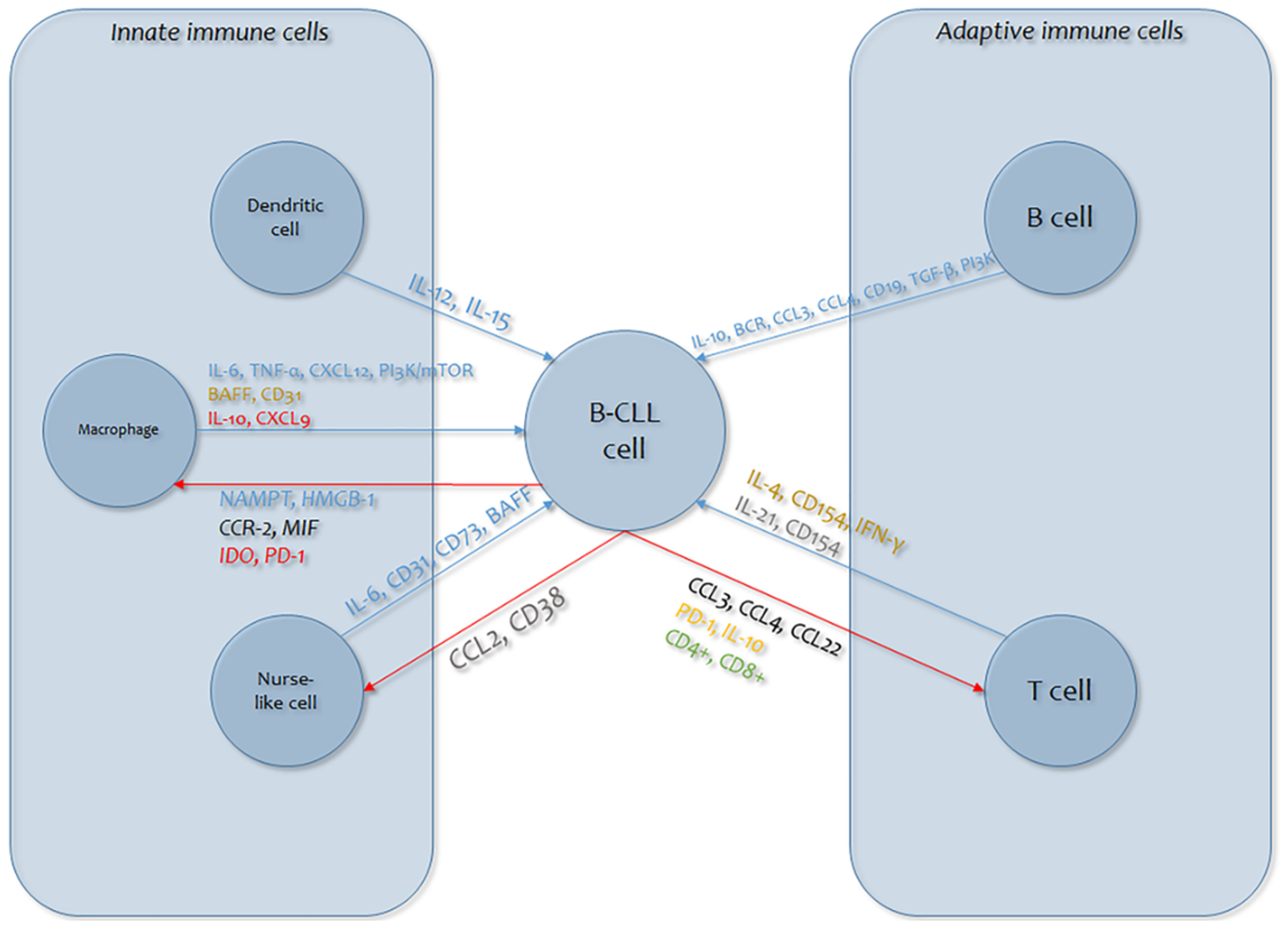
1.1. General Considerations of Immunological Alterations in Chronic Lymphocytic Leukemia
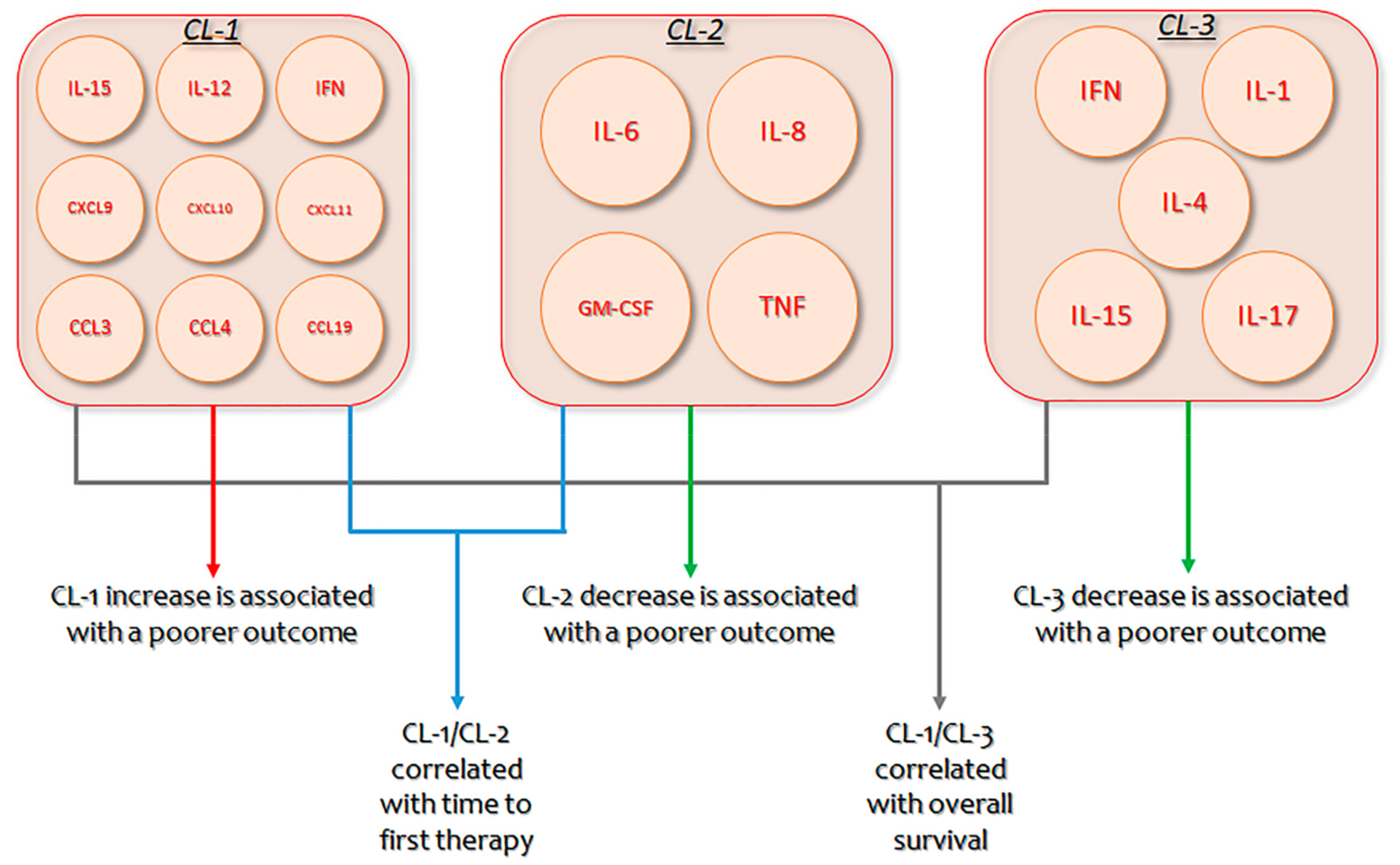
1.2. Effects of Cytokines on the Onset, Progression, and Complications of Chronic Lymphocytic Leukemia
2. Future Perspectives
3. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [Green Version]
- Haseeb, M.; Anwar, M.A.; Choi, S. Molecular Interactions Between Innate and Adaptive Immune Cells in Chronic Lymphocytic Leukemia and Their Therapeutic Implications. Front. Immunol. 2018, 9, e2720. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.P.; Contesti, J.; Huergo-Zapico, L.; Lopez-Soto, A.; Fernandez-Guizan, A.; Acebes-Huerta, A.; Gonzalez-Huerta, A.J.; Gonzalez, E.; Fernandez-Alvarez, C.; Gonzalez, S. Prognostic significance of CD8 and CD4 T cells in chronic lymphocytic leukemia. Leuk. Lymphoma 2010, 51, 1829–1836. [Google Scholar] [CrossRef]
- Palmer, S.; Hanson, C.A.; Zent, C.S.; Porrata, L.F.; LaPlant, B.; Geyer, S.M.; Markovic, S.N.; Call, T.G.; Bowen, D.A.; Diane, F.; et al. Prognostic importance of T and NK-cells in a consecutive series of newly diagnosed patients with chronic lymphocytic leukaemia. Br. J. Haematol. 2008, 141, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Huergo-Zapico, L.; Acebes-Huerta, A.; Gonzalez-Rodriguez, A.P.; Contesti, J.; Gonzalez-Garcıa, E.; Payer, A.R.; Villa-Alvarez, M.; Fernandez-Guizan, A.; Lopez-Soto, A.; Gonzalez, S. Expansion of NK cells and reduction of NKG2D expression in chronic lymphocytic leukemia. Correlation with progressive disease. PLoS ONE 2014, 9, e108326. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; DiPaolo, R.A.; Andersson, D.M.; Zhao, D.M.; Stephens, G.L.; Thornton, A.M. The lifestyle of naturally occurring CD4 + CD25 + Foxp3 + regulatory T cells. Immunol. Rev. 2006, 212, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.; Kochanek, M.; Darabi, K.; Popov, A.; Jensen, M.; Endl, E.; Knolle, P.A.; Thomas, R.K.; von Bergwelt-Baildo, M.; Debey, S.; et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood 2005, 106, 2018–2025. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, V.; Perry, C.; Polliack, A. Expansion of regulatory T cells in B chronic lymphocytic leukemia: Enhanced ‘brakes’ on host immunity. Leuk. Lymphoma 2009, 50, 687–688. [Google Scholar] [CrossRef]
- Jak, M.; Mous, R.; Remmerswaal, E.B.; Spijker, R.; Jaspers, A.; Yagüe, A.; Eldering, E.; Van Lier, R.A.; Van Oers, M.H. Enhanced formation and survival of CD4+ CD25hi Foxp3+ T-cells in chronic lymphocytic leukemia. Leuk. Lymphoma 2009, 50, 788–801. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Hoffbrand, A.V.; Panayiotidis, P.; Reittie, J.; Ganeshaguru, K. Autocrine and paracrine growth loops in chronic lymphocytic leukaemia. Semin. Hematol. 1993, 30, 306–317. [Google Scholar] [PubMed]
- Pistoia, V. Production of cytokines by human B cells in health and disease. Immunol. Today 1997, 18, 343–350. [Google Scholar] [CrossRef]
- Mainou-Fowler, T.; Copplestone, J.A.; Prentice, A.G. Effect of interleukins on the proliferation and survival of B cell chronic lymphocytic leukaemia cells. J. Clin Pathol. 1995, 48, 482–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainou-Fowler, T.; Prentice, A.G. Modulation of apoptosis by cytokines in B-cell chronic lymphocytic leukaemia. Leuk. Lymphoma 1996, 21, 369–378. [Google Scholar] [CrossRef]
- Trentin, L.; Zambello, R.; Agostini, C.; Enthammer, C.; Cerutti, A.; Adami, F. Expression and regulation of tumor factor necrosis, interleukin-2, and hematopoietic growth factor receptors in B-cell chronic lymphocytic leukaemia. Blood 1994, 84, 4249–4256. [Google Scholar] [CrossRef] [Green Version]
- Trentin, L.; Zambello, R.; Facco, M. Interleukin-15: A novel cytokine with regulatory properties on normal and neoplastic B lymphocytes. Leuk. Lymphoma 1997, 27, 35–42. [Google Scholar] [CrossRef]
- van Kooten, C.; Rensink, I.; Aarden, L. Interleukin-4 inhibits both paracrine and autocrine tumor necrosis factor-–induced proliferation of B chronic lymphocytic leukaemia cells. Blood 1992, 80, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, E.; Llamas, P.; Vargas, J.A.; Durantez, A. Citocinas y leucemia linfatica cronica B. Sangre 1997, 42, 39–46. [Google Scholar]
- Castejon, R.; Vargas, J.A.; Romero, Y.; Briz, M.; Munoz, R.M.; Durantez, A. Modulation of apoptosis by cytokines in B-cell chronic lymphocytic leukaemia. Clin. Cytom. 1999, 38, 224–230. [Google Scholar] [CrossRef]
- Ghamlouch, H.; Ouled-Haddou, H.; Damaj, G.; Royer, B.; Gubler, B.; Marolleau, P. A Combination of Cytokines Rescues Highly Purified Leukemic CLL B-Cells from Spontaneous Apoptosis In Vitro. PLoS ONE 2013, 8, e60370. [Google Scholar] [CrossRef]
- Francis, S.; Karanth, M.; Pratt, G.; Starczynski, J.; Hooper, L.; Fegan, C.; Pepper, C.; Valcarcel, D.; Milligan, D.W.; Delgado, J. The effect of immunoglobulin VH gene mutation status and other prognostic factors on the incidence of major infections in patients with chronic lymphocytic leukemia. Cancer 2006, 107, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Morrison, V.A. Infectious complications of chronic lymphocytic leukemia: Pathogenesis, spectrum of infection, preventive approaches. Best Pract. Res. Clin. Haematol. 2010, 23, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Musolino, C.; Di Cesare, E.; Alonci, A.; Allegra, A.; Orlando, A.; Grosso, P.; Squadrito, G. Serum levels of CD8 antigen and soluble interleukin 2 receptors in patients with B cell chronic lymphocytic leukemia. Acta. Haematol. 1991, 85, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.W.; Tsuda, H.; Takatsuki, K. Interleukin-2 prevents programmed cell death in chronic lymphocytic leukaemia cells. Int. J. Hematol. 1993, 58, 83–92. [Google Scholar]
- Okada, H.; Banchereau, J.; Lotze, M.T. Interleukin-4. In The Cytokine Handbook, Vol. I; Thompson, A.W., Lotze, M.T., Eds.; Academic Press: London, UK, 2003; pp. 227–262. [Google Scholar]
- Fink, S.R.; Paternoster, S.F.; Smoley, S.A.; Flynn, H.C.; Geyer, S.M.; Shanafelt, T.D.; Lee, Y.K.; Jelinek, D.F.; Kay, N.E.; Dewald, G.W. Fluorescent-labeled DNA probes applied to novel biological aspects of B-cell chronic lymphocytic leukemia. Leuk. Res. 2005, 29, 253–262. [Google Scholar] [CrossRef]
- Kay, N.E.; Bone, N.D.; Tschumper, R.C.; Howell, K.H.; Geyer, S.M.; Dewald, G.W.; Hanson, C.A.; Jelinek, D.F. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia 2002, 16, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Kay, N.E.; Bone, N.D.; Lee, Y.K.; Jelinek, D.F.; Leland, P.; Battle, T.E.; Frank, D.A.; Puri, R.K. A recombinant IL-4 Pseudomonas exotoxin inhibits protein synthesis and overcomes apoptosis resistance in human CLL B cells. Leuk. Res. 2005, 29, 1009–1018. [Google Scholar] [CrossRef]
- Dancescu, M.; Rubio-Trujillo, M.; Biron, G.; Bron, D.; Delespesse, G.; Sarfati, M. Interleukin 4 protects chronic lymphocytic leukemic B cells from death by apoptosis and upregulates Bcl-2 expression. J. Exp. Med. 1992, 176, 1319–1326. [Google Scholar] [CrossRef]
- Douglas, R.S.; Capocasale, R.J.; Lamb, R.J.; Nowell, P.C.; Moore, J.S. Chronic lymphocytic leukemia B cells are resistant to the apoptotic effects of Transforming Growth Factor-β. Blood 1997, 89, 941–947. [Google Scholar] [CrossRef]
