Are Glucosylceramide-Related Sphingolipids Involved in the Increased Risk for Cancer in Gaucher Disease Patients? Review and Hypotheses

 , , ,
, , ,
Abstract
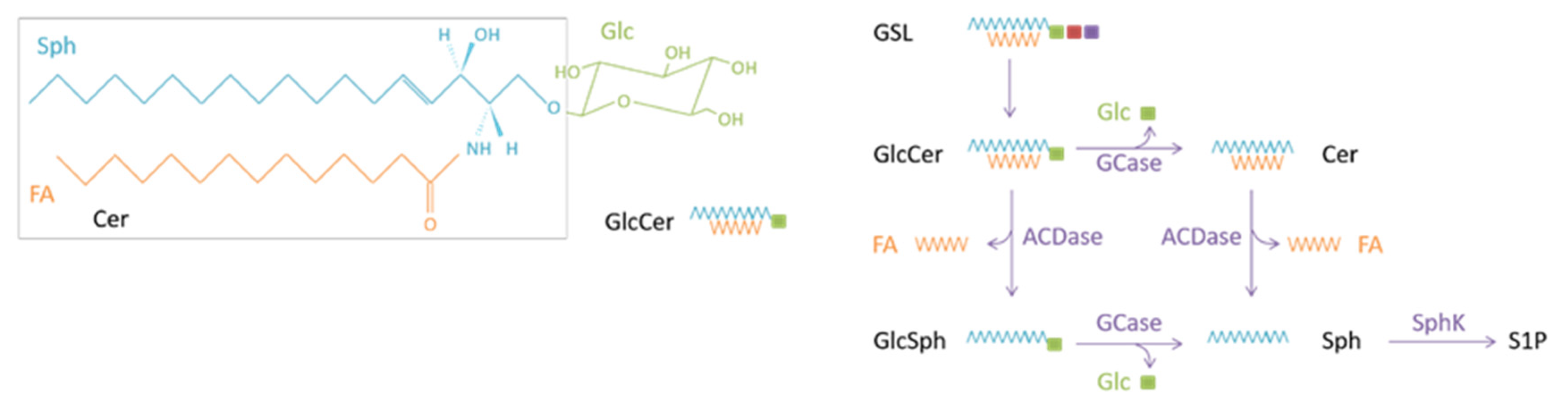
:1. Introduction: Glucosylceramide and Gaucher Disease
2. An increased Risk of Cancer in Patients with Gaucher Disease
3. What Could Be the Relationship between Glucosylceramide, Gaucher Disease and Cancer?
4. Increased Melanoma Growth in Gaucher Disease
5. Concluding Remarks and Outlook
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Hait, N.C.; Maiti, A. The Role of Sphingosine-1-Phosphate and Ceramide-1-Phosphate in Inflammation and Cancer. Mediat. Inflamm. 2017, 2017, 4806541. [Google Scholar] [CrossRef]
- Hakomori, S.-I.; Handa, K. GM3 and cancer. Glycoconj. J. 2015, 32, 1–8. [Google Scholar] [CrossRef]
- Groux-Degroote, S.; Rodríguez-Walker, M.; Dewald, J.H.; Daniotti, J.L.; Delannoy, P. Gangliosides in Cancer Cell Signaling. Prog. Mol. Biol. Transl. Sci. 2018, 156, 197–227. [Google Scholar]
- Hakomori, S. Structure and function of glycosphingolipids and sphingolipids: Recollections and future trends. Biochim. Biophys. Acta 2008, 1780, 325–346. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Alam, S.; Konantz, M.; Liang, C.-Y.; Kohler, R.S.; Everest-Dass, A.V.; Huang, Y.-L.; Rimmer, N.; Fedier, A.; Schötzau, A.; et al. Transition of Mesenchymal and Epithelial Cancer Cells Depends on α1-4 Galactosyltransferase-Mediated Glycosphingolipids. Cancer Res. 2018, 78, 2952–2965. [Google Scholar] [CrossRef] [Green Version]
- Levade, T.; Andrieu-Abadie, N.; Micheau, O.; Legembre, P.; Ségui, B. Sphingolipids modulate the epithelial-mesenchymal transition in cancer. Cell Death Discov. 2015, 1, 15001. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Lavie, Y.; Cao, H.; Bursten, S.L.; Giuliano, A.E.; Cabot, M.C. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J. Biol. Chem. 1996, 271, 19530–19536. [Google Scholar] [CrossRef] [Green Version]
- Lavie, Y.; Cao, H.T.; Volner, A.; Lucci, A.; Han, T.Y.; Geffen, V.; Giuliano, A.E.; Cabot, M.C. Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. J. Biol. Chem. 1997, 272, 1682–1687. [Google Scholar] [CrossRef] [Green Version]
- Morad, S.A.F.; Cabot, M.C. The Onus of Sphingolipid Enzymes in Cancer Drug Resistance. Adv. Cancer Res. 2018, 140, 235–263. [Google Scholar]
- Wegner, M.-S.; Gruber, L.; Mattjus, P.; Geisslinger, G.; Grösch, S. The UDP-glucose ceramide glycosyltransferase (UGCG) and the link to multidrug resistance protein 1 (MDR1). BMC Cancer 2018, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Astudillo, L.; Therville, N.; Colacios, C.; Ségui, B.; Andrieu-Abadie, N.; Levade, T. Glucosylceramidases and malignancies in mammals. Biochimie 2016, 125, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Kobayashi, S.; Hirabayashi, Y.; Murakami-Murofushi, K. Cholesterol glucosylation is catalyzed by transglucosylation reaction of β-glucosidase 1. Biochem. Biophys. Res. Commun. 