Increased Expression of Micro-RNA-23a Mediates Chemoresistance to Cytarabine in Acute Myeloid Leukemia

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
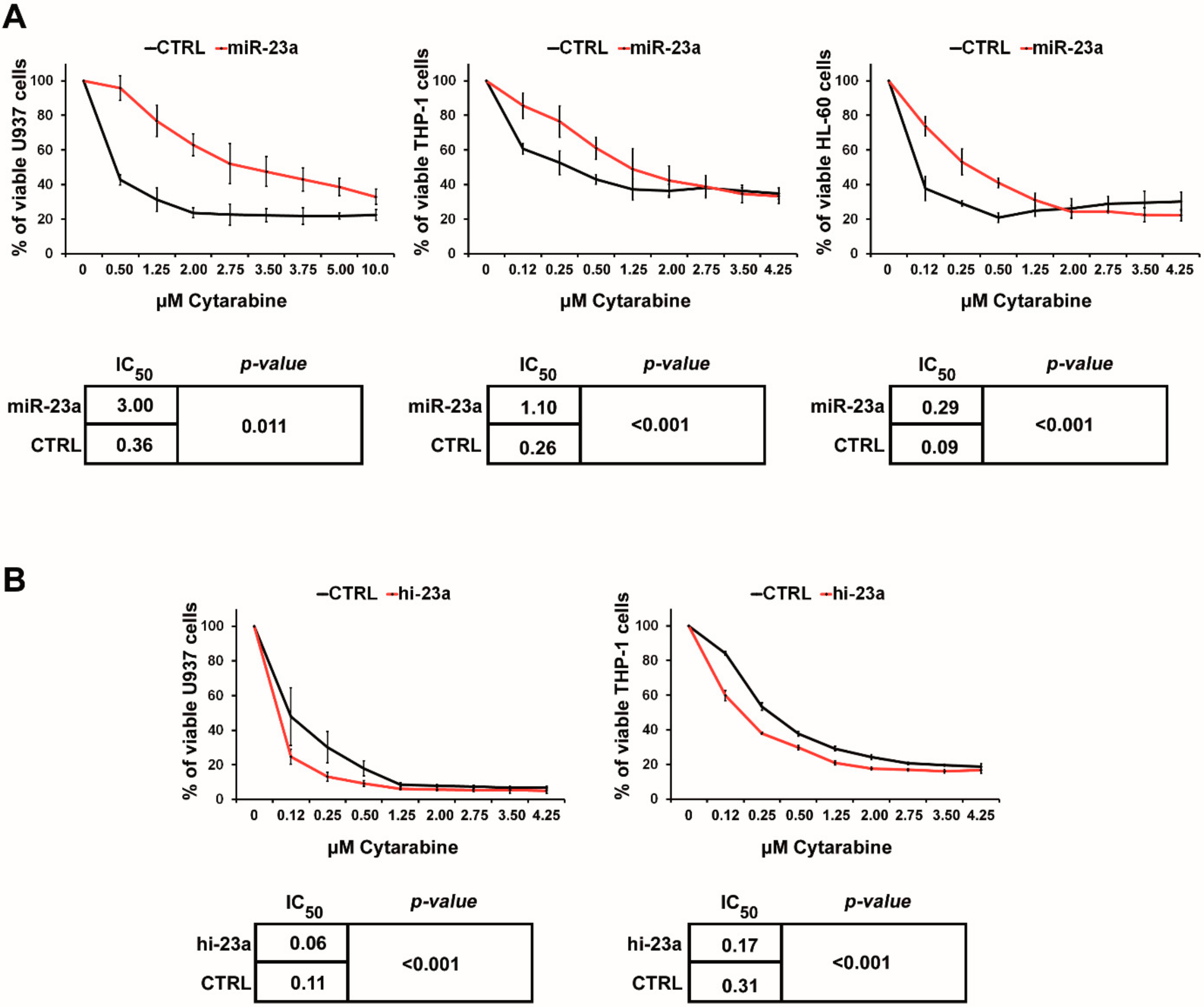
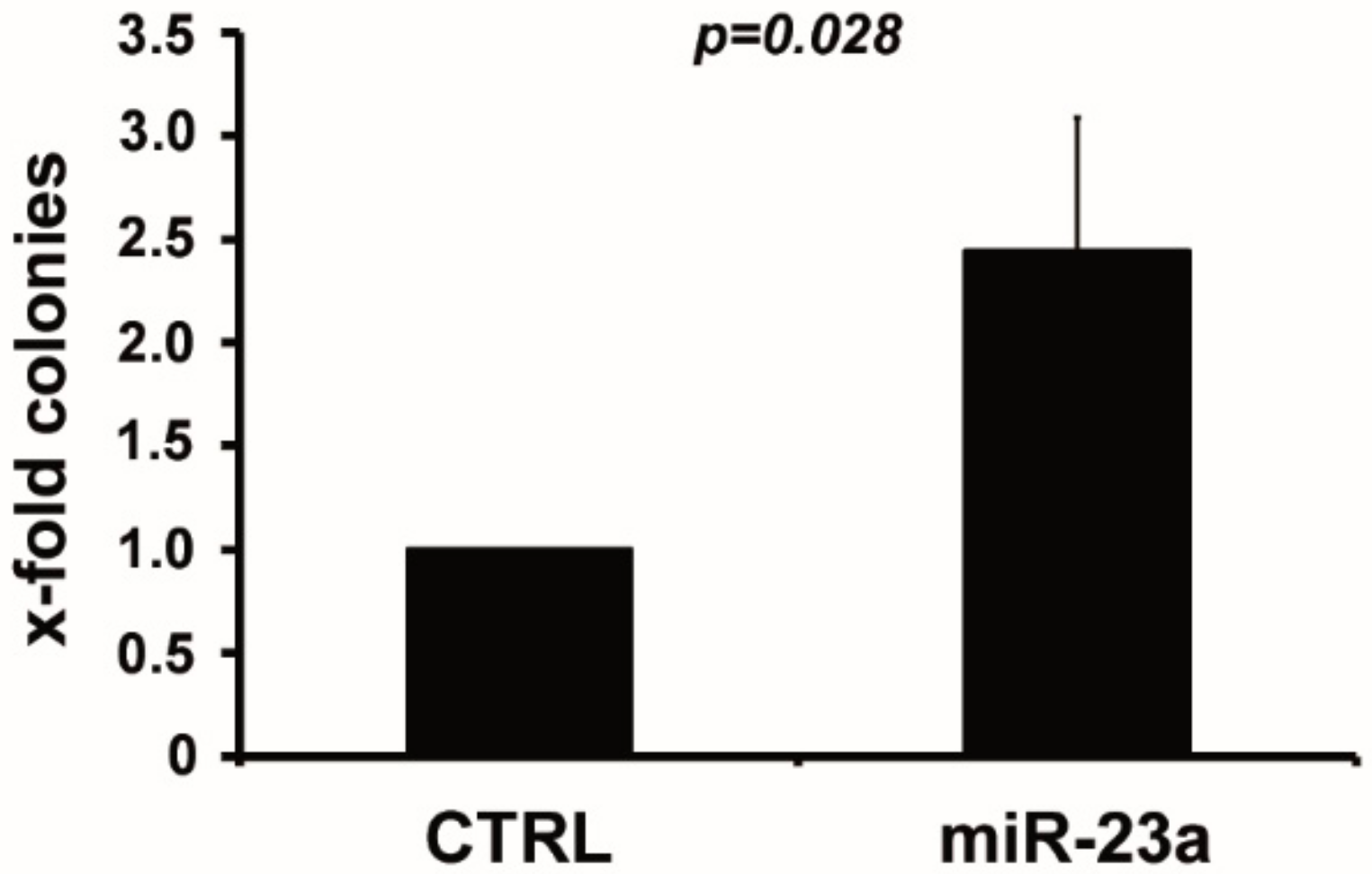
2.1. miR-23a Mediates Resistance to AraC
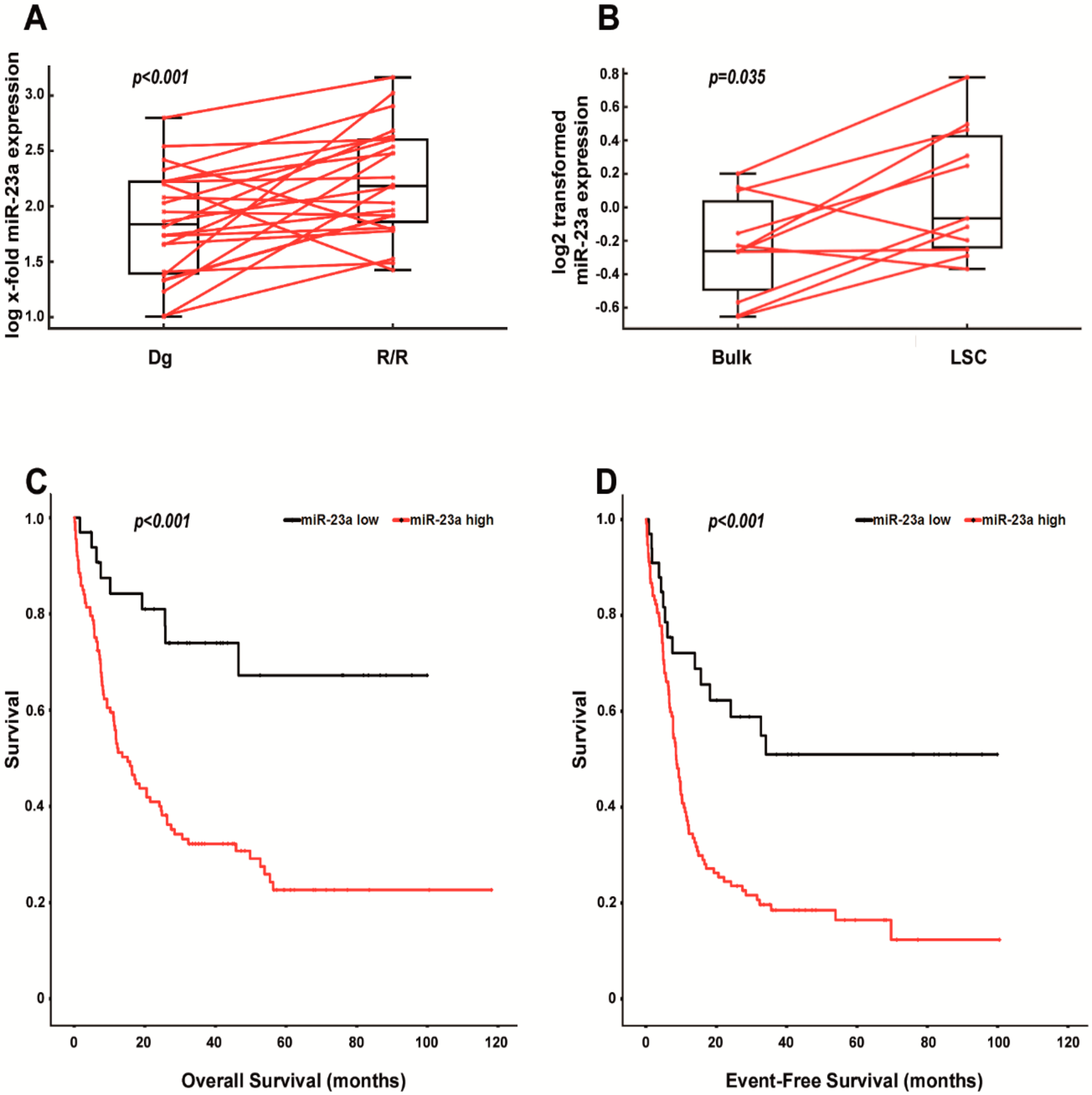
2.2. Increased Expression of miR-23a Correlates with Relapsed/Refractory AML, with the Leukemic Stem Cell Pool and with Shorter Survival in AraC-Treated Patients
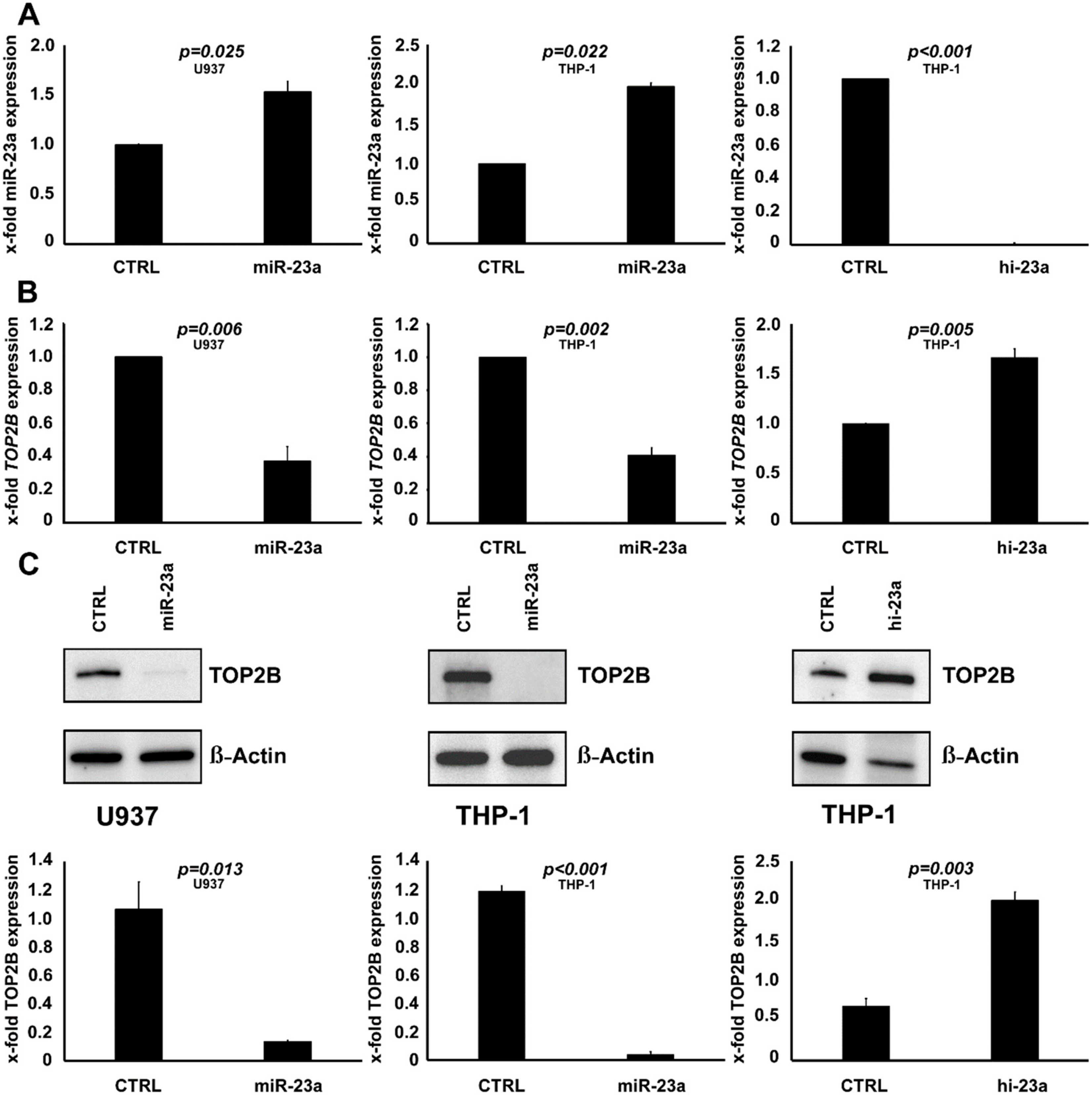
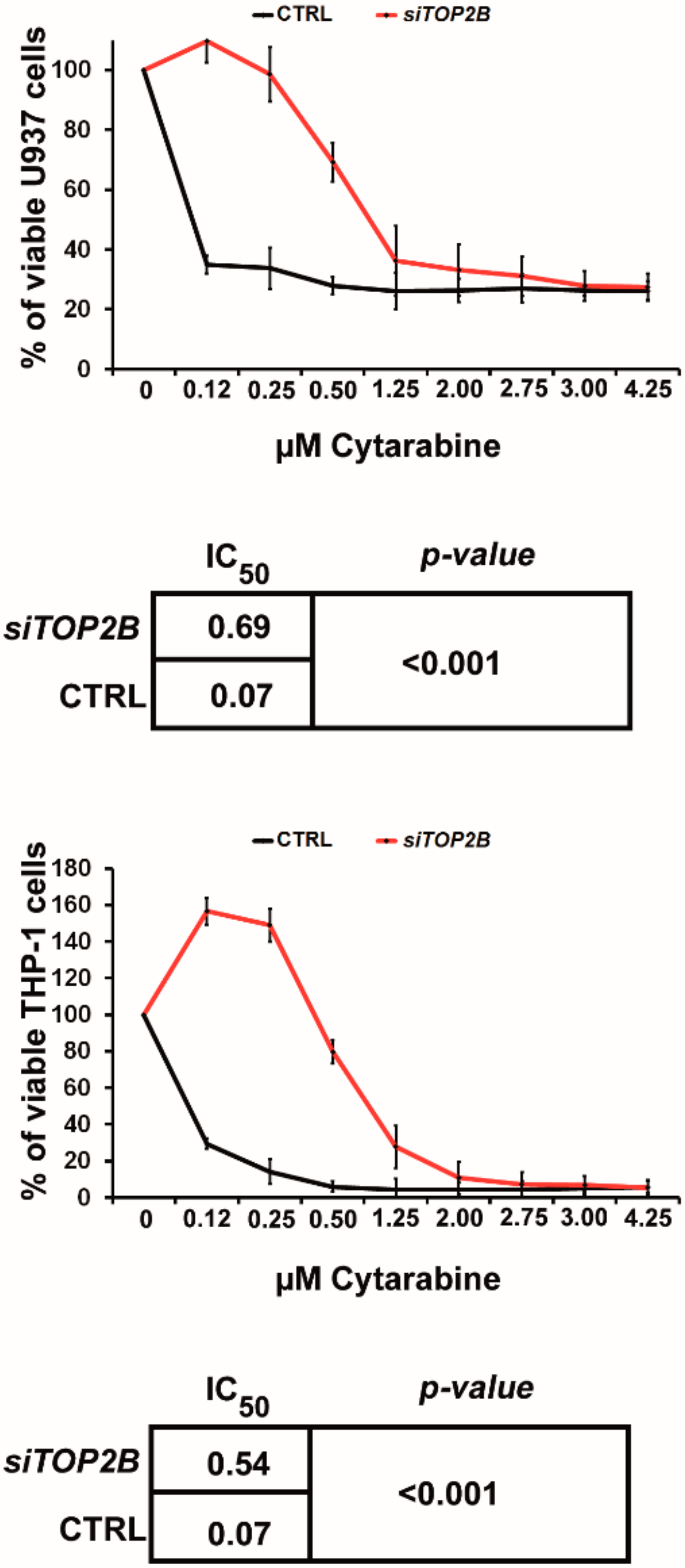
2.3. TOP2B Is Regulated by miR-23a and Affects the Sensitivity to AraC as Well
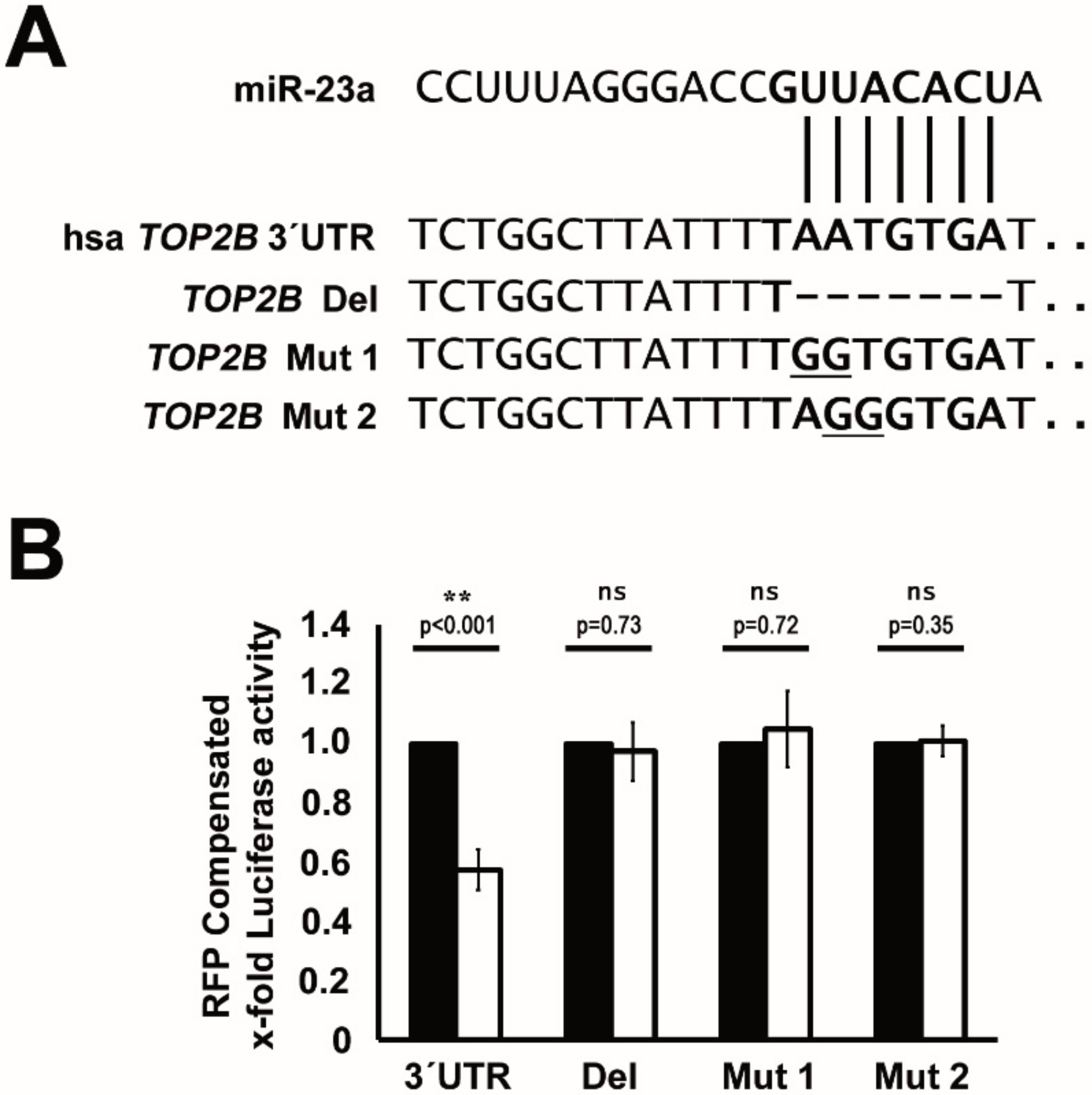
2.4. TOP2B Is a Direct Target of miR-23a
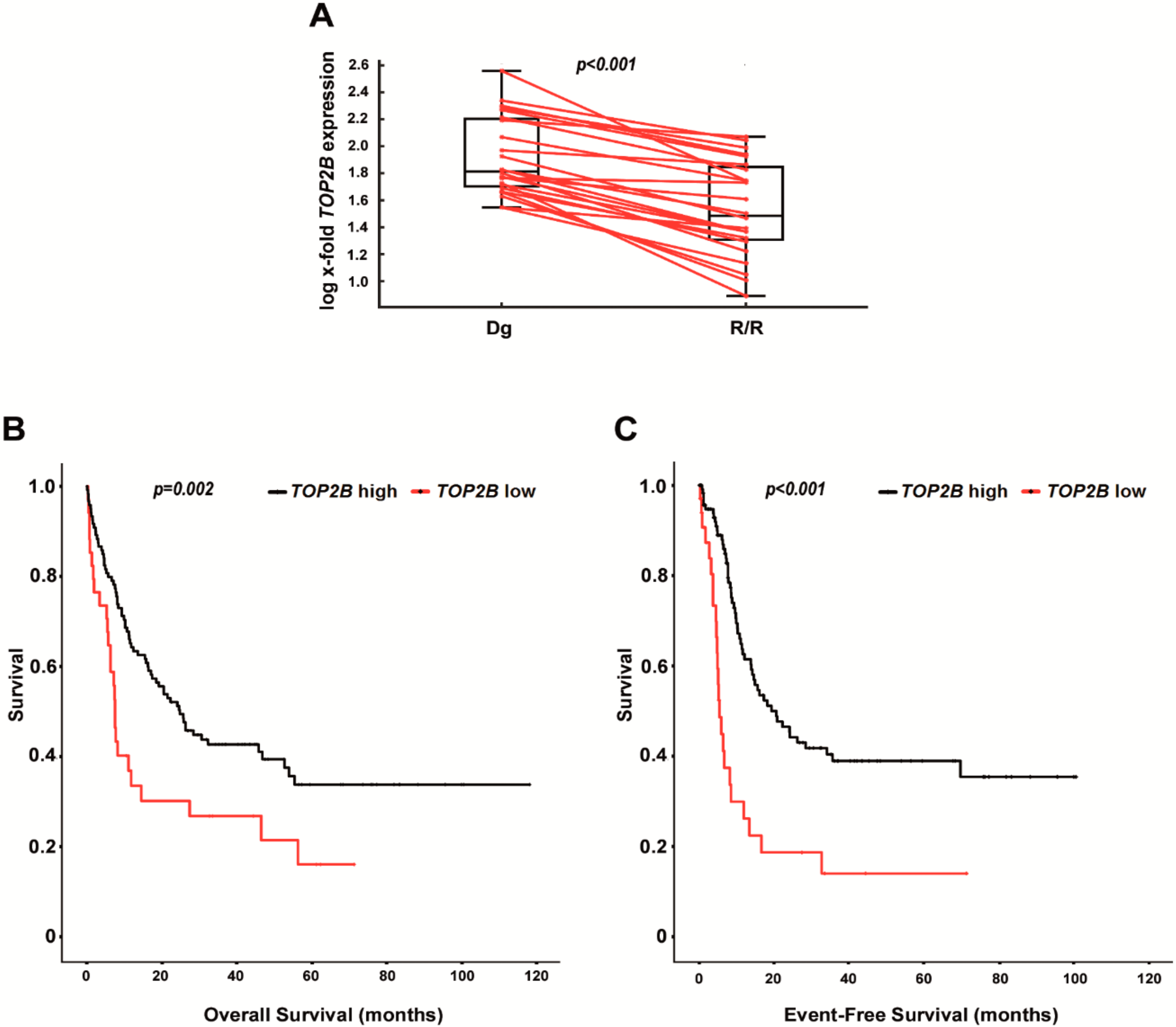
2.5. Decreased Expression of TOP2B Correlates with Relapsed/Refractory AML and with Shorter Survival in AraC-Treated Patients
3. Discussion
4. Materials and Methods
4.1. Patient Samples and Cell Lines
4.2. Cell Culture, Lentiviral Transduction and Transfection
4.3. qPCR and Immunoblot Analysis
4.4. Chemosensitivity Assays
4.5. Luciferase Reporter Assays
4.6. Database Retrieval and Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koreth, J.; Schlenk, R.; Kopecky, K.J.; Honda, S.; Sierra, J.; Djulbegovic, B.J.; Wadleigh, M.; DeAngelo, D.J.; Stone, R.M.; Sakamaki, H.; et al. Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia in First Complete Remission: Systematic Review and Meta-Analysis of Prospective Clinical Trials. JAMA 2009, 301, 2349–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magina, K.N.; Pregartner, G.; Zebisch, A.; Wolfler, A.; Neumeister, P.; Greinix, H.T.; Berghold, A.; Sill, H. Cytarabine Dose in the Consolidation Treatment of AML: A Systematic Review and Meta-Analysis. Blood 2017, 130, 946–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zebisch, A.; Hatzl, S.; Pichler, M.; Wolfler, A.; Sill, H. Therapeutic Resistance in Acute Myeloid Leukemia: The Role of Non-Coding RNAs. Int. J. Mol. Sci. 2016, 17, E2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliusson, G.; Lazarevic, V.; Horstedt, A.S.; Hagberg, O.; Hoglund, M.; Swedish Acute Leukemia Registry Group. Acute Myeloid Leukemia in the Real World: Why Population-Based Registries are Needed. Blood 2012, 119, 3890–3899. [Google Scholar] [CrossRef] [Green Version]