- Coscia, M.; Pantaleoni, F.; Riganti, C.; Vitale, C.; Rigoni, M.; Peola, S.; Castella, B.; Foglietta, M.; Griggio, V.; Drandi, D.; et al. IGHV unmutated cells are more prone to spontaneous apoptosis and subject to environmental prosurvival signals than mutated CLL B cells. Leukemia 2011, 25, 828–837. [Google Scholar] [CrossRef]
- Steele, A.J.; Prentice, A.G.; Cwynarski, K.; Hoffbrand, A.V.; Hart, S.M.; Lowdell, M.W.; Samuel, E.R.; Wickremasinghe, R.G. The JAK3-selective inhibitor PF-956980 reverses the resistance to cytotoxic agents induced by interleukin-4 treatment of chronic lymphocytic leukemia cells: Potential for reversal of cytoprotection by the microenvironment. Blood 2010, 116, 4569–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panayiotidis, P.; Ganeshaguru, K.; Jabbar, S.A.B.; Hoffbrand, A.V. Interleukin 4 inhibits apoptotic cell death and loss of bcl-2 protein in B-cell chronic lymphocytic leukaemia cells in vitro. Br. J. Haematol. 1993, 85, 39–45. [Google Scholar] [CrossRef]
- Frankfurt, O.S.; Byrnes, J.J.; Villa, L. Protection from apoptotic cell death by interleukin-4 is increased in previously treated chronic lymphocytic leukemia patients. Leuk. Res. 1997, 21, 9–16. [Google Scholar] [CrossRef]
- Weinkove, R.; Brooks, C.R.; Carter, J.M.; Hermans, I.F.; Ronchese, F. Functional invariant natural killer T-cell and CD1d axis in chronic lymphocytic leukemia: Implications for immunotherapy. Haematologica 2013, 98, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-Q.; Jia, L.; Yu-Ting Li, Y.-T.; Farren, T.; Agrawal, S.G.; Feng-Ting Liu, F.-T. Increased autocrine interleukin-6 production is significantly associated with worse clinical out come in patients with chronic lymphocytic leukemia. J. Cell Physiol. 2019, 234, 13994–14006. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, T. Interleukin-6: From basic science to medicine—40 years in immunology. Annu. Rev. Immunol. 2005, 23, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Ebnet, K.; Vestweber, D. Molecular mechanisms that control eukocyte extravasation: The selectins and the chemokines. Histochem. Cell Biol. 1999, 112, 1–23. [Google Scholar] [CrossRef]
- Aderka, D.; Maor, Y.; Novick, D.; Engelmann, H.; Kahn, Y.; Levo, Y.; Wallach, D.; Revel, M. Interleukin-6 inhibits the proliferation of B-chronic lymphocytic leukemia cells that is induced by tumor necrosis factor-α or-β. Blood 1993, 81, 2076–2084. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Shi, Y.; McCaw, L.; Li, Y.J.; Zhu, F.; Gorczynski, R.; Duncan, G.S.; Yang, B.; Ben-David, Y.; Spaner, D.E. Microenvironmental interleukin-6 suppresses toll-like receptor signaling in human leukemia cells through miR-17/19A. Blood 2015, 126, 766–778. [Google Scholar] [CrossRef] [Green Version]
- Di Celle, P.F.; Mariani, S.; Riera, L.; Stacchini, A.; Reato, G.; Foa, R. Interleukin-8 induces the accumulation of B-cell chronic lymphocytic leukemia cells by prolonging survival in an autocrine fashion. Blood 1996, 87, 4382–4389. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.; Korsmeyer, S.J. Molecular thanaptosis: A discourse on the bcl-2 family and cell death. Blood 1996, 88, 386–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.E.; Hori, Y.; Fan, T.P. Interleukin-8 stimulates angiogenesis in rats. Inflammation 1993, 17, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Bellail, A.C.; van Meir, E.G. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-Oncology 2005, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Isaacson, P.G. Classification of lymphoid neoplasms: The microscope as a tool for disease discovery. Blood 2008, 112, 4384–4399. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Lai, R.; Lin, Q.; Lau, E.; Thomazy, D.M.; Calame, D.; Ford, R.J.; Kwak, L.W.; Kirken, R.A.; Amin, H.M. Autocrine release of interleukin-9 promotes Jak3-dependent survival of ALK anaplastic large-cell lymphoma cells. Blood 2006, 108, 2407–2415. [Google Scholar] [CrossRef] [Green Version]
- Renauld, J.C.; Vink, A.; Louahed, J.; Van Snick, J. Interleukin-9 is a major anti-apoptotic factor for thymic lymphomas. Blood 1995, 85, e13001305. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Feng, L.; Qu, H.; Lu, K.; Li, P.; Lv, X.; Wang, X. Overexpression of IL-9 induced by STAT3 phosphorylation is mediated by miR-155 and miR-21 in chronic lymphocytic leukemia. Oncol. Rep. 2018, 39, 3064–3072. [Google Scholar] [CrossRef]
- Steel, J.C.; Waldmann, T.A.; Morris, J.C. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol. Sci. 2012, 33, 35–41. [Google Scholar] [CrossRef] [Green Version]
- de Totero, D.; Meazza, R.; Capaia, M.; Fabbi, M.; Azzarone, B.; Balleari, E.; Gobbi, M.; Cutrona, G.; Ferrarini, M.; Ferrini, S. The opposite effects of IL-15 and IL-21 on CLL B cells correlate with differential activation of the JAK/ STAT and ERK1/2 pathways. Blood 2008, 111, 517–524. [Google Scholar] [CrossRef]
- Laprevotte, E.; Voisin, G.; Ysebaert, L.; Klein, C.; Daugrois, C.; Laurent, G.; Fournie, J.J.; Quillet-Mary, A. Recombinant Human IL-15 Trans-Presentation by B Leukemic Cells from Chronic Lymphocytic Leukemia Induces Autologous NK Cell Proliferation Leading to Improved Anti-CD20 Immunotherapy. J. Immunol. 2013, 191, 3634–3640. [Google Scholar] [CrossRef] [Green Version]
- Moga, E.; Cantó, E.; Vidal, S.; Juarez, C.; Sierra, J.; Briones, J. Interleukin-15 enhances rituximab-dependent cytotoxicity against chronic lymphocytic leukemia cells and overcomes transforming growth factor beta-mediated immunosuppression. Exp. Hematol. 2011, 39, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; McCaw, L.; Spaner, D.E.; Gorczynski, R.M. Targeting the IL-17/IL-6 axis can alter growth of Chronic Lymphocytic Leukemia in vivo/in vitro. Leuk. Res. 2018, 66, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Ghalamfarsa, G.; Memarian, A.; Asgarian-Omran, H.; Razavi, S.M.; Sarrafnejad, A.; Shokri, F. Downregulation of IL-17-producing T cells is associated with regulatory T cell expansion and disease progression in chronic lymphocytic leukemia. Tumor Biol. 2013, 34, 929–940. [Google Scholar] [CrossRef] [PubMed]
- de Totero, D.; Meazza, R.; Zupo, S.; Cutrona, G.; Matis, S.; Colombo, M.; Balleari, E.; Pierri, I.; Fabbi, M.; Capaia, M.; et al. Interleukin-21 receptor (IL-21R) is up-regulated by CD40 triggering and mediates proapoptotic signals in chronic lymphocytic leukemia B cells. Blood 2006, 107, 3708–3715. [Google Scholar] [CrossRef] [Green Version]
- De Cecco, L.; Capaia, M.; Zupo, S.; Cutrona, G.; Matis, S.; Brizzolara, A.; Orengo, A.M.; Croce, M.; Marchesi, E.; Ferrarini, M. Interleukin 21 Controls mRNA and MicroRNA Expression in CD40 Activated Chronic Lymphocytic Leukemia Cells. PLoS ONE 2015, 10, e0134706. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.D.; Skrumsager, B.K.; Cebon, J.; Nicholaou, T.; Barlow, J.W.; Moller, N.P.; Skak, K.; Lundsgaard, D.; Frederiksen, K.S.; Thygesen, P.; et al. An open-label, two-arm, phase I trial of recombinant human interleukin-21 in patients with metastatic melanoma. Clin. Cancer Res. 2007, 13, 3630–3636. [Google Scholar] [CrossRef] [Green Version]