2013, 441, 838–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderjagt, D.J.; Fry, D.E.; Glew, R.H. Human glucocerebrosidase catalyses transglucosylation between glucocerebroside and retinol. Biochem. J. 1994, 300, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, O.; Mansson, J.-E.; Hakänsson, G.; Svennerholm, L. The occurrence of psychosine and other glycolipids in spleen and liver from the three major types of Gaucher’s disease. Biochim. Biophys. Acta 1982, 712, 453–463. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Ranes, B.; Brignol, N.; Hamler, R.; Clark, S. The origins of glucosylsphingosine in Gaucher disease. Mol. Genet. Metab. 2013, 108, S40–S41. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Marques, A.R.A.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef] [Green Version]
- Mistry, P.K.; Batista, J.L.; Andersson, H.C.; Balwani, M.; Burrow, T.A.; Charrow, J.; Kaplan, P.; Khan, A.; Kishnani, P.S.; Kolodny, E.H.; et al. Transformation in pretreatment manifestations of Gaucher disease type 1 during two decades of alglucerase/imiglucerase enzyme replacement therapy in the International Collaborative Gaucher Group (ICGG) Gaucher Registry. Am. J. Hematol. 2017, 92, 929–939. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Belmatoug, N.; Di Rocco, M.; Fraga, C.; Giraldo, P.; Hughes, D.; Lukina, E.; Maison-Blanche, P.; Merkel, M.; Niederau, C.; Plӧckinger, U.; et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe. Eur. J. Intern. Med. 2017, 37, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Joshua, H.; Djaldetti, M.; Hazaz, B.; Pinkhas, J. Nonsecretory IgD-kappa multiple myeloma in a patient with Gaucher’s disease. Scand. J. Haematol. 1979, 22, 179–184. [Google Scholar] [CrossRef]
- Bruckstein, A.H.; Karanas, A.; Dire, J.J. Gaucher’s disease associated with Hodgkin’s disease. Am. J. Med. 1980, 68, 610–613. [Google Scholar] [CrossRef]
- Burstein, Y.; Rechavi, G.; Rausen, A.R.; Frisch, B.; Spirer, Z. Association of Gaucher’s disease and lymphoid malignancy in 2 children. Scand. J. Haematol. 1985, 35, 445–447. [Google Scholar] [CrossRef]
- Chang-Lo, M.; Yam, L.T.; Rubenstone, A.I.; Schwartz, S.O. Gaucher’s disease associated with chronic lymphocytic leukaemia, gout and carcinoma. J. Pathol. 1975, 116, 203–207. [Google Scholar] [CrossRef]
- Corbett, G.M.; Darbyshire, P.J.; Besley, G.T.; Parker, A.C. Adult Gaucher disease in association with acute leukaemia. Postgrad. Med. J. 1987, 63, 899–900. [Google Scholar] [CrossRef]
- Garfinkel, D.; Sidi, Y.; Ben-Bassat, M.; Salomon, F.; Hazaz, B.; Pinkhas, J. Coexistence of Gaucher’s disease and multiple myeloma. Arch. Intern. Med. 1982, 142, 2229–2230. [Google Scholar] [CrossRef]
- Lee, R.E. The pathology of Gaucher disease. Prog. Clin. Biol. Res. 1982, 95, 177–217. [Google Scholar]