- Juliusson, G.; Antunovic, P.; Derolf, A.; Lehmann, S.; Mollgard, L.; Stockelberg, D.; Tidefelt, U.; Wahlin, A.; Hoglund, M. Age and Acute Myeloid Leukemia: Real World Data on Decision to Treat and Outcomes from the Swedish Acute Leukemia Registry. Blood 2009, 113, 4179–4187. [Google Scholar] [CrossRef] [Green Version]
- Bakker, E.; Qattan, M.; Mutti, L.; Demonacos, C.; Krstic-Demonacos, M. The Role of Microenvironment and Immunity in Drug Response in Leukemia. Biochim. Biophys. Acta 2016, 1863, 414–426. [Google Scholar] [CrossRef]
- Rashidi, A.; Uy, G.L. Targeting the Microenvironment in Acute Myeloid Leukemia. Curr. Hematol. Malign Rep. 2015, 10, 126–131. [Google Scholar] [CrossRef]
- Tabe, Y.; Konopleva, M. Role of Microenvironment in Resistance to Therapy in AML. Curr. Hematol. Malign Rep. 2015, 10, 96–103. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechman, E.R.; Gentner, B.; Ng, S.W.; Schoof, E.M.; van Galen, P.; Kennedy, J.A.; Nucera, S.; Ciceri, F.; Kaufmann, K.B.; Takayama, N.; et al. MiR-126 Regulates Distinct Self-Renewal Outcomes in Normal and Malignant Hematopoietic Stem Cells. Cancer Cell 2016, 29, 214–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of Therapeutic Targets for Quiescent, Chemotherapy-Resistant Human Leukemia Stem Cells. Sci. Transl. Med. 2010, 2, 17ra9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.; Majeti, R. Biology and Relevance of Human Acute Myeloid Leukemia Stem Cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, A.; Chan, S.M.; Thomas, D.; Majeti, R. Biology and Clinical Relevance of Acute Myeloid Leukemia Stem Cells. Semin. Hematol. 2015, 52, 150–164. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and Other Non-Coding RNAs as Targets for Anticancer Drug Development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Pichler, M.; Calin, G.A. MicroRNAs in Cancer: From Developmental Genes in Worms to their Clinical Application in Patients. Br. J. Cancer 2015, 113, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Zebisch, A.; Caraffini, V.; Sill, H. RAF Kinase Inhibitor Protein in Myeloid Leukemogenesis. Int. J. Mol. Sci. 2019, 5756. [Google Scholar] [CrossRef] [Green Version]