- Jahrsdörfer, B.; Blackwell, S.E.; Wooldridge, J.E.; Huang, J.; Andreski, M.W.; Jacobus, L.S.; Taylor, C.M.; Weiner, G.J. B-chronic lymphocytic leukemia cells and other B cells can produce granzyme B and gain cytotoxic potential after interleukin-21-based activation. Blood 2006, 108, 2712–2719. [Google Scholar] [CrossRef] [Green Version]
- Jahrsdörfer, B.; Wooldridge, J.E.; Blackwell, S.E.; Taylor, C.M.; Griffith, T.S.; Link, B.K.; Weiner, G.J. Immunostimulatory oligodeoxynucleotides induce apoptosis of B cell chronic lymphocytic leukemia cells. J. Leukoc. Biol. 2005, 77, 378–387. [Google Scholar] [CrossRef]
- Browning, R.L.; Mo, X.; Muthusamy, N.; Byrd, J.C. CpG oligodeoxynucleotide CpG-685 upregulates functional interleukin-21 receptor on chronic lymphocytic leukemia B cells through an NF-κB mediated pathway. Oncotarget 2015, 6, e15931. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Moseman, E.A.; Farra, M.A.; Bachanova, V.; Weisdorf, D.J.; Blazar, B.R.; Chen, W. Toll-like receptor 9 signaling by CpG-B oligodeoxynucleotides induces an apop-totic pathway in human chronic lymphocytic leukemia B cells. Blood 2010, 115, 5041–5052. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, A.P.; Joller, N.; Michaud, M.; Liu, S.M.; Kuchroo, V.K.; Grusby, M.J. IL-21 promotes CD8+ CTL activity via the transcription factor T-bet. J. Immunol. 2013, 190, 3977–3984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bard, J.D.; Gelebart, P.; Anand, M.; Amin, H.M.; Lai, R. Aberrant expression of IL-22 receptor 1 and autocrine IL-22 stimulation contribute to tumorigenicity in ALK+ anaplastic large cell lymphoma. Leukemia 2008, 22, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Dumoutier, L.; de Meester, C.; Tavernier, J.; Renauld, J.C. New activation modus of STAT3: A tyrosine-less region of the interleukin-22 receptor recruits STAT3 by interacting with its coiled-coil domain. J. Biol. Chem. 2009, 284, 26377–26384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyng, H.; Landsverk, K.S.; Kristiansen, E.; DeAngelis, P.M.; Ree, A.H.; Myklebost, O. Response of malignant B lymphocytes to ionizing radiation: Gene expression and genotype. Int. J. Cancer 2005, 115, 935–942. [Google Scholar] [CrossRef]
- Gelebart, P.; Zak, Z.; Dien-Bard, J.; Anand, M.; Lai, R. Interleukin 22 signaling promotes cell growth in mantle cell lymphoma. Transl. Oncol. 2011, 4, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Cutrona, G.; Tripodo, C.; Matis, S.; Recchia, A.G.; Massucco, C.; Fabbi, M.; Colombo, M.; Emionite, L.; Sangaletti, S.; Gulino, A.; et al. Microenvironmental regulation of the IL-23R/IL-23 axis overrides chronic lymphocytic leukemia indolence. Sci. Transl. Med. 2018, 10, eaal1571. [Google Scholar] [CrossRef] [Green Version]
- Foa, R.; Massaia, M.; Cardona, S.; Tos, A.G.; Bianchi, A.; Attisano, C.; Guarini, A.; di Celle, P.F.; Fierro, M.T. Production of tumor necrosis factor-alpha by B-cell chronic lymphocytic leukaemia cells: A possible regulatory role of TNF in the progression of the disease. Blood 1990, 76, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Jabbar, S.A.B.; Hoffbrand, V.; Wickremashinghe, G. Regulation of transcription factors NFB and AP-1 following tumor necrosis factor-treatment of cells from B cell chronic lymphocytic leukaemia patients. Br. J. Haematol. 1994, 86, 496–504. [Google Scholar] [CrossRef]
- Choudhry, H.; Helmi, N.; Abdulaal, W.H.; Zeyadi, M.; Zamzami, M.A.; Wu, W.; Mahmoud, M.M.; Warsi, M.K.; Rasool, M.; Jamal, M.S. Prospects of IL-2 in Cancer Immunotherapy. Biomed. Res. Int. 2018, 2018, e9056173. [Google Scholar] [CrossRef] [Green Version]
- Mortara, L.; Balza, E.; Bruno, A.; Poggi, A.; Orecchia, P.; Carnemoll, B. Anti-cancer Therapies Employing IL-2 Cytokine Tumor Targeting: Contribution of Innate, Adaptive and Immunosuppressive Cells in the Anti-tumor Efficacy. Front. Immunol. 2018, 9, e2905. [Google Scholar] [CrossRef] [Green Version]
- Villa-Alvarez, M.; Lorenzo-Herrero, S.; Gonzalez-Rodriguez, A.P.; Lopez-Soto, A.; Payer, A.R.; Gonzalez-Garcia, E.; Huergo-Zapico, L.; Gonzalez, S. Ig-like transcript 2 (ILT2) suppresses T cell function in chronic lymphocytic leukemia. Oncoimmunology 2017, 10, e1353856. [Google Scholar] [CrossRef] [PubMed]
- Gallego, A.; Vargas, J.A.; Castejon, R.; Citores, M.J.; Romero, Y.; Millan, I.; Durantez, A. Production of intracellular IL-2, TNF-α, and IFN-γ by T cells in B-CLL. Cytom. Part. B Clin. Cytom. 2003, 56, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, W.; Harris, D.P.; Sprague, F.; Mousseau, B.; Makris, M.; Kusser, K.; Honjo, T.; Mohrs, K.; Mohrs, M.; Randall, T.; et al. Cytokine-producing effector B cells regulate type 2 immunity to H. polygyrus. Immunity 2009, 30, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Rothstein, T.L. IL-4 upregulates Igαand Igβprotein, resulting in augmented IgM maturation and B cell receptor–triggered B cell activation. J. Immunol. 2013, 191, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messner, B.; Stütz, A.M.; Albrecht, B.; Peiritsch, S.; Woisetschläger, M. Cooperation of binding sites for STAT6 and NF kappa B/rel in the IL-4-induced up-regulation of the human IgE germline promoter. J. Immunol. 1997, 159, 3330–3337. [Google Scholar] [PubMed]
- Shen, C.H.; Stavnezer, J. Interaction of stat6 and NF-kappaB: Direct association and synergistic activation of interleukin-4-induced transcription. Mol. Cell. Biol. 1998, 18, 3395–3404. [Google Scholar] [CrossRef] [Green Version]
- Zamorano, J.; Mora, A.L.; Boothby, M.; Keegan, A.D. NF-kappa B activation plays an important role in the IL-4-induced protection from apoptosis. Int. Immunol. 2001, 13, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Thieu, V.T.; Nguyen, E.T.; McCarthy, B.P.; Bruns, H.A.; Kapur, R.; Chang, C.H.; Kaplan, M.H. IL-4-stimulated NF-kappa B activity is required for Stat6 DNA binding. J. Leukoc. Biol. 2007, 82, 370–379. [Google Scholar] [CrossRef]
- Ruiz-Lafuente, N.; Alcaraz-García, M.-J.; Sebastián-Ruiz, S.; García-Serna, A.-M.; Gómez Espuch, J.; Moraleda, J.-M.; Minguela, A.; García-Alonso, A.M.; Parrado, A. IL-4 Up Regulates MiR-21 and the MiRNAs Hosted in the CLCN5 Gene in Chronic Lymphocytic Leukemia. PLoS ONE 2015, 10, e0124936. [Google Scholar] [CrossRef] [Green Version]
- Lezina, L.; Spriggs, R.V.; Beck, D.; Jones, C.; Dudek, K.M.; Bzura, A.; Jones, G.D.D.; Packham, G.; Willis, A.E.; Wagner, S.D. CD40L/IL-4–stimulated CLL demonstrates variation in translational regulation of DNA damage response genes including ATM. Blood Adv. 2018, 15, e1869. [Google Scholar] [CrossRef]
- Fluckiger, A.C.; Rossi, J.F.; Bussel, A.; Bryon, P.; Banchereau, J.; Defrance, T. Responsiveness of chronic lymphocytic leukemia B cells activated via surface Igs or CD40 to B-cell tropic factors. Blood 1992, 80, 3173–3181. [Google Scholar] [CrossRef] [Green Version]
- Willimott, S.; Beck, D.; Ahearne, M.J.; Adams, V.C.; Wagner, S.D. Cap-translation inhibitor, 4EGI-1, restores sensitivity to ABT-737 apoptosis through cap dependent and -independent mechanisms in chronic lymphocytic leukemia. Clin. Cancer Res. 2013, 19, 3212–3223. [Google Scholar] [CrossRef] [Green Version]