- Marti, G.E.; Ryan, E.T.; Papadopoulos, N.M.; Filling-Katz, M.; Barton, N.; Fleischer, T.A.; Rick, M.; Gralnick, H.R. Polyclonal B-cell lymphocytosis and hypergammaglobulinemia in patients with Gaucher disease. Am. J. Hematol. 1988, 29, 189–194. [Google Scholar] [CrossRef]
- De Fost, M.; Out, T.A.; de Wilde, F.A.; Tjin, E.P.M.; Pals, S.T.; van Oers, M.H.J.; Boot, R.G.; Aerts, J.F.M.G.; Maas, M.; Vom Dahl, S.; et al. Immunoglobulin and free light chain abnormalities in Gaucher disease type I: Data from an adult cohort of 63 patients and review of the literature. Ann. Hematol. 2008, 87, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef]
- Shiran, A.; Brenner, B.; Laor, A.; Tatarsky, I. Increased risk of cancer in patients with Gaucher disease. Cancer 1993, 72, 219–224. [Google Scholar] [CrossRef]
- Arends, M.; van Dussen, L.; Biegstraaten, M.; Hollak, C.E.M. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br. J. Haematol. 2013, 161, 832–842. [Google Scholar] [CrossRef]
- Zimran, A.; Liphshitz, I.; Barchana, M.; Abrahamov, A.; Elstein, D. Incidence of malignancies among patients with type I Gaucher disease from a single referral clinic. Blood Cells Mol. Dis. 2005, 34, 197–200. [Google Scholar] [CrossRef]
- Rosenbloom, B.E.; Weinreb, N.J.; Zimran, A.; Kacena, K.A.; Charrow, J.; Ward, E. Gaucher disease and cancer incidence: A study from the Gaucher Registry. Blood 2005, 105, 4569–4572. [Google Scholar] [CrossRef]
- De Fost, M.; Vom Dahl, S.; Weverling, G.J.; Brill, N.; Brett, S.; Häussinger, D.; Hollak, C.E.M. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol. Dis. 2006, 36, 53–58. [Google Scholar] [CrossRef]
- Taddei, T.H.; Kacena, K.A.; Yang, M.; Yang, R.; Malhotra, A.; Boxer, M.; Aleck, K.A.; Rennert, G.; Pastores, G.M.; Mistry, P.K. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am. J. Hematol. 2009, 84, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, V.; Lischuk, A.; Haims, A.; Lackman, R.; Brooks, J.S.; Mankin, H.; Mistry, P.K. Case series and literature review of skeletal tumors and their incidence in the Gaucher disease population. Am. J. Hematol. 2016, 91, 736–741. [Google Scholar] [CrossRef] [Green Version]
- Landgren, O.; Turesson, I.; Gridley, G.; Caporaso, N.E. Risk of malignant disease among 1525 adult male US Veterans with Gaucher disease. Arch. Intern. Med. 2007, 167, 1189–1194. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, D.H.; Flaks-Manov, N.; Benis, A.; Gabay, H.; DiBonaventura, M.; Rosenbaum, H.; Joseph, A.; Bachrach, A.; Leventer-Roberts, M. Population-based cohort of 500 patients with Gaucher disease in Israel. BMJ Open 2019, 9, e024251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stirnemann, J.; (Service de Médecine Interne, Hôpitaux Universitaires de Genève, Genève, Switzerland). Personal communication, 2019.