- Barth, D.A.; Slaby, O.; Klec, C.; Juracek, J.; Drula, R.; Calin, G.A.; Pichler, M. Current Concepts of Non-Coding RNAs in the Pathogenesis of Non-Clear Cell Renal Cell Carcinoma. Cancers 2019, 11, 1580. [Google Scholar] [CrossRef] [Green Version]
- Klec, C.; Prinz, F.; Pichler, M. Involvement of the Long Noncoding RNA NEAT1 in Carcinogenesis. Mol. Oncol. 2019, 13, 46–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolle, M.A.; Prinz, F.; Calin, G.A.; Pichler, M. Current Concepts of Non-Coding RNA Regulation of Immune Checkpoints in Cancer. Mol. Asp. Med. 2019, 70, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Vincent, K.; Pichler, M.; Fodde, R.; Berindan-Neagoe, I.; Slack, F.J.; Calin, G.A. Junk DNA and the Long Non-Coding RNA Twist in Cancer Genetics. Oncogene 2015, 34, 5003–5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.W.; Liu, N.; Chen, S.; Wang, Y.E.; Sun, K.L.; Xu, Z.M.; Fu, W.N. Upregulation of microRNA-23a Regulates Proliferation and Apoptosis by Targeting in Laryngeal Carcinoma. Oncol. Lett. 2015, 10, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, R.; Dubey, R.; Saini, N. Cooperative and Individualistic Functions of the microRNAs in the miR-23a~27a~24-2 Cluster and its Implication in Human Diseases. Mol. Cancer 2010, 9, 232. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.C.; Ye, H.; Zeng, Z.; Chin, Y.E.; Huang, Y.N.; Fu, G.H. The NF-kappaB p65/miR-23a-27a-24 Cluster is a Target for Leukemia Treatment. Oncotarget 2015, 6, 33554–33567. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Wang, S.; Zhao, Y.; Du, F.; Wang, W.; Lv, P.; Qi, L. Long Noncoding RNA NEAT1 Modulates Cell Proliferation and Apoptosis by Regulating miR-23a-3p/SMC1A in Acute Myeloid Leukemia. J. Cell. Physiol. 2019, 234, 6161–6172. [Google Scholar] [CrossRef]