- Bojarska-Junak, A.; Waldowska, M.; Woś, J.; Chocholska, S.; Hus, I.; Tomczak, W.; Dzik, M.; Hus, M.; Rolińsk, J. Intracellular IL-4 and IFN-γ expression in iNKT cells from patients with chronic lymphocytic leukemia. Oncol. Lett. 2018, 15, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Burger, J.A.; Blum, K.A.; Coleman, M.; Wierda, W.G.; Jones, J.A.; Zhao, W.; Heerema, N.A.; et al. Three-year follow-up of treatment naıve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015, 125, 2497–2506. [Google Scholar] [CrossRef]
- Mockridge, C.I.; Potter, K.N.; Wheatley, I.; Neville, L.A.; Packham, G.; Stevenson, F.K. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood 2007, 109, 4424–4431. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Hernandez, M.A.; Blunt, M.D.; Dobson, R.; Yeomans, A.; Thirdborough, S.; Larrayoz, M.; Smith, L.D.; Linley, A.; Strefford, J.C.; Davies, A.; et al. IL-4 enhances expression and function of surface IgM in CLL cells. Blood 2016, 127, 3015–3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 trans-signaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef] [Green Version]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Jain, P.; Veletic, I.; Ferrajoli, A.; Burger, J.; Thompson, P.; Jain, N.; et al. Activation of the B-cell receptor successively activates NFkappaB and STAT3 in chronic lymphocytic leukemia cells. Int. J. Cancer 2017, 141, 2076–2081. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.T.; Jia, L.; Wang, P.; Farren, T.; Li, H.; Hao, X.; Agrawal, S.G. CD126 and targeted therapy with tocilizumab in chronic lymphocytic leukemia. Clin. Cancer Res. 2016, 22, 2462–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Dozmorov, I.; Li, W.; Yancopoulos, S.; Sison, C.; Centola, M.; Jain, P.; Allen, S.L.; Kolitz, J.E.; Rai, K.R.; et al. Identification of outcome correlated cytokine clusters in chronic lymphocytic leukemia. Blood 2011, 19, 5201–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fayad, L.; Keating, M.J.; Reuben, J.M.; O’Brien, S.; Lee, B.N.; Lerner, S.; Kurzrock, R. Interleukin-6 and interleukin-10 levels in chronic lymphocytic leukemia: Correlation with phenotypic characteristics and outcome. Blood 2001, 97, 256–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzio, M.; Scielzo, C.; Bertilaccio, M.T.; Frenquelli, M.; Ghia, P.; Caligaris-Cappio, F. Expression and function of toll like receptors in chronic lymphocytic leukaemia cells. Br. J. Haematol. 2009, 144, 507–516. [Google Scholar] [CrossRef]
- Spaner, D.E.; Masellis, A. Toll-like receptor agonists in the treatment of chronic lymphocytic leukemia. Leukemia 2007, 21, 53–60. [Google Scholar] [CrossRef]
- Spaner, D.E.; Shi, Y.; White, D.; Mena, J.; Hammond, C.; Tomic, J.; He, L.; Tomai, M.A.; Miller, R.L.; Booth, J.; et al. Immunomodulatory effects of toll-like receptor-7 activation on chronic lymphocytic leukemia cells. Leukemia 2006, 20, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; White, D.; He, L.; Miller, R.L.; Spaner, D.E. Toll-like receptor-7 tolerizes malignant B cells and enhances killing by cytotoxic agents. Cancer Res. 2007, 67, 1823–1831. [Google Scholar] [CrossRef] [Green Version]
- Spaner, D.E.; Miller, R.L.; Mena, J.; Grossman, L.; Sorrenti, V.; Shi, Y. Regression of lymphomatous skin deposits in a chronic lymphocytic leukemia patient treated with the toll-like receptor-7/8 agonist, imiquimod. Leuk. Lymphoma 2005, 46, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M.; Loetscher, P.; Moser, B. Interleukin-8 and the chemokine family. Int. J. Immunopharmacol. 1995, 17, 103–108. [Google Scholar] [CrossRef]
- di Celle, P.F.; Carbone, A.; Marchis, D.; Zhou, D.; Sozzani, S.; Zupo, S.; Pini, M.; Mantovani, A.; Foa, R. Cytokine gene expression in B-cell chronic lymphocytic leukemia: Evidence of constitutive interleukin-8 (IL-8) mRNA expression and secretion of biologically active IL-8 protein. Blood 1994, 84, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierda, W.G.; Johnson, M.M.; Do, K.A.; Manshouri, T.; Dey, A.; O’Brien, S.; Giles, F.J.; Kantarjian, H.; Thomas, D.; Faderl, S.; et al. Plasma interleukin 8 level predicts for survival in chronic lymphocytic leukaemia. Br. J. Haematol. 2003, 120, 452–456. [Google Scholar] [CrossRef]
- Podaza, E.; Sabbione, F.; Risnik, D.; Borge, M.; Almejún, M.B.; Colado, A.; Fernández-Grecco, H.; Cabrejo, M.; Bezares, R.F.; Trevani, A.; et al. Neutrophils from chronic lymphocytic leukemia patients exhibit an increased capacity to release extracellular traps (NETs). Cancer Immunol. Immunother. 2017, 66, 77–89. [Google Scholar] [CrossRef]
- Buechele, C.; Baessler, T.; Wirths, S.; Schmohl, J.U.; Schmiedel, B.J.; Salih, H.R. Glucocorticoid-induced TNFR-related protein (GITR) ligand modulates cytokine release and NK cell reactivity in chronic lymphocytic leukemia (CLL). Leukemia 2012, 26, 991–1000. [Google Scholar] [CrossRef]
- Binsky, I.; Haran, M.; Starlets, D.; Gore, Y.; Lantner, F.; Harpaz, N.; Leng, L.; Goldenberg, D.M.; Shvidel, L.; Berrebi, A.; et al. IL-8 secreted in a macrophage migration-inhibitory factor- and CD74-dependent manner regulates B cell chronic lymphocytic leukemia survival. PNAS 2007, 104, 13408–13413. [Google Scholar] [CrossRef] [Green Version]
- Molica, S.; Vitelli, G.; Levato, D.; Levato, L.; Dattilo, A.; Gandolfo, G.M. Clinico-biological implications of increased serum levels of interleukin-8 in B-cell chronic lymphocytic leukemia. Haematologica 1999, 84, 208–211. [Google Scholar]
- Ghobrial, I.M.; Bone, N.D.; Stenson, M.J.; Novak, A.; Hedin, K.E.; Kay, N.E.; Ansell, S.M. Expression of the chemokine receptors CXCR4 and CCR7 and disease progression in B-cell chronic lymphocytic leukemia/small lymphocytic lymphoma. Mayo Clinic Proc. 2004, 79, 318–325. [Google Scholar] [CrossRef]
- Levidou, G.; Sachanas, S.; Pangalis, G.A.; Kalpadakis, C.; Yiakoumis, X.; Moschogiannis, M.; Sepsa, A.; Lakiotaki, E.; Milionis, V.; Kyrtsonis, M.C.; et al. Immunohistochemical analysis of IL-6, IL-8/CXCR2 axis, Tyr p-STAT-3, and SOCS-3 in lymph nodes from patients with chronic lymphocytic leukemia: Correlation between microvascular characteristics and prognostic significance. BioMed Res. Int. 2014, 251479. [Google Scholar] [CrossRef] [Green Version]
- Baggiolini, M.; Dewald, B.; Moser, B. Interleukin-8 and related chemotactic cytokines–CXC and CC chemokines. Adv. Immunol. 1994, 55, 97–179. [Google Scholar] [PubMed]
- Beliakova-Bethell, N.; Massanella, M.; White, C.; Lada, S.; Du, P.; Vaida, F.; Blanco, J.; Spina, C.A.; Woelk, C.H. The effect of cell subset isolation method on gene expression in leukocytes. Cytom. Part. A 2014, 85, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Calzetti, F.; Tamassia, N.; Arruda-Silva, F.; Gasperini, S.; Cassatella, M.A. The importance of being “pure” neutrophils. J. Allergy Clin. Immunol. 2017, 139, 352–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Wu, P.; Anthes, J.C.; Siegel, M.I.; Egan, R.W.; Billah, M.M. Interleukin-10 inhibits interleukin-8 production in human neutrophils. Blood 1994, 83, 2678–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risnik, D.; Podaza, E.; Almejún, M.B.; Colado, A.; Elías, E.E.; Bezares, R.F.; Fernández-Grecco, H.; Cranco, S.; Sánchez Ávalos, J.C.; Borge, M.; et al. Revisiting the role of interleukin-8 in chronic lymphocytic leukemia. Sci. Rep. 2017, 7, e15714. [Google Scholar] [CrossRef] [Green Version]