- Stirnemann, J.; Vigan, M.; Hamroun, D.; Heraoui, D.; Rossi-Semerano, L.; Berger, M.G.; Rose, C.; Camou, F.; de Roux-Serratrice, C.; Grosbois, B.; et al. The French Gaucher’s disease registry: Clinical characteristics, complications and treatment of 562 patients. Orphanet J. Rare Dis. 2012, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.M.; Stein, P.; Mullaly, S.; Bar, M.; Jain, D.; Pastores, G.M.; Mistry, P.K. Expanding spectrum of the association between Type 1 Gaucher disease and cancers: A series of patients with up to 3 sequential cancers of multiple types--correlation with genotype and phenotype. Am. J. Hematol. 2010, 85, 340–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boven, L.A.; van Meurs, M.; Boot, R.G.; Mehta, A.; Boon, L.; Aerts, J.M.; Laman, J.D. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am. J. Clin. Pathol. 2004, 122, 359–369. [Google Scholar] [CrossRef]
- Allen, M.J.; Myer, B.J.; Khokher, A.M.; Rushton, N.; Cox, T.M. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: Increased release of interleukin-6 and interleukin-10. QJM 1997, 90, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelakakis, H.; Spanou, C.; Kondyli, A.; Dimitriou, E.; Van Weely, S.; Hollak, C.E.; Van Oers, M.H.; Aerts, J.M. Plasma tumor necrosis factor-a (TNF-a) levels in Gaucher disease. Biochim. Biophys. Acta 1996, 1317, 219–222. [Google Scholar] [CrossRef] [Green Version]
- Hollak, C.E.; Evers, L.; Aerts, J.M.; van Oers, M.H. Elevated levels of M-CSF, sCD14 and IL8 in type 1 Gaucher disease. Blood Cells Mol. Dis. 1997, 23, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Kitatani, K.; Wada, M.; Perry, D.; Usui, T.; Sun, Y.; Obeid, L.M.; Yaegashi, N.; Grabowski, G.A.; Hannun, Y.A. Activation of p38 Mitogen-Activated Protein Kinase in Gaucher’s Disease. PLoS ONE 2015, 10, e0136633. [Google Scholar] [CrossRef]
- Sun, Y.; Ran, H.; Liou, B.; Quinn, B.; Zamzow, M.; Zhang, W.; Bielawski, J.; Kitatani, K.; Setchell, K.D.R.; Hannun, Y.A.; et al. Isofagomine in vivo effects in a neuronopathic Gaucher disease mouse. PLoS ONE 2011, 6, e19037. [Google Scholar] [CrossRef]
- Kitatani, K.; Sheldon, K.; Rajagopalan, V.; Anelli, V.; Jenkins, R.W.; Sun, Y.; Grabowski, G.A.; Obeid, L.M.; Hannun, Y.A. Involvement of acid beta-glucosidase 1 in the salvage pathway of ceramide formation. J. Biol. Chem. 2009, 284, 12972–12978. [Google Scholar] [CrossRef] [Green Version]
- Kitatani, K.; Idkowiak-Baldys, J.; Bielawski, J.; Taha, T.A.; Jenkins, R.W.; Senkal, C.E.; Ogretmen, B.; Obeid, L.M.; Hannun, Y.A. Protein kinase C-induced activation of a ceramide/protein phosphatase 1 pathway leading to dephosphorylation of p38 MAPK. J. Biol. Chem. 2006, 281, 36793–36802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitatani, K.; Sheldon, K.; Anelli, V.; Jenkins, R.W.; Sun, Y.; Grabowski, G.A.; Obeid, L.M.; Hannun, Y.A. Acid beta-glucosidase 1 counteracts p38delta-dependent induction of interleukin-6: Possible role for ceramide as an anti-inflammatory lipid. J. Biol. Chem. 2009, 284, 12979–12988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, M.; Limgala, R.P.; Changsila, E.; Kamath, R.; Ioanou, C.; Goker-Alpan, O. Gaucheromas: When macrophages promote tumor formation and dissemination. Blood Cells Mol. Dis. 2018, 68, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Yeung, O.W.H.; Lo, C.-M.; Ling, C.-C.; Qi, X.; Geng, W.; Li, C.-X.; Ng, K.T.P.; Forbes, S.J.; Guan, X.-Y.; Poon, R.T.P.; et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef]
- Xu, Y.-H.; Quinn, B.; Witte, D.; Grabowski, G.A. Viable mouse models of acid beta-glucosidase deficiency: The defect in Gaucher disease. Am. J. Pathol. 2003, 163, 2093–2101. [Google Scholar] [CrossRef]
- Pandey, M.K.; Rani, R.; Zhang, W.; Setchell, K.; Grabowski, G.A. Immunological cell type characterization and Th1-Th17 cytokine production in a mouse model of Gaucher disease. Mol. Genet. Metab. 2012, 106, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Jabre, N.A.; Xu, Y.-H.; Zhang, W.; Setchell, K.D.R.; Grabowski, G.A. Gaucher disease: Chemotactic factors and immunological cell invasion in a mouse model. Mol. Genet. Metab. 2014, 111, 163–171. [Google Scholar] [CrossRef]
- Lukas, J.; Cozma, C.; Yang, F.; Kramp, G.; Meyer, A.; Neßlauer, A.-M.; Eichler, S.; Böttcher, T.; Witt, M.; Bräuer, A.; et al. Glucosylsphingosine Causes Hematological and Visceral Changes in Mice—Evidence for a Pathophysiological Role in Gaucher Disease. Int. J. Mol. Sci. 2017, 18, 2192. [Google Scholar] [CrossRef] [Green Version]
- Nagata, M.; Izumi, Y.; Ishikawa, E.; Kiyotake, R.; Doi, R.; Iwai, S.; Omahdi, Z.; Yamaji, T.; Miyamoto, T.; Bamba, T.; et al. Intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc. Natl. Acad. Sci. USA 2017, 114, E3285–E3294. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.; McEachern, K.A.; Chuang, W.-L.; Hutto, E.; Siegel, C.S.; Shayman, J.A.; Grabowski, G.A.; Scheule, R.K.; Copeland, D.P.; Cheng, S.H. Improved management of lysosomal glucosylceramide levels in a mouse model of type 1 Gaucher disease using enzyme and substrate reduction therapy. J. Inherit. Metab. Dis. 2010, 33, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Boddupalli, C.S.; Verma, R.; Liu, J.; Yang, R.; Pastores, G.M.; Mistry, P.K.; Dhodapkar, M.V. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood 2015, 125, 1256–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Sng, J.; Boddupalli, C.S.; Seckinger, A.; Chesi, M.; Fulciniti, M.; Zhang, L.; Rauniyar, N.; Lopez, M.; Neparidze, N.; et al. Antigen-mediated regulation in monoclonal gammopathies and myeloma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; Mckay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef]
- Mizukami, H.; Mi, Y.; Wada, R.; Kono, M.; Yamashita, T.; Liu, Y.; Werth, N.; Sandhoff, R.; Sandhoff, K.; Proia, R.L. Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J. Clin. Invest. 2002, 109, 1215–1221. [Google Scholar] [CrossRef]
- Pavlova, E.V.; Wang, S.Z.; Archer, J.; Dekker, N.; Aerts, J.M.F.G.; Karlsson, S.; Cox, T.M. B cell lymphoma and myeloma in murine Gaucher’s disease. J. Pathol. 2013, 231, 88–97. [Google Scholar] [CrossRef]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.-L.; Mehal, W.Z.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Halene, S.; Yang, M.; Iqbal, J.; Yang, R.; Mehal, W.Z.; Chuang, W.-L.; Jain, D.; Yuen, T.; Sun, L.; et al. Gaucher disease gene GBA functions in immune regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 10018–10023. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, E.V.; Archer, J.; Wang, S.; Dekker, N.; Aerts, J.M.; Karlsson, S.; Cox, T.M. Inhibition of UDP-glucosylceramide synthase in mice prevents Gaucher disease-associated B-cell malignancy. J. Pathol. 2015, 235, 113–124. [Google Scholar] [CrossRef]
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.-H.; et al. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.V.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatti, M.; Motta, M.; Di Bartolomeo, S.; Scarpa, S.; Cianfanelli, V.; Cecconi, F.; Salvioli, R. Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts. Hum. Mol. Genet. 2012, 21, 5159–5173. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Grönke, S.; Castillo-Quan, J.I.; Woodling, N.S.; Li, L.; Sirka, E.; Gegg, M.; Mills, K.; Hardy, J.; Bjedov, I.; et al. A Drosophila Model of Neuronopathic Gaucher Disease Demonstrates Lysosomal-Autophagic Defects and Altered mTOR Signalling and Is Functionally Rescued by Rapamycin. J. Neurosci. 2016, 36, 11654–11670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panicker, L.M.; Miller, D.; Park, T.S.; Patel, B.; Azevedo, J.L.; Awad, O.; Masood, M.A.; Veenstra, T.D.; Goldin, E.; Stubblefield, B.K.; et al. Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc. Natl. Acad. Sci. USA 2012, 109, 18054–18059. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.A.; Voit, A.; Srikanth, M.P.; Thayer, J.A.; Kingsbury, T.J.; Jacobson, M.A.; Lipinski, M.M.; Feldman, R.A.; Awad, O. mTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. Dis. Model Mech. 2019, 12, dmm038596. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Malta, C.D.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.J.; La Spada, A.R. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: Molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol. Dis. 2019, 122, 83–93. [Google Scholar] [CrossRef]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Miller, D.; Zeng, X.; Sgambato, J.A.; Lipinski, M.M.; Feldman, R.A. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 2015, 24, 5775–5788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [Green Version]
- Park, S.M.; Ou, J.; Chamberlain, L.; Simone, T.M.; Yang, H.; Virbasius, C.-M.; Ali, A.M.; Zhu, L.J.; Mukherjee, S.; Raza, A.; et al. U2AF35(S34F) Promotes Transformation by Directing Aberrant ATG7 Pre-mRNA 3′ End Formation. Mol. Cell 2016, 62, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kroemer, G. Defective Autophagy Initiates Malignant Transformation. Mol. Cell 2016, 62, 473–474. [Google Scholar] [CrossRef]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.-B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 2019, 10, 1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Pópulo, H.; Soares, P.; Faustino, A.; Rocha, A.S.; Silva, P.; Azevedo, F.; Lopes, J.M. mTOR pathway activation in cutaneous melanoma is associated with poorer prognosis characteristics. Pigment Cell Melanoma Res. 2011, 24, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Blanz, J.; Saftig, P. Parkinson’s disease: Acid-glucocerebrosidase activity and alpha-synuclein clearance. J. Neurochem. 2016, 139 (Suppl. 1), 198–215. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Barkhuizen, M.; Anderson, D.G.; Grobler, A.F. Advances in GBA-associated Parkinson’s disease—Pathology, presentation and therapies. Neurochem. Int. 2016, 93, 6–25. [Google Scholar] [CrossRef] [PubMed]
- Wahabi, K.; Perwez, A.; Rizvi, M.A. Parkin in Parkinson’s Disease and Cancer: A Double-Edged Sword. Mol. Neurobiol. 2018, 55, 6788–6800. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Li, X.; Jankovic, J. The association between Parkinson’s disease and melanoma. Int. J. Cancer 2011, 128, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Leyva, I.; Chi-Ahumada, E.; Mejía, M.; Castanedo-Cazares, J.P.; Eng, W.; Saikaly, S.K.; Carrizales, J.; Levine, T.D.; Norman, R.A.; Jimenez-Capdeville, M.E. The Presence of Alpha-Synuclein in Skin from Melanoma and Patients with Parkinson’s Disease. Mov. Disord. Clin. Pract. 2017, 4, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israeli, E.; Yakunin, E.; Zarbiv, Y.; Hacohen-Solovich, A.; Kisos, H.; Loeb, V.; Lichtenstein, M.; Ben-Gedalya, T.; Sabag, O.; Pikarsky, E.; et al. α-Synuclein Expression Selectively Affects Tumorigenesis in Mice Modeling Parkinson’s Disease. PLoS ONE 2011, 6, e19622. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, S.; Vijayan, B.; Kishore, A.; Thekkuveettil, A. Autophagy enhancement is rendered ineffective in presence of α-synuclein in melanoma cells. J. Cell Commun. Signal. 2017, 11, 381–394. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef] [Green Version]
- Maor, G.; Rapaport, D.; Horowitz, M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 2019, 28, 1768–1781. [Google Scholar] [CrossRef]
- Gatto, E.M.; Da Prat, G.; Etcheverry, J.L.; Drelichman, G.; Cesarini, M. Parkinsonisms and Glucocerebrosidase Deficiency: A Comprehensive Review for Molecular and Cellular Mechanism of Glucocerebrosidase Deficiency. Brain Sci. 2019, 9, 30. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, I.; Horowitz, M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 2005, 14, 2387–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, I.; Rapaport, D.; Horowitz, M. Interaction between parkin and mutant glucocerebrosidase variants: Apossible link between Parkinson disease and Gaucher disease. Hum. Mol. Genet. 2010, 19, 3771–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendikov-Bar, I.; Rapaport, D.; Larisch, S.; Horowitz, M. Parkin-mediated ubiquitination of mutant glucocerebrosidase leads to competition with its substrates PARIS and ARTS. Orphanet J. Rare Dis. 2014, 9, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Wang, H.-W.; Bowman, R.L.; Joyce, J.A. STAT3 and STAT6 Signaling Pathways Synergize to Promote Cathepsin Secretion from Macrophages via IRE1α Activation. Cell Rep. 2016, 16, 2914–2927. [Google Scholar] [CrossRef] [Green Version]
- Cabasso, O.; Paul, S.; Dorot, O.; Maor, G.; Krivoruk, O.; Pasmanik-Chor, M.; Mirzaian, M.; Ferraz, M.; Aerts, J.; Horowitz, M. Drosophila melanogaster Mutated in its GBA1b Ortholog Recapitulates Neuronopathic Gaucher Disease. J. Clin. Med. 2019, 8, 1420. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, W.; Xu, Y.-H.; Quinn, B.; Dasgupta, N.; Liou, B.; Setchell, K.D.R.; Grabowski, G.A. Substrate compositional variation with tissue/region and Gba1 mutations in mouse models--implications for Gaucher disease. PLoS ONE 2013, 8, e57560. [Google Scholar] [CrossRef] [Green Version]
- Chipeaux, C.; de Person, M.; Burguet, N.; Billette de Villemeur, T.; Rose, C.; Belmatoug, N.; Héron, S.; Le Van Kim, C.; Franco, M.; Moussa, F. Optimization of ultra-high pressure liquid chromatography—Tandem mass spectrometry determination in plasma and red blood cells of four sphingolipids and their evaluation as biomarker candidates of Gaucher’s disease. J. Chromatogr. A 2017, 1525, 116–125. [Google Scholar] [CrossRef]
- Dekker, N.; van Dussen, L.; Hollak, C.E.M.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.M.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astudillo, L. Rôle des sphingolipides dans la cancérogenèse: L’exemple de la maladie de Gaucher. Ph.D. Thesis, Université Toulouse III - Paul Sabatier, Toulouse, France, 2016. [Google Scholar]
- Xu, Y.-H.; Jia, L.; Quinn, B.; Zamzow, M.; Stringer, K.; Aronow, B.; Sun, Y.; Zhang, W.; Setchell, K.D.; Grabowski, G.A. Global gene expression profile progression in Gaucher disease mouse models. BMC Genom. 2011, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Barak, V.; Acker, M.; Nisman, B.; Kalickman, I.; Abrahamov, A.; Zimran, A.; Yatziv, S. Cytokines in Gaucher’s disease. Eur. Cytokine Netw. 1999, 10, 205–210. [Google Scholar] [PubMed]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Mistry, P.K.; Liu, J.; Sun, L.; Chuang, W.-L.; Yuen, T.; Yang, R.; Lu, P.; Zhang, K.; Li, J.; Keutzer, J.; et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. USA 2014, 111, 4934–4939. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Type of Population | Number of Patients | Age of Patients (Years) | Incidence and Type of Cancer, and Relative Risk for the Indicated Malignancy (in Brackets, 95% CI) | Overall Relative Risk for Cancer (in Brackets, 95% CI) | Reference |
|---|---|---|---|---|---|
| Ashkenazi Jewish (Israel) | 48 | 54 ± 20 | Patients with any cancer: 20.8% Mean age at cancer diagnosis: 57 ± 18 RR for hematologic cancers: 14.7 [5.2–41.7] | 3.6 [1.7–7.5] | Shiran et al. 1993 [33] |
| Caucasian, non-Jewish (22.7%) Ashkenazi Jewish (23.2%) Unreported (39.8%) | 2742 | 0–14: 25.6% 15–44: 47% 45–54: 12.2% 55–64: 7.5% >65: 7.7% | Patients with any cancer: 4.6% (9.1% in Ashkenazi Jewish) RR for myeloma: 5.9 [2.8–10.8] RR for hematologic cancers: 1.23 [0.73–1.90] | 0.79 [0.67–0.94] | Rosenbloom et al. 2005 [36] |
| Ashkenazi Jewish | 505 | Males: 38.7 ± 21 Females: 37 ± 21 | Patients with any cancer: 4.0% | Males: 0.6 [0.12–1.1] Females: 1.4 [0.7–2.2] | Zimran et al. 2005 [35] |
| Mostly non-Ashkenazi Jewish (Netherlands and Germany) | 131 | 50 ± 14 | Patients with any cancer: 10.7% Median age at cancer diagnosis: 52 RR for hematologic cancers: 12.7 [2.6–37.0] RR for myeloma: 51.1 [6.2–184] RR for liver carcinoma: 141.3 [17.1–510.5] | 2.5 [1.1–4.7] | de Fost et al. 2006 [37] |
| US male veterans (African Americans: 11.7%) | 1525 | African Americans: 46.0 Whites: 49.7 | Patients with any cancer: 9% RR for non-Hodgkin lymphoma: 2.54 [1.32–4.88] RR for melanoma: 3.07 [1.28–7.38] RR for pancreatic cancer: 2.37 [1.13–4.98] | 0.91 [0.76–1.08] | Landgren et al. 2007 [40] |
| Mostly Ashkenazi Jewish | 403 | 44 (for homozygous N370S patients) | Patients with any cancer: 12.5% RR for myeloma: 25 [9.17–54.4] RR for other hematologic cancers: 3.45 [1.49–6.79] RR for breast cancer: 1.84 [0.84–3.49] RR for melanoma: 2.26 [0.62–5.79] | 1.80 [1.32–2.40] | Taddei et al. 2009 [38] |
| Mostly Jewish (Israel) | 500 | 0–17: 9.4% 18–24: 4.4% 25–34: 11.4% 35–44: 20.6% 45–54: 12.4% 55–64: 16.0% 65–74: 14.6% >75: 11.2% | Patients with any cancer: 22.2% Patients with myeloma: 1% | ND | Jaffe et al. 2019 [41] |
| Caucasian (France) | 658 | 50.8 | Patients with any cancer: 4.1% Patients with hematologic cancers: 1.5% | ND | Stirnemann (unpublished) [42] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubot, P.; Astudillo, L.; Therville, N.; Sabourdy, F.; Stirnemann, J.; Levade, T.; Andrieu-Abadie, N. Are Glucosylceramide-Related Sphingolipids Involved in the Increased Risk for Cancer in Gaucher Disease Patients? Review and Hypotheses. Cancers 2020, 12, 475. https://doi.org/10.3390/cancers12020475
Dubot P, Astudillo L, Therville N, Sabourdy F, Stirnemann J, Levade T, Andrieu-Abadie N. Are Glucosylceramide-Related Sphingolipids Involved in the Increased Risk for Cancer in Gaucher Disease Patients? Review and Hypotheses. Cancers. 2020; 12(2):475. https://doi.org/10.3390/cancers12020475
Chicago/Turabian StyleDubot, Patricia, Leonardo Astudillo, Nicole Therville, Frédérique Sabourdy, Jérôme Stirnemann, Thierry Levade, and Nathalie Andrieu-Abadie. 2020. "Are Glucosylceramide-Related Sphingolipids Involved in the Increased Risk for Cancer in Gaucher Disease Patients? Review and Hypotheses" Cancers 12, no. 2: 475. https://doi.org/10.3390/cancers12020475