- Hatzl, S.; Geiger, O.; Kuepper, M.K.; Caraffini, V.; Seime, T.; Furlan, T.; Nussbaumer, E.; Wieser, R.; Pichler, M.; Scheideler, M.; et al. Increased Expression of miR-23a Mediates a Loss of Expression in the RAF Kinase Inhibitor Protein RKIP. Cancer Res. 2016, 76, 3644–3654. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, X.; Liao, D.; Wang, X.; Wu, Z.; Nie, J.; Bai, M.; Fu, X.; Mei, Q.; Han, W. Elevated microRNA-23a Expression Enhances the Chemoresistance of Colorectal Cancer Cells with Microsatellite Instability to 5-Fluorouracil by Directly Targeting ABCF1. Curr. Protein Pept. Sci. 2015, 16, 301–309. [Google Scholar] [CrossRef]
- Peng, F.; Zhang, H.; Du, Y.; Tan, P. MiR-23a Promotes Cisplatin Chemoresistance and Protects Against Cisplatin-Induced Apoptosis in Tongue Squamous Cell Carcinoma Cells through Twist. Oncol. Rep. 2015, 33, 942–950. [Google Scholar] [CrossRef]
- Yu, Z.W.; Zhong, L.P.; Ji, T.; Zhang, P.; Chen, W.T.; Zhang, C.P. MicroRNAs Contribute to the Chemoresistance of Cisplatin in Tongue Squamous Cell Carcinoma Lines. Oral Oncol. 2010, 46, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Ichikawa, D.; Kawaguchi, T.; Takeshita, H.; Miyamae, M.; Ohashi, T.; Okajima, W.; Imamura, T.; Kiuchi, J.; Arita, T.; et al. Plasma microRNA Profiles: Identification of miR-23a as a Novel Biomarker for Chemoresistance in Esophageal Squamous Cell Carcinoma. Oncotarget 2016, 7, 62034–62048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, R.A.; Capizzi, R.L.; Rosner, G.L.; Oliver, L.K.; Smith, S.J.; Schiffer, C.A.; Silver, R.T.; Peterson, B.A.; Weiss, R.B.; Omura, G.A. Clinical Pharmacology of Cytarabine in Patients with Acute Myeloid Leukemia: A Cancer and Leukemia Group B Study. Cancer Chemother. Pharmacol. 1995, 36, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Sadovnik, I.; Eisenwort, G.; Bauer, K.; Herrmann, H.; Gleixner, K.V.; Schulenburg, A.; Rabitsch, W.; Sperr, W.R.; Wolf, D. Immunotherapy-Based Targeting and Elimination of Leukemic Stem Cells in AML and CML. Int. J. Mol. Sci. 2019, 20, 4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Dweep, H.; Gretz, N. MiRWalk2.0: A Comprehensive Atlas of microRNA-Target Interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem Cell Gene Expression Programs Influence Clinical Outcome in Human Leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Uuskula-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II Beta Interacts with Cohesin and CTCF at Topological Domain Borders. Genome Biol. 2016, 17, 182. [Google Scholar] [CrossRef] [Green Version]
- Jahid, S.; Sun, J.; Edwards, R.A.; Dizon, D.; Panarelli, N.C.; Milsom, J.W.; Sikandar, S.S.; Gumus, Z.H.; Lipkin, S.M. MiR-23a Promotes the Transition from Indolent to Invasive Colorectal Cancer. Cancer Discov. 2012, 2, 540–553. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Zhou, X.; Li, S.; Qin, Y.; Chen, Y.; Liu, H. Inhibition of miR-23a Increases the Sensitivity of Lung Cancer Stem Cells to Erlotinib through PTEN/PI3K/Akt Pathway. Oncol. Rep. 2017, 38, 3064–3070. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tan, H.Y.; Feng, Y.G.; Zhang, C.; Chen, F.; Feng, Y. MicroRNA-23a in Human Cancer: Its Roles, Mechanisms and Therapeutic Relevance. Cancers 2018, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombret, H.; Itzykson, R. How and when to Decide between Epigenetic Therapy and Chemotherapy in Patients with AML. Hematol. Am. Soc. Hematol. Educ. Program. 2017, 2017, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleyer, L.; Dohner, H.; Dombret, H.; Seymour, J.F.; Schuh, A.C.; Beach, C.L.; Swern, A.S.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; et al. Azacitidine for Front-Line Therapy of Patients with AML: Reproducible Efficacy Established by Direct Comparison of International Phase 3 Trial Data with Registry Data from the Austrian Azacitidine Registry of the AGMT Study Group. Int. J. Mol. Sci. 2017, 18, 415. [Google Scholar] [CrossRef]
- Pleyer, L.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; Sill, H.; Schlick, K.; Thaler, J.; Halter, B.; Machherndl-Spandl, S.; Zebisch, A.; et al. Azacitidine Front-Line in 339 Patients with Myelodysplastic Syndromes and Acute Myeloid Leukaemia: Comparison of French-American-British and World Health Organization Classifications. J. Hematol. Oncol. 2016, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.E.; Wong, J.J.; Rasko, J.E. MicroRNAs in Myeloid Malignancies. Br. J. Haematol. 2013, 162, 162–176. [Google Scholar] [CrossRef]
- Austin, C.A.; Lee, K.C.; Swan, R.L.; Khazeem, M.M.; Manville, C.M.; Cridland, P.; Treumann, A.; Porter, A.; Morris, N.J.; Cowell, I.G. TOP2B: The First Thirty Years. Int. J. Mol. Sci. 2018, 19, 2765. [Google Scholar] [CrossRef] [Green Version]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as Anticancer Targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Pourquier, P.; Takebayashi, Y.; Urasaki, Y.; Gioffre, C.; Kohlhagen, G.; Pommier, Y. Induction of Topoisomerase I Cleavage Complexes by 1-Beta -D-Arabinofuranosylcytosine (Ara-C) in Vitro and in Ara-C-Treated Cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1885–1890. [Google Scholar] [CrossRef] [Green Version]
- Song, J.H.; Kweon, S.H.; Kim, H.J.; Lee, T.H.; Min, W.S.; Kim, H.J.; Kim, Y.K.; Hwang, S.Y.; Kim, T.S. High TOP2B/TOP2A Expression Ratio at Diagnosis Correlates with Favourable Outcome for Standard Chemotherapy in Acute Myeloid Leukaemia. Br. J. Cancer 2012, 107, 108–115. [Google Scholar] [CrossRef]
- Caraffini, V.; Geiger, O.; Rosenberger, A.; Hatzl, S.; Perfler, B.; Berg, J.L.; Lim, C.; Strobl, H.; Kashofer, K.; Schauer, S.; et al. Loss of RAF Kinase Inhibitor Protein is Involved in Myelomonocytic Differentiation and Aggravates RAS-Driven Myeloid Leukemogenesis. Haematologica 2020, 105, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochazka, K.T.; Pregartner, G.; Rucker, F.G.; Heitzer, E.; Pabst, G.; Wolfler, A.; Zebisch, A.; Berghold, A.; Dohner, K.; Sill, H. Clinical Implications of Subclonal TP53 Mutations in Acute Myeloid Leukemia. Haematologica 2019, 104, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caraffini, V.; Perfler, B.; Berg, J.L.; Uhl, B.; Schauer, S.; Kashofer, K.; Ghaffari-Tabrizi-Wizsy, N.; Strobl, H.; Wolfler, A.; Hoefler, G.; et al. Loss of RKIP is a Frequent Event in Myeloid Sarcoma and Promotes Leukemic Tissue Infiltration. Blood 2018, 131, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Lal, R.; Lind, K.; Heitzer, E.; Ulz, P.; Aubell, K.; Kashofer, K.; Middeke, J.M.; Thiede, C.; Schulz, E.; Rosenberger, A.; et al. Somatic TP53 Mutations Characterize Preleukemic Stem Cells in Acute Myeloid Leukemia. Blood 2017, 129, 