- Hornakova, T.; Staerk, J.; Royer, Y.; Flex, E.; Tartaglia, M.; Constantinescu, S.N.; Knoops, L.; Renauld, J.C. Acute lymphoblastic leukemia-associated JA- K1 mutants activate the Janus kinase/STAT pathway via interleukin-9 receptor alpha homodimers. J. Biol. Chem. 2009, 284, e67736781. [Google Scholar] [CrossRef] [Green Version]
- Noelle, R.J.; Nowak, E.C. Cellular sources and immune functions of interleukin-9. Nat. Rev. Immunol. 2010, 10, 683–687. [Google Scholar] [CrossRef]
- Demoulin, J.-B.; Renauld, J.-C. Interleukin 9 and its receptor: An overview of structure and function. Int. Rev. Immunol. 1998, 16, 345–364. [Google Scholar] [CrossRef]
- Renauld, J.C.; Druez, C.; Kermouni, A.; Houssiau, F.; Uyttenhove, C.; Van Roost, E.; Van Snick, J. Expression cloning of the murine and human interleukin 9 receptor cDNAs. Proc. Natl. Acad. Sci. 1992, 89, 5690–5694. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. Innate lymphoid cells: New paradigm in immunology of inflammation. Immunol. Lett. 2014, 157, 23–37. [Google Scholar] [CrossRef]
- Shang, Y.; Kakinuma, S.; Amasaki, Y.; Nishimura, M.; Kobayashi, Y.; Shimada, Y. Aberrant activation of interleukin-9 receptor and downstream Stat3/5 in primary T-cell lymphomas in vivo in susceptible B6 and resistant C3H mice. In Vivo 2008, 22, 713–720. [Google Scholar]
- Bittner, C.; Merz, H.; Krokowski, M.; Briese, J.; Wiedemann, G.J.; Feller, A.C. New immunotherapeutic approaches for the treatment of anaplastic large cell lymphoma in a mouse model. Verh. Dtsch. Ges. Pathol. 2000, 84, 187–198. [Google Scholar]
- Renauld, J.C.; van der Lugt, N.; Vink, A.; van Roon, M.; Godfraind, C.; Warnier, G.; Merz, H.; Feller, A.; Berns, A.; Van Snick, J. Thymic lymphomas in interleukin 9 transgenic mice. Oncogene 1994, 9, 1327–1332. [Google Scholar]
- Tete, S.; Saggini, A.; Maccauro, G.; Rosati, M.; Conti, F.; Cianchetti, E.; Tripodi, D.; Toniato, E.; Fulcheri, M.; Salini, V.; et al. Interleukin-9 and mast cells. J. Biol. Regul. Homeost. Agents 2012, 26, 319–326. [Google Scholar] [PubMed]
- Lu, D.; Qin, Q.; Lei, R.; Hu, B.; Qin, S. Targeted blockade of interleukin 9 inhibits tumor growth in murine model of pancreatic cancer. Adv. Clin. Exp. Med. 2019, 28, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Lv, X.; Li, P.; Lu, K.; Wang, X. Role of high expression of IL-9 in prognosis of CLL. Int. J. Clin. Exp. Pathol. 2014, 7, 716–721. [Google Scholar]
- Abbassy, H.A.; Aboelwafa, R.A.; Ghallab, O.M. Evaluation of Interleukin-9 Expression as a Potential Therapeutic Target in Chronic Lymphocytic Leukemia in a Cohort of Egyptian Patients. Indian J. Hematol. Blood Transfus. 2017, 33, 477–482. [Google Scholar] [CrossRef]
- Bialecka, M.; Klodowska-Duda, G.; Kurzawski, M.; Slawek, J.; Opala, G.; Bialecki, P.; Safranow, K.; Droździk, M. Interleukin-10 gene polymorphism in Parkinson’s disease patients. Arch. Med. Res. 2007, 38, 858–863. [Google Scholar] [CrossRef]
- Kamper, E.F.; Papaphilis, A.D.; Angelopoulou, M.K.; Kopeikina, L.T.; Siakantaris, M.P.; Pangalis, G.A.; Stavridis, J.C. Serum levels of tetranectin, intracellular adhesion molecule-1 and interleukin-10 in B-chronic lymphocytic leukemia. Clin. Biochem. 1999, 32, 639–645. [Google Scholar] [CrossRef]
- Bessler, H.; Bergman, M.; Salman, H. Factor(s) released from irradiated B-CLL cells induce apoptosis in leukemic lymphocytes. Cancer Lett. 2002, 179, 103–108. [Google Scholar] [CrossRef]
- Pang, N.; Zhang, R.; Li, J.; Zhang, Z.; Yuan, H.; Chen, G.; Zhao, F.; Wang, L.; Cao, H.; Qu, J.; et al. Increased IL-10/IL-17 ratio is aggravated along with the prognosis of patients with chronic lymphocytic leukemia. Int. Immunopharmacol. 2016, 40, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Montfort, A.; Capasso, M. B regulatory cells in cancer. Trends Immunol. 2013, 34, 169–173. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Weinberg, J.B.; Yoshizaki, A.; Horikawa, M.; Bryant, J.M.; Iwata, Y.; Matsushita, T.; Matta, K.M.; Chen, Y.; Venturi, G.M.; et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia 2013, 27, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Sindhava, V.J.; Bondada, S. Multiple regulatory mechanisms control B-1 B cell activation. Front. Immunol. 2012, 3, e372. [Google Scholar] [CrossRef] [Green Version]
- Satterthwaite, A.B.; Cheroutre, H.; Khan, W.N.; Sideras, P.; Witte, O.N. Btk dosage determines sensitivity to B cell antigen receptor cross-linking. Proc. Natl. Acad. Sci. USA 1997, 94, 13152–13157. [Google Scholar] [CrossRef] [Green Version]
- Alhakeem, S.S.; Sindhava, V.J.; McKenna, M.K.; Gachuki, B.W.; Byrd, J.C.; Muthusamy, N.; Bonda, S. Role of B cell receptor signaling in IL-10 production by normal and malignant B-1 cells. Ann. New York Acad. Sci. 2015, 1362, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Vincent, F.B.; Saulep-Easton, D.; Figgett, W.A.; Fairfax, K.A.; Mackay, F. The BAFF/APRIL system: Emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013, 24, 203–215. [Google Scholar]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Novak, A.J.; Bram, R.J.; Kay, N.E.; Jelinek, D.F. Aberrant expression of B595 lymphocyte stimulator by B-chronic lymphocytic leukemia cells: A mechanism for survival. Blood 2002, 100, 2973–2979. [Google Scholar] [CrossRef] [Green Version]
- Saulep-Easton, D.; Vincent, F.B.; Quah, P.S.; Wei, A.; Ting, S.B.; Croce, C.M.; Tam, C.; Mackay, F. The BAFF receptor TACI controls IL-10 production by regulatory B cells and CLL B cells. Leukemia 2016, 30, 163–172. [Google Scholar] [CrossRef]
- Yen Chong, S.; Lin, Y.C.; Czarneski, J.; Zhang, M.; Coffman, F.; Kashanchi, F.; Raveche, E. Cell cycle effects of IL-10 on malignant B-1 cells. Genes Immun. 2001, 2, 239–247. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, B.A.; Mansour, A.; Lin, Y.C.; Kotenko, S.; Raveche, E. RNA interference of IL-10 in leukemic B-1 cells. Cancer Immun. 2004, 4, e6. [Google Scholar]
- Sjoberg, J.; Aguilar-Santelises, M.; Sjogren, A.-M.; Pisa, E.K.; Ljungdahl, A.; Bjorkholm, M.; Jondan, M.; Mellstedt, H.; Pisa, P. Interleukin-10 m-RNA expression in B-cell chronic lymphocytic leukaemia inversely correlates with progress of disease. Br. J. Haematol. 1996, 92, 393–400. [Google Scholar] [CrossRef]
- Wang, X.; Lupardus, P.; Laporte, S.L.; Garcia, K.C. Structural biology of shared cytokine receptors. Annu. Rev. Immunol. 2009, 27, 29–60. [Google Scholar] [CrossRef] [Green Version]
- Meazza, R.; Azzarone, B.; Orengo, A.M.; Ferrini, S. Role of common-gamma chain cytokines in NK cell development and function: Perspectives for immunotherapy. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef]
- Becknell, B.; Caligiuri, M.A. Interleukin-2, interleukin-15, and their roles in human natural killer cells. Adv. Immunol. 2005, 86, 209–239. [Google Scholar]
