Epstein-Barr Virus-Associated Post-Transplant Lymphoproliferative Disorders after Hematopoietic Stem Cell Transplantation: Pathogenesis, Risk Factors and Clinical Outcomes


Abstract
:1. Introduction
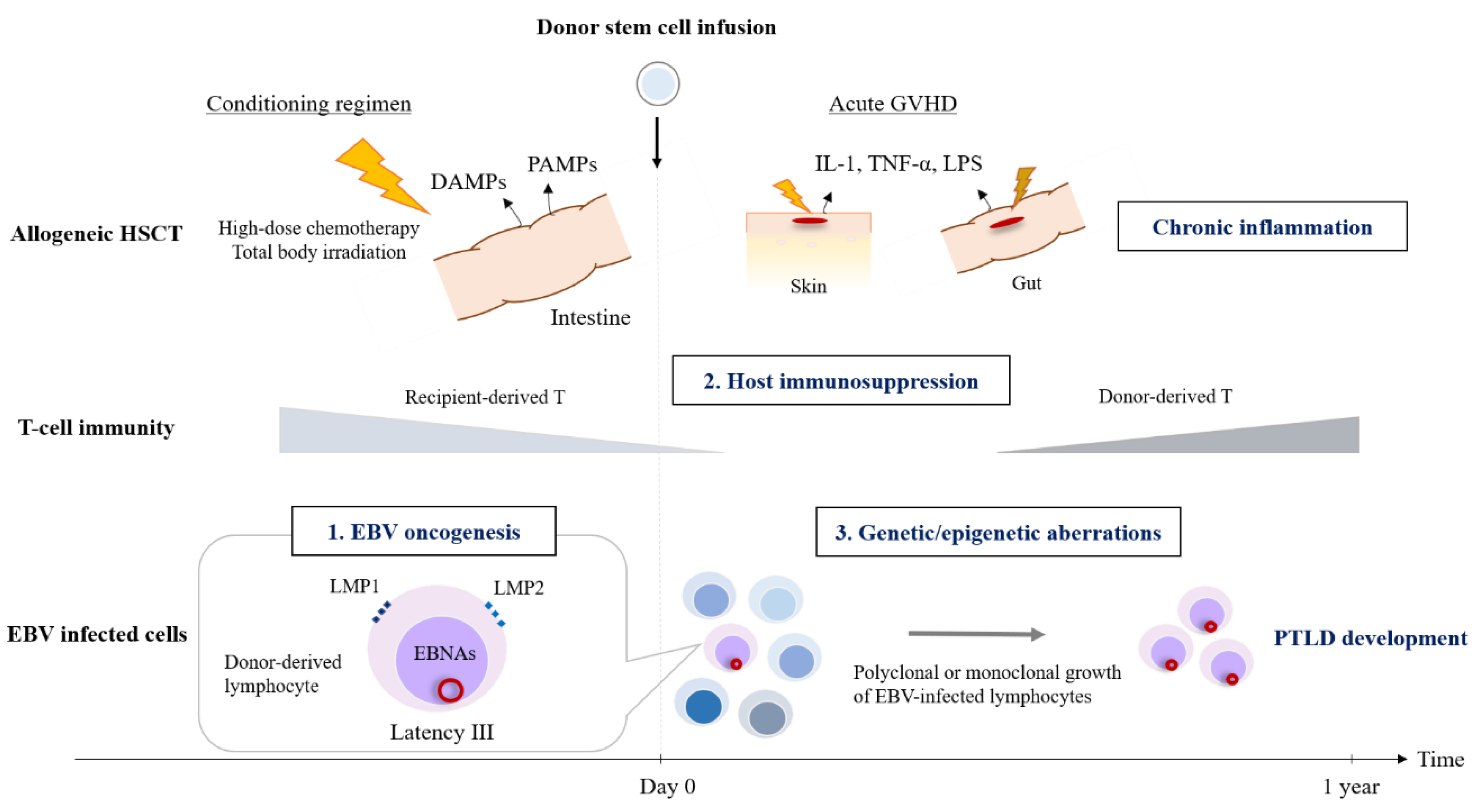
2. Pathogenesis
2.1. EBV Infection and Latent Status
2.2. EBV-Induced Oncogenesis
2.3. Hematopoietic Stem Cell Transplantation Setting
2.4. Genetic or Epigenetic Alternations
3. Epidemiology
4. Risk Factors
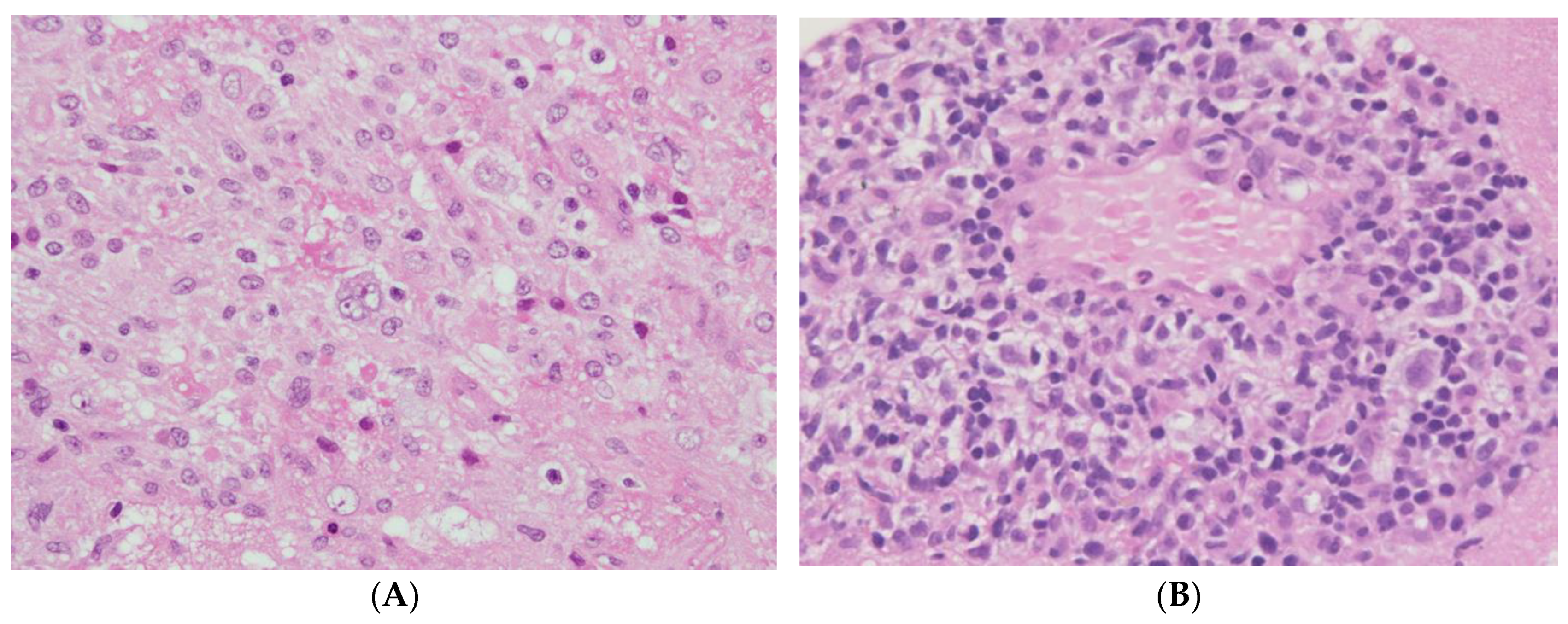
5. Clinical Presentation
6. Treatments
6.1. Propylaxis
6.2. Pre-Emptive Therapy
6.3. Targeted Therapy
6.3.1. Rituximab
6.3.2. Chemotherapy
6.3.3. Adoptive Immunotherapy
6.3.4. Possible Future Therapy
6.3.5. Reduction of Immunosuppression
6.3.6. Other Strategy
6.3.7. Management for Rare Cases
6.3.8. Treatment Response Evaluation
7. Prognosis
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cohen, J.I. Epstein-Barr virus infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Penn, I.; Hammond, W.; Brettschneider, L.; Starzl, T.E. Malignant lymphomas in transplantation patients. Transplant. Proc. 1969, 1, 106–112. [Google Scholar]
- Starzl, T.E.; Nalesnik, M.A.; Porter, K.A.; Ho, M.; Iwatsuki, S.; Griffith, B.P.; Rosenthal, J.T.; Hakala, T.R.; Shaw, B.W., Jr.; Hardesty, R.L.; et al. Reversibility of lymphomas and lymphoproliferative lesions developing under cyclosporin-steroid therapy. Lancet 1984, 1, 583–587. [Google Scholar] [CrossRef] [Green Version]
- Dierickx, D.; Tousseyn, T.; Sagaert, X.; Fieuws, S.; Wlodarska, I.; Morscio, J.; Brepoels, L.; Kuypers, D.; Vanhaecke, J.; Nevens, F.; et al. Single-center analysis of biopsy-confirmed posttransplant lymphoproliferative disorder: incidence, clinicopathological characteristics and prognostic factors. Leuk. Lymphoma 2013, 54, 2433–2440. [Google Scholar] [CrossRef]
- LaCasce, A.S. Post-transplant lymphoproliferative disorders. Oncologist 2006, 11, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Luskin, M.R.; Heil, D.S.; Tan, K.S.; Choi, S.; Stadtmauer, E.A.; Schuster, S.J.; Porter, D.L.; Vonderheide, R.H.; Bagg, A.; Heitjan, D.F.; et al. The Impact of EBV Status on Characteristics and Outcomes of Posttransplantation Lymphoproliferative Disorder. Am. J. Transplant. 2015, 15, 2665–2673. [Google Scholar] [CrossRef]