2587–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zebisch, A.; Lal, R.; Muller, M.; Lind, K.; Kashofer, K.; Girschikofsky, M.; Fuchs, D.; Wolfler, A.; Geigl, J.B.; Sill, H. Acute Myeloid Leukemia with TP53 Germ Line Mutations. Blood 2016, 128, 2270–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zebisch, A.; Cerroni, L.; Beham-Schmid, C.; Sill, H. Therapy-Related Leukemia Cutis: Case Study of an Aggressive Disorder. Ann. Hematol. 2003, 82, 705–707. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Milewska, M.; Romano, D.; Herrero, A.; Guerriero, M.L.; Birtwistle, M.; Quehenberger, F.; Hatzl, S.; Kholodenko, B.N.; Segatto, O.; Kolch, W.; et al. Mitogen-Inducible Gene-6 Mediates Feedback Inhibition from Mutated BRAF Towards the Epidermal Growth Factor Receptor and Thereby Limits Malignant Transformation. PLoS ONE 2015, 10, e0129859. [Google Scholar] [CrossRef]
- Zebisch, A.; Wolfler, A.; Fried, I.; Wolf, O.; Lind, K.; Bodner, C.; Haller, M.; Drasche, A.; Pirkebner, D.; Matallanas, D.; et al. Frequent Loss of RAF Kinase Inhibitor Protein Expression in Acute Myeloid Leukemia. Leukemia 2012, 26, 1842–1849. [Google Scholar] [CrossRef] [Green Version]
- Auner, H.W.; Zebisch, A.; Schimek, M.G.; Bodner, C.; Hiden, K.; Linkesch, W.; Haas, O.A.; Beham-Schmid, C.; Sill, H. High Expression of the Sister-Chromatid Separation Regulator and Proto-Oncogene hSecurin Occurs in a Subset of Myeloid Leukaemias but is Not Implicated in the Pathogenesis of Aneuploidy. Leukemia 2004, 18, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Zebisch, A.; Haller, M.; Hiden, K.; Goebel, T.; Hoefler, G.; Troppmair, J.; Sill, H. Loss of RAF Kinase Inhibitor Protein is a Somatic Event in the Pathogenesis of Therapy-Related Acute Myeloid Leukemias with C-RAF Germline Mutations. Leukemia 2009, 23, 1049–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zebisch, A.; Staber, P.B.; Delavar, A.; Bodner, C.; Hiden, K.; Fischereder, K.; Janakiraman, M.; Linkesch, W.; Auner, H.W.; Emberger, W.; et al. Two Transforming C-RAF Germ-Line Mutations Identified in Patients with Therapy-Related Acute Myeloid Leukemia. Cancer Res. 2006, 66, 3401–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruopp, M.D.; Perkins, N.J.; Whitcomb, B.W.; Schisterman, E.F. Youden Index and Optimal Cut-Point Estimated from Observations Affected by a Lower Limit of Detection. Biom. J. 2008, 50, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR-23a | ||||
|---|---|---|---|---|
| Parameter | Variable | Hazard Ratio | 95% CI | p-Value |
| OS | miR-23a high | 2.862 | 1.385–5.913 | 0.005 |
| WBC, G/L | 1.000 | 1.000–1.001 | 0.637 | |
| Age at diagnosis | 1.019 | 1.002–1.035 | 0.024 | |
| Cytogenetics | 1.418 | 1.013–1.984 | 0.042 | |
| EFS | miR-23a high | 1.854 | 1.023–3.363 | 0.042 |
| WBC, G/L | 1.000 | 1.000–1.001 | 0.279 | |
| Age at diagnosis | 1.011 | 0.996–1.025 | 0.144 | |
| Cytogenetics | 1.549 | 1.130–2.110 | 0.006 | |
| TOP2B | ||||
|---|---|---|---|---|
| Parameter | Variable | Hazard Ratio | 95% CI | p-Value |
| OS | TOP2B high | 0.581 | 0.367–0.922 | 0.021 |
| WBC, G/L | 1.000 | 1.000–1.001 | 0.092 | |
| Age at diagnosis | 1.031 | 1.015–1.047 | <0.001 | |
| Cytogenetics | 1.844 | 1.328–2.561 | <0.001 | |
| EFS | TOP2B high | 0.399 | 0.245–0.650 | <0.001 |
| WBC, G/L | 1.000 | 1.000–1.001 | 0.067 | |
| Age at diagnosis | 1.009 | 0.994–1.025 | 0.232 | |
| Cytogenetics | 1.513 | 1.056–2.168 | 0.024 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatzl, S.; Perfler, B.; Wurm, S.; Uhl, B.; Quehenberger, F.; Ebner, S.; Troppmair, J.; Reinisch, A.; Wölfler, A.; Sill, H.; et al. Increased Expression of Micro-RNA-23a Mediates Chemoresistance to Cytarabine in Acute Myeloid Leukemia. Cancers 2020, 12, 496. https://doi.org/10.3390/cancers12020496
Hatzl S, Perfler B, Wurm S, Uhl B, Quehenberger F, Ebner S, Troppmair J, Reinisch A, Wölfler A, Sill H, et al. Increased Expression of Micro-RNA-23a Mediates Chemoresistance to Cytarabine in Acute Myeloid Leukemia. Cancers. 2020; 12(2):496. https://doi.org/10.3390/cancers12020496
Chicago/Turabian StyleHatzl, Stefan, Bianca Perfler, Sonja Wurm, Barbara Uhl, Franz Quehenberger, Susanne Ebner, Jakob Troppmair, Andreas Reinisch, Albert Wölfler, Heinz Sill, and et al. 2020. "Increased Expression of Micro-RNA-23a Mediates Chemoresistance to Cytarabine in Acute Myeloid Leukemia" Cancers 12, no. 2: 496. https://doi.org/10.3390/cancers12020496