- Stonier, S.W.; Schluns, K.S. Trans-presentation: A novel mechanism regulating IL-15 delivery and responses. Immunol. Lett. 2010, 127, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Huntington, N.D.; Alves, N.L.; Legrand, N.; Lim, A.; Strick-Marchand, H.; Mention, G.; Plet, A.; Weijer, K.; Jacques, Y.; Becker, P.D. IL-15 transpresentation promotes both human T-cell reconstitution and T-cell-dependent antibody responses in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 6217–6222. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.A.; Liou, Y.H.; Wang, S.W.; Ko, K.L.; Jiang, S.T.; Liao, N.S. Different NK cell developmental events require different levels of IL-15 trans-presentation. J. Immunol. 2011, 187, 1212–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehniger, T.A.; Caligiuri, M.A. Interleukin 15: Biology and relevance to human disease. Blood 2001, 97, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Jakobisiak, M.; Golab, J.; Lasek, W. Interleukin 15 as a promising candidate for tumor immunotherapy. Cytokine Growth Factor Rev. 2011, 22, 99–108. [Google Scholar]
- Bhat, R.; Watzl, C. Serial killing of tumor cells by human natural killer cells–enhancement by therapeutic antibodies. PLoS ONE 2007, 2, e326–e332. [Google Scholar] [CrossRef]
- Wu, Z.; Xu, Y. IL-15R alpha-IgG1-Fc enhances IL-2 and IL-15 anti-tumor action through NK and CD8+ T cells proliferation and activation. J. Mol. Cell Biol. 2010, 2, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Bernasconi, N.L.; Traggiai, E.; Lanzavecchia, A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002, 298, 2199–2202. [Google Scholar] [CrossRef]
- Mongini, P.K.A.; Gupta, R.; Boyle, E.; Nieto, J.; Lee, H.; Stein, J.; Bandovic, J.; Stankovic, T.; Barrientos, J.; Kolitz, J.E.; et al. TLR-9 and IL-15 Synergy Promotes the In Vitro Clonal Expansion of Chronic Lymphocytic Leukemia B Cells. J. Immunol. 2015, 195, 901–923. [Google Scholar] [CrossRef] [Green Version]
- Lotz, M.; Ranheim, E.; Kipps, T.J. Transforming growth factor beta as endogenous growth inhibitor of chronic lymphocytic leukemia B cells. J. Exp. Med. 1994, 179, 999–1004. [Google Scholar] [CrossRef]
- Kremer, J.P.; Reisbach, G.; Nerl, C.; Dormer, P. B-cell chronic lym-phocytic leukaemia cells express and release transforming growth factor-beta. Br. J. Haematol. 1992, 80, 480–487. [Google Scholar] [CrossRef]
- Trotta, R.; Dal Col, J.; Yu, J.; Ciarlariello, D.; Thomas, B.; Zhang, X.; Allard, J., 2nd; Wei, M.; Mao, H.; Byrd, J.C.; et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cyto-toxicity in human NK cells. J. Immunol. 2008, 181, 3784–3792. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.; Fuller, J.A.; Christianson, G.; Krupke, D.M.; Troutt, A.B. IL-15 mediates anti-tumor effects after cyclophosphamide injection of tumor-bearing mice and enhances adoptive immunotherapy: The potential role of NK cell subpopulations. Cell Immunol. 1997, 179, 66–73. [Google Scholar] [CrossRef]
- Ma, X.; Reynolds, S.L.; Baker, B.J.; Li, X.; Benveniste, E.N.; Qin, H. IL-17 enhancement of the IL-6 signaling cascade in astrocytes. J. Immunol. 2010, 184, 4898–4906. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Dong, W. IL-17 cytokines in immunity and inflammation. Emerging Microbes Infect. 2013, 2, 1–5. [Google Scholar]
- Sherry, B.; Jain, P.; Chiu, P.Y.; Leung, L.; Allen, S.L.; Kolitz, J.E.; Rai, K.R.; Barrientos, J.; Liang, S.; Hatwin, R.; et al. Identification and characterization of distinct IL-17F expression patterns and signaling pathways in chronic lymphocytic leukemia and normal B lymphocytes. Immunol. Res. 2015, 63, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Niu, Q.; Jiang, N.; Li, J.; Zheng, Q.; Jia, Y. Increased frequencies of Th17 in the peripheral blood of patients with chronic lymphocytic leukemia: A one year follow-up. Pak. J. Med. Sci. 2014, 30, 1128–1133. [Google Scholar]
- Hus, I.; Bojarska-Junak, A.; Chocholska, S.; Tomczak, W.; Woś, J.; Dmoszyńska, A.; Roliński, J. Th17/IL-17A might play a protective role in chronic lymphocytic leukemia immunity. PLoS ONE 2013, 8, e78091. [Google Scholar] [CrossRef] [Green Version]
- Habib, T.; Nelson, A.; Kaushanksy, K. IL-21: A novel IL-2-family lymphokine that modulates B, T, and natural killer cell responses. J. Allergy Clin. Immunol. 2003, 112, 1033–1045. [Google Scholar] [CrossRef]
- Strengell, M.; Saraneva, T.; Foster, D.; Julkunen, I.; Matikainen, S. IL-21 up-regulates the expression of genes associated with innate immunity and Th1 response. J. Immunol. 2002, 169, 3600–3605. [Google Scholar] [CrossRef] [Green Version]
- Spolski, R.; Leonard, W.J. Interleukin-21: Basic biology and implications for cancer and autoimmunity. Annu Rev. Immunol. 2008, 26, 57–79. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.S.; Wurster, A.L.; Grusby, M.J. Biology of IL-21 and the IL-21 receptor. Immunol Rev. 2004, 202, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Parrish-Novak, J.; Dillon, S.R.; Nelson, A.; Hammond, A.; Sprecher, C.; Gross, J.A.; Johnston, J.; Madden, K.; Xu, W.; West, J.; et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 2000, 408, 57–63. [Google Scholar] [CrossRef]
- Gowda, A.; Roda, J.; Hussain, S.R.; Ramanunni, A.; Joshi, T.; Schmidt, S.; Zhang, X.; Lehman, A.; Jarjoura, D.; Carson, W.E.; et al. IL-21 mediates apoptosis through up-regulation of the BH3 family member BIM and enhances both direct and antibody-dependent cellular cytotoxicity in primary chronic lymphocytic leukemia cells in vitro. Blood 2008, 111, 4723–4730. [Google Scholar] [CrossRef] [Green Version]
- Lamprecht, B.; Kreher, S.; Anagnostopoulos, I.; Jöhrens, K.; Monteleone, G.; Jundt, F.; Stein, H.; Janz, M.; Dörken, B.; Mathas, S. Aberrant expression of the Th2 cytokine IL-21 in Hodgkin lymphoma cells regulates STAT3 signaling and attracts Treg cells via regulation of MIP-3α. Blood 2008, 112, 3339–3347. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Eidenschenk, C.; Ouyang, W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013, 252, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Witte, E.; Wallace, E.; Döcke, W.D.; Kunz, S.; Asadullah, K.; Volk, H.D.; Sterry, W.; Sabat, R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur. J. Immunol. 2006, 36, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Tachiiri, A.; Imamura, R.; Wang, Y.; Fukui, M.; Umemura, M.; Suda, T. Genomic structure and inducible expression of the IL-22 receptor alpha chain in mice. Genes Immun. 2003, 4, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yao, Y.; Yao, M.; Fu, P.; Wang, W. Interleukin-22 promotes triple negative breast cancer cells migration and paclitaxel resistance through JAK-STAT3/MAPKs/AKT signaling pathways. Biochem. Biophys. Res. Commun. 2018, 503, 1605–1609. [Google Scholar] [CrossRef]
- Nagalakshmi, M.L.; Rascle, A.; Zurawski, S.; Menon, S.; de Waal Malefyt, R. Interleukin-22 activates STAT3 and induces IL-10 by colon epithelial cells. Int. Immunopharmacol. 2004, 4, 679–691. [Google Scholar] [CrossRef]
- Yu, S.; Liu, C.; Zhang, L.; Shan, B.; Tian, T.; Hu, Y. Elevated Th22 cells correlated with Th17 cells in peripheral blood of patients with acute myeloid leukemia. Int. J. Mol. Sci. 2014, 15, 1927–1945. [Google Scholar] [CrossRef] [Green Version]
- Heiba, N.M.; Elshazly, S.A. Plasma interleukin-22 and its cellular receptor (IL-22RA1) expression in chronic lymphocytic leukemia. Egypt. J. Haematol. 2013, 38, 123–129. [Google Scholar]
- Mohammadi, M.; Kaghazian, M.; Rahmani, O.; Ahmadi, K. Overexpression of interleukins IL-17 and IL-8 with poor prognosis in colorectal cancer induces metastasis. Tumour Biol. 2016, 37, 7501–7505. [Google Scholar] [CrossRef] [PubMed]
- Kouzegaran, S.; Siroosbakht, S.; Farsad, B.F.; Rezakhaniha, B.; Dormanesh, B.; Behnod, V.; Tanha, A.S. Elevated IL-17A and IL-22 Regulate Expression of Inducible CD38 and Zap-70 in Chronic Lymphocytic Leukemia. Cytom. Part. B 2018, 94, 143–147. [Google Scholar] [CrossRef]
- Gangemi, S.; Allegra, A.; Alonci, A.; Pace, E.; Ferraro, M.; Cannavò, A.; Penna, G.; Saitta, S.; Gerace, D.; Musolino, C. Interleukin 22 is increased and correlated with CD38 expression in patients with B-chronic lymphocytic leukemia. Blood Cells Mol. Dis. 2013, 50, 39–40. [Google Scholar] [CrossRef]
- Jamroziak, K.; Balcerczak, E.; Smolewski, P.; Robey, R.W.; Cebula, B.; Panczyk, M. MDR1 (ABCB1) gene polymorphism C3435T is associated with P-glycoprotein activity in B-cell chronic lymphocytic leukemia. Pharmacol. Rep. 2006, 58, 720–728. [Google Scholar] [PubMed]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupé, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 2005, 5, 521–531. [Google Scholar] [CrossRef]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef]
- Teng, M.W.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 cytokines: From discovery to targeted therapies for immunemediated inflammatory diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef]
- Cocco, C.; Canale, S.; Frasson, C.; Di Carlo, E.; Ognio, E.; Ribatti, D.; Prigione, I.; Basso, G.; Airoldi, I. Interleukin-23 acts as antitumor agent on childhood B-acute lymphoblastic leukemia cells. Blood 2010, 116, 3887–3898. [Google Scholar] [CrossRef] [Green Version]
- Cocco, C.; Morandi, F.; Airoldi, I. Interleukin-27 and interleukin-23 modulate human plasmacell functions. J. Leukoc. Biol. 2011, 89, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Cocco, C.; Di Carlo, E.; Zupo, S.; Canale, S.; Zorzoli, A.; Ribatti, D.; Morandi, F.; Ognio, E.; IAiroldi, I. Complementary IL-23 and IL-27 anti-tumor activities cause strong inhibition of human follicular and diffuse large B-cell lymphoma growth in vivo. Leukemia 2012, 26, 1365–1374. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, N.; Airoldi, I. Novel insights into the role of interleukin-27 and interleukin-23 in human malignant and normal plasma cells. Clin. Cancer Res. 2011, 17, 6963–6970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Hu, Z. The enigmatic processing and secretion of interleukin-33. Cell Mol. Immunol. 2010, 7, 260–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musolino, C.; Allegra, A.; Profita, M.; Alonci, A.; Saitta, S.; Bonanno, A.; Gerace, D.; Calabrò, L.; Gangemi, S. Reduction in IL-33 plasma levels might be involved in T cell dysregulation in chronic lymphocytic leukemia. Acta. Haematol. 2014, 131, 165–166. [Google Scholar] [CrossRef]
- Podhorecka, M.; Dmoszynska, A.; Rolinski, J.; Wasik, E. T type 1/type 2 subsets balance in B cell chronic lymphocytic leukemia – the three-color flow cytometry analysis. Leuk. Res. 2002, 26, 657–660. [Google Scholar] [CrossRef]
- Huang, L.; Chen, S.; Zha, X.; Yang, L.; Li, B.; Yu, Z.; Wang, L.; Li, Y. Expression feature of CD3, FcεRIγ, and Zap-70 in patients with chronic lymphocytic leukemia. Hematology 2012, 17, 71–75. [Google Scholar] [CrossRef]
- Rosati, E.; Sabatini, R.; Tabilio, A.; Di Ianni, M.; Bartoli, A.; Marconi, P. B chronic lymphocytic leukemia cells exert an in vitro cytotoxicity mediated by TNFα. Leuk. Res. 2005, 29, 829–839. [Google Scholar] [CrossRef]
- Jetovic-Stoimenov, T.; Kocic, G.; Pavlovic, D.; Macukanovic-Golubovic, L.; Marjanovic, G.; Djordjevic, V.; Tosic, N.; Pavlovic, S. Polymorphisms of tumor-necrosis factor-alpha –308 and lymphotoxin-alpha +250: Possible modulation of susceptibility to apoptosis in chronic lymphocytic leukemia and non-Hodgkin lymphoma mononuclear cells. Leuk. Lymphoma 2008, 49, 2163–2169. [Google Scholar] [CrossRef]
- Waage, A.; Liabakk, N.; Lien, E.; Lamvik, J.; Espevik, T. p55 and p75 tumor necrosis factor receptors in patients with chronic lymphocytic leukemia. Blood 1992, 80, 2577–2583. [Google Scholar] [CrossRef] [Green Version]
- Ferraioli, A.; Keating, M.J.; Taghi, M.; Gilles, F.J.; Dey, A.; Estrov, Z.; Koller, C.A.; Kurzrock, R.; Thomas, D.A.; Faderl, S.; et al. The clinical significance of tumor necrosis factor-alpha plasma level in patients having chronic lymphocytic leukemia. Blood 2002, 100, 1215–1219. [Google Scholar] [CrossRef]
- Adami, F.; Guarini, A.; Pini, M.; Siviero, F.; Sancetta, R.; Massaia, M.; Trentin, L.; Foà, R.; Semenzato, G. Serum levels of tumour necrosis factor-alpha in patients with B-cell chronic lymphocytic leukaemia. Eur. J. Cancer 1994, 30, 1259–1263. [Google Scholar] [CrossRef]
- Bojarska-Junak, A.; Hus, I.; Szczepanek, E.W.; Dmoszynska, A.; Rolinski, J. Peripheral blood and bone marrow TNF and TNF receptors in early and advanced stages of B-CLL in correlation with ZAP-70 protein and CD38 antigen. Leuk. Res. 2008, 32, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Digel, W.; Porzsolt, F.; Schmid, M.; Herrmann, F.; Lesslauer, W.; Brockhaus, M. High levels of circulating soluble receptors for tumor necrosis factor in hairy cell leukemia and type B chronic lymphocytic leukemia. J. Clin. Invest. 1992, 89, 1690–1693. [Google Scholar] [CrossRef] [PubMed]
- Digel, W.; Stefanic, M.; Schöniger, W.; Buck, C.; Raghavachar, A.; Frickhofen, N.; Heimpel, H.; Porzsolt, F. Tumor necrosis factor induces proliferation of neoplastic B cells from chronic lymphocytic leukemia. Blood 1989, 73, 1242–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordingley, F.T.; Bianchi, A.; Hoffbrand, A.V.; Reittie, J.E.; Heslop, H.E.; Vyakarnam, A.; Turner, M.; Meager, A.; Brenner, M.K. Tumour necrosis factor as an autocrine tumour growth factor for chronic B-cell malignancies. Lancet 1988, 1, 969–971. [Google Scholar] [CrossRef]
- Woyach, J.A.; Lin, T.S.; Lucas, M.S.; Heerema, N.; Moran, M.E.; Cheney, C.; Lucas, D.M.; Wei, L.; Caligiuri, M.A.; Byrd, J.C. A phase I/II study of rituximab and etanercept in patients with chronic lymphocytic leukemia and small lymphocytic lymphoma. Leukemia 2009, 23, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Meinhardt, G.; Wendtner, C.M.; Hallek, M. Molecular pathogenesis of chronic lymphocytic leukemia: Factors and signaling pathways regulating cell growth and survival. J. Mol. Med. 1999, 77, 282–293. [Google Scholar] [CrossRef]
- Zhu, W.; Shiojima, I.; Ito, Y.; Li, Z.; Ikeda, H.; Yoshida, M.; Naito, A.T.; Nishi, J.; Ueno, H.; Umezawa, A. IGFBP-4 is an inhibitor of canonical Wnt signalling required for cardiogenesis. Nature 2008, 7202, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Djurdjevic, P.; Zelen, I.; Ristic, P.; Baskic, D.; Popovic, S.; Arsenijevic, N. Role of Decreased Production of Interleukin-10 and Interferon-Gamma in Spontaneous Apoptosis of B-Chronic Lymphocytic Leukemia Lymphocytes In Vitro. Arch. Med. Res. 2009, 40, 357–363. [Google Scholar] [CrossRef]
- Mahadevan, D.; Choi, J.; Cooke, L.; Simons, B.; Riley, C.; Klinkhammer, T.; Sud, R.; Maddipoti, S.; Hehn, S.; Garewal, H.; et al. Gene Expression and Serum Cytokine Profiling of Low Stage CLL Identify WNT/PCP, Flt-3L/Flt-3, andCXCL9/CXCR3as Regulators of Cell Proliferation, Survival, and Migration. Hum. Genom. Proteom. 2009, 2009, e453634. [Google Scholar]