- Corcoran, L.M.; Tarlinton, D.M. Regulation of germinal center responses, memory B cells and plasma cell formation-an update. Curr. Opin. Immunol. 2016, 39, 59–67. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV the prototypical human tumor virus—Just how bad is it? J. Allergy Clin. Immunol. 2005, 116, 251–261. [Google Scholar] [CrossRef]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef]
- Morscio, J.; Tousseyn, T. Recent insights in the pathogenesis of post-transplantation lymphoproliferative disorders. World J. Transplant. 2016, 6, 505–516. [Google Scholar] [CrossRef]
- Murata, T.; Sato, Y.; Kimura, H. Modes of infection and oncogenesis by the Epstein-Barr virus. Rev. Med. Virol. 2014, 24, 242–253. [Google Scholar] [CrossRef]
- Middeldorp, J.M.; Pegtel, D.M. Multiple roles of LMP1 in Epstein-Barr virus induced immune escape. Semin. Cancer Biol. 2008, 18, 388–396. [Google Scholar] [CrossRef]
- Chen, M.R. Epstein-barr virus, the immune system, and associated diseases. Front. Microbiol. 2011, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Terrin, L.; Dolcetti, R.; Corradini, I.; Indraccolo, S.; Dal Col, J.; Bertorelle, R.; Bonaldi, L.; Esposito, G.; De Rossi, A. hTERT inhibits the Epstein-Barr virus lytic cycle and promotes the proliferation of primary B lymphocytes: Implications for EBV-driven lymphomagenesis. Int. J. Cancer 2007, 121, 576–587. [Google Scholar] [CrossRef] [Green Version]
- Terrin, L.; Dal Col, J.; Rampazzo, E.; Zancai, P.; Pedrotti, M.; Ammirabile, G.; Bergamin, S.; Rizzo, S.; Dolcetti, R.; De Rossi, A. Latent membrane protein 1 of Epstein-Barr virus activates the hTERT promoter and enhances telomerase activity in B lymphocytes. J. Virol. 2008, 82, 10175–10187. [Google Scholar] [CrossRef] [Green Version]
- Capello, D.; Rossi, D.; Gaidano, G. Post-transplant lymphoproliferative disorders: Molecular basis of disease histogenesis and pathogenesis. Hematol. Oncol. 2005, 23, 61–67. [Google Scholar] [CrossRef]
- Petrara, M.R.; Freguja, R.; Gianesin, K.; Zanchetta, M.; De Rossi, A. Epstein-Barr virus-driven lymphomagenesis in the context of human immunodeficiency virus type 1 infection. Front. Microbiol. 2013, 4, 311. [Google Scholar] [CrossRef] [Green Version]
- Shallis, R.M.; Terry, C.M.; Lim, S.H. Changes in intestinal microbiota and their effects on allogeneic stem cell transplantation. Am. J. Hematol. 2018, 93, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Rinaldi, A.; Kwee, I.; Poretti, G.; Mensah, A.; Pruneri, G.; Capello, D.; Rossi, D.; Zucca, E.; Ponzoni, M.; Catapano, C.; et al. Comparative genome-wide profiling of post-transplant lymphoproliferative disorders and diffuse large B-cell lymphomas. Br. J. Haematol. 2006, 134, 27–36. [Google Scholar] [CrossRef]
- Djokic, M.; Le Beau, M.M.; Swinnen, L.J.; Smith, S.M.; Rubin, C.M.; Anastasi, J.; Carlson, K.M. Post-transplant lymphoproliferative disorder subtypes correlate with different recurring chromosomal abnormalities. Genes Chromosomes Cancer 2006, 45, 313–318. [Google Scholar] [CrossRef]
- Poirel, H.A.; Bernheim, A.; Schneider, A.; Meddeb, M.; Choquet, S.; Leblond, V.; Charlotte, F.; Davi, F.; Canioni, D.; Macintyre, E.; et al. Characteristic pattern of chromosomal imbalances in posttransplantation lymphoproliferative disorders: Correlation with histopathological subcategories and EBV status. Transplantation 2005, 80, 176–184. [Google Scholar] [CrossRef]
- Rinaldi, A.; Capello, D.; Scandurra, M.; Greiner, T.C.; Chan, W.C.; Bhagat, G.; Rossi, D.; Morra, E.; Paulli, M.; Rambaldi, A.; et al. Single nucleotide polymorphism-arrays provide new insights in the pathogenesis of post-transplant diffuse large B-cell lymphoma. Br. J. Haematol. 2010, 149, 569–577. [Google Scholar] [CrossRef]
- Ferreiro, J.F.; Morscio, J.; Dierickx, D.; Vandenberghe, P.; Gheysens, O.; Verhoef, G.; Zamani, M.; Tousseyn, T.; Wlodarska, I. EBV-Positive and EBV-Negative Posttransplant Diffuse Large B Cell Lymphomas Have Distinct Genomic and Transcriptomic Features. Am. J. Transplant. 2016, 16, 414–425. [Google Scholar] [CrossRef]