- Allegra, A.; Innao, V.; Gerace, D.; Vaddinelli, D.; Musolino, C. Adoptive immunotherapy for hematological malignancies: Current status and new insights in chimeric antigen receptor T cells. Blood Cells Mol. Dis. 2016, 62, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Russo, S.; Gerace, D.; Calabrò, L.; Maisano, V.; Innao, V.; Musolino, C. Vaccination strategies in lymphoproliferative disorders: Failures and successes. Leuk. Res. 2015, 39, 1006–1019. [Google Scholar] [CrossRef]
- Jaime-Ramirez, A.C.; McMichael, E.; Kondadasula, S.V.; Skinner, C.C.; Mundy-Bosse, B.L.; Luedke, E.; Jones, N.B.; Mani, A.; Roda, J.; Karpa, V.; et al. NK Cell–Mediated Antitumor Effects of a Folate-Conjugated Immunoglobulin are Enhanced by Cytokines. Cancer Immunol. Res. 2016, 4, 323–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Pennati, A.; Cohen, J.B.; Wu, Y.; Ng, S.; Wu, J.H.; Flowers, C.R.; Galipeau, J. GIFT4 fusokine converts leukemic B cells into immune helper cells. J. Transl. Med. 2016, 14, e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fas, S.C.; Baumann, S.; Zhu, J.Y.; Giaisi, M.; Treiber, M.K.; Mahlknecht, U.; Krammer, P.H.; Li-Weber, M. Wogonin sensitizes resistant malignant cells to TNFalpha- and TRAIL-induced apoptosis. Blood 2006, 108, 3700–3706. [Google Scholar] [CrossRef]
- Li-Weber, M. Targeting apoptosis pathways in cancer by Chinese medicine. Cancer Lett. 2013, 332, 304–312. [Google Scholar] [CrossRef]
- Dürr, C.; Hanna, B.S.; Schulz, A.; Lucas, F.; Zucknick, M.; Benner, A.; Clear, A.; Ohl, S.; Öztürk, S.; Zenz, T.; et al. Tumor necrosis factor receptor signaling is a driver of chronic lymphocytic leukemia that can be therapeutically targeted by the flavonoid wogonin. Haematologica 2018, 103, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.N.; Gao, H.; Cohen, E.N.; Badoux, X.; Wierda, W.G.; Estrov, Z.; Faderl, S.H.; Keating, M.J.; Ferrajoli, A.; Reuben, J.M. Treatment with Lenalidomide Modulates T-Cell Immunophenotype and Cytokine Production in Patients with Chronic Lymphocytic Leukemia. Cancer 2011, 117, 3999–4008. [Google Scholar] [CrossRef] [Green Version]
- Gangemi, S.; Allegra, A.; Aguennouz, M.; Alonci, A.; Speciale, A.; Cannavò, A.; Cristani, M.; Russo, S.; Spatari, G.; Alibrandi, A.; et al. Relationship between advanced oxidation protein products, advanced glycation end products, and S-nitrosylated proteins with biological risk and MDR-1 polymorphisms in patients affected by B-chronic lymphocytic leukemia. Cancer Invest. 2012, 30, 20–26. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Alonci, A.; Saija, A.; Russo, S.; Cannavò, A.; Cristani, M.; Centorrino, R.; Saitta, S.; Alibrandi, A.; et al. Carbonyl group serum levels are associated with CD38 expression in patients with B chronic lymphocytic leukemia. Clin. Biochem. 2011, 44, 1487–1490. [Google Scholar] [CrossRef] [PubMed]
- Djurdjevic, P.; Ristic, P.; Jovanovic, I.; Jakovljevic, V.; Baskic, D.; Popovic, S.; Arsenijevic, N. Oxidative stress accelerates spontaneous apoptosis of B-chronic lymphocytic leukemia lymphocytes. J. BUON 2009, 14, 281–287. [Google Scholar] [PubMed]
- Schulz, A.; Toedt, G.; Zenz, T.; Stilgenbauer, S.; Lichter, P.; Seiffert, M. Inflammatory cytokines and signaling pathways are associated with survival of primary chronic lymphocytic leukemia cells in vitro: A dominant role of CCL2. Haematologica 2011, 96, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, U.; Dalla-Favera, R. New insights into the pathogenesis of chronic lymphocytic leukemia. Semin. Cancer Biol. 2010, 20, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Circosta, P.; Scielzo, C.; Vallario, A.; Camporeale, A.; Granziero, L.; Caligaris-Cappio, F. Differential effects on CLL cell survival exerted by different microenvironmental elements. Curr. Top. Microbiol. Immunol. 2005, 294, 135–145. [Google Scholar]
- Scielzo, C.; Ten Hacken, E.; Bertilaccio, M.T.; Muzio, M.; Calissano, C.; Ghia, P.; Caligaris-Cappio, F. How the microenvironment shapes chronic lymphocytic leukemia: The cytoskeleton connection. Leuk. Lymphoma 2010, 51, 1371–1374. [Google Scholar] [CrossRef]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 14, 3367–3375. [Google Scholar] [CrossRef] [Green Version]
- Schrottner, P.; Leick, M.; Burger, M. The role of chemokines in B cell chronic lymphocytic leukaemia: Pathophysiological aspects and clinical impact. Ann. Hematol. 2010, 89, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Kipps, T.J. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk. Lymphoma 2002, 43, 461–466. [Google Scholar] [CrossRef]
- Ten Hacken, E.; Burger, J.A. Microenvironment interactions and B-cell receptor signaling in chronic lymphocytic leukemia: Implications for disease pathogenesis and treatment. Biochim. Biophys. Acta. 2016, 1863, 401–413. [Google Scholar] [CrossRef] [PubMed]
- van Attekum, M.H.; Eldering, E.; Kater, A.P. Chronic lymphocytic leukemia cells are active participants in microenvironmental cross-talk. Haematologica 2017, 102, 1469–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Cytokine | Action | Mechanism | Reference |
|---|---|---|---|
| IL-2 | Protumoral | Reduced apoptosis | [23] |
| Increased growth | [24] | ||
| IL-4 | Protumoral | Reduced apoptosis | [25,26,27,28,29,30,31,32] |
| Stimulation of STAT | [33] | ||
| Increase miR-21-5p | [34] | ||
| iNKT alteration | [35] | ||
| IL-6 | Protumoral | Stimulation of STAT3 | [36] |
| Stimulation of NF-kB | [36] | ||
| Modulation of VEGF | [37,38] | ||
| Antitumoral | Action on Toll-like receptor | [39,40] | |
| IL-8 | Protumoral | Increase of BCL-2 | [41,42] |
| Angiogenesis | [43,44] | ||
| IL-9 | Protumoral | Increased proliferation | [45] |
| Decreased apoptosis | [46] | ||
| Increased JAK/STAT | [47,48] | ||
| IL-10 | Protumoral | Increased proliferation | [49,50] |
| Antitumoral | Action on NK cells | [51,52] | |
| IL-17 | Protumoral | Increased IL-6 | [53] |
| Antitumoral | Action on immunity | [54] | |
| IL-21 | Antitumoral | Increased apoptosis | [55,56,57,58,59,60] |
| Increased proliferation of cytotoxic T cells | [61,62] | ||
| IL-22 | Protumoral | Stimulation of STAT3 | [63,64,65,66] |
| Reduced apoptosis | [63,64,65,66] | ||
| IL-23 | Protumoral | Unclear | [67] |
| TNF-alpha | Protumoral | Increased proliferation | [68,69] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, A.; Musolino, C.; Tonacci, A.; Pioggia, G.; Casciaro, M.; Gangemi, S. Clinico-Biological Implications of Modified Levels of Cytokines in Chronic Lymphocytic Leukemia: A Possible Therapeutic Role. Cancers 2020, 12, 524. https://doi.org/10.3390/cancers12020524
Allegra A, Musolino C, Tonacci A, Pioggia G, Casciaro M, Gangemi S. Clinico-Biological Implications of Modified Levels of Cytokines in Chronic Lymphocytic Leukemia: A Possible Therapeutic Role. Cancers. 2020; 12(2):524. https://doi.org/10.3390/cancers12020524
Chicago/Turabian StyleAllegra, Alessandro, Caterina Musolino, Alessandro Tonacci, Giovanni Pioggia, Marco Casciaro, and Sebastiano Gangemi. 2020. "Clinico-Biological Implications of Modified Levels of Cytokines in Chronic Lymphocytic Leukemia: A Possible Therapeutic Role" Cancers 12, no. 2: 524. https://doi.org/10.3390/cancers12020524