- Menter, T.; Juskevicius, D.; Alikian, M.; Steiger, J.; Dirnhofer, S.; Tzankov, A.; Naresh, K.N. Mutational landscape of B-cell post-transplant lymphoproliferative disorders. Br. J. Haematol. 2017, 178, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Morscio, J.; Dierickx, D.; Ferreiro, J.F.; Herreman, A.; Van Loo, P.; Bittoun, E.; Verhoef, G.; Matthys, P.; Cools, J.; Wlodarska, I.; et al. Gene expression profiling reveals clear differences between EBV-positive and EBV-negative posttransplant lymphoproliferative disorders. Am. J. Transplant. 2013, 13, 1305–1316. [Google Scholar] [CrossRef]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [Green Version]
- Koon, H.B.; Ippolito, G.C.; Banham, A.H.; Tucker, P.W. FOXP1: A potential therapeutic target in cancer. Expert Opin. Ther. Targets 2007, 11, 955–965. [Google Scholar] [CrossRef]
- Bea, S.; Zettl, A.; Wright, G.; Salaverria, I.; Jehn, P.; Moreno, V.; Burek, C.; Ott, G.; Puig, X.; Yang, L.; et al. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood 2005, 106, 3183–3190. [Google Scholar] [CrossRef]
- Gascoyne, D.M.; Banham, A.H. The significance of FOXP1 in diffuse large B-cell lymphoma. Leuk. Lymphoma 2017, 58, 1037–1051. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Sandey, M.; DeInnocentes, P.; Bird, R.C. Tumor suppressor gene p16/INK4A/CDKN2A-dependent regulation into and out of the cell cycle in a spontaneous canine model of breast cancer. J. Cell. Biochem. 2013, 114, 1355–1363. [Google Scholar] [CrossRef]
- LaPak, K.M.; Burd, C.E. The molecular balancing act of p16(INK4a) in cancer and aging. Mol. Cancer Res. 2014, 12, 167–183. [Google Scholar] [CrossRef] [Green Version]
- Jardin, F.; Jais, J.P.; Molina, T.J.; Parmentier, F.; Picquenot, J.M.; Ruminy, P.; Tilly, H.; Bastard, C.; Salles, G.A.; Feugier, P.; et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: A GELA study. Blood 2010, 116, 1092–1104. [Google Scholar] [CrossRef] [Green Version]
- Leonard, S.; Wei, W.; Anderton, J.; Vockerodt, M.; Rowe, M.; Murray, P.G.; Woodman, C.B. Epigenetic and transcriptional changes which follow Epstein-Barr virus infection of germinal center B cells and their relevance to the pathogenesis of Hodgkin’s lymphoma. J. Virol. 2011, 85, 9568–9577. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, H.A.; Naresh, K.N. Posttransplant lymphoproliferative disorders. Adv. Hematol. 2012, 2012, 230173. [Google Scholar] [CrossRef]
- Manez, R.; Breinig, M.C.; Linden, P.; Wilson, J.; Torre-Cisneros, J.; Kusne, S.; Dummer, S.; Ho, M. Posttransplant lymphoproliferative disease in primary Epstein-Barr virus infection after liver transplantation: The role of cytomegalovirus disease. J. Infect. Dis. 1997, 176, 1462–1467. [Google Scholar] [CrossRef] [Green Version]
- Jox, A.; Rohen, C.; Belge, G.; Bartnitzke, S.; Pawlita, M.; Diehl, V.; Bullerdiek, J.; Wolf, J. Integration of Epstein-Barr virus in Burkitt’s lymphoma cells leads to a region of enhanced chromosome instability. Ann. Oncol. 1997, 8, S131–S135. [Google Scholar] [CrossRef]
- Ambinder, R.F. Gammaherpesviruses and “Hit-and-Run” oncogenesis. Ame. J. Pathol. 2000, 156, 1–3. [Google Scholar] [CrossRef]
- Doycheva, I.; Amer, S.; Watt, K.D. De Novo Malignancies After Transplantation: Risk and Surveillance Strategies. Med. Clin. North Am. 2016, 100, 551–567. [Google Scholar] [CrossRef]
- Cockfield, S.M. Identifying the patient at risk for post-transplant lymphoproliferative disorder. Transplant. Infect. Dis. 2001, 3, 70–78. [Google Scholar] [CrossRef]
- Opelz, G.; Dohler, B. Lymphomas after solid organ transplantation: A collaborative transplant study report. Am. J. Transplant. 2004, 4, 222–230. [Google Scholar] [CrossRef]
- Allen, U.D.; Preiksaitis, J.K. Post-transplant lymphoproliferative disorders, Epstein-Barr virus infection, and disease in solid organ transplantation: Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13652. [Google Scholar] [CrossRef]
- Romero, S.; Montoro, J.; Guinot, M.; Almenar, L.; Andreu, R.; Balaguer, A.; Beneyto, I.; Espi, J.; Gomez-Codina, J.; Iacoboni, G.; et al. Post-transplant lymphoproliferative disorders after solid organ and hematopoietic stem cell transplantation. Leuk. Lymphoma 2019, 60, 142–150. [Google Scholar] [CrossRef]
- Kinch, A.; Cavelier, L.; Bengtsson, M.; Baecklund, E.; Enblad, G.; Backlin, C.; Thunberg, U.; Sundstrom, C.; Pauksens, K. Donor or recipient origin of posttransplant lymphoproliferative disorders following solid organ transplantation. Am. J. Transplant. 2014, 14, 2838–2845. [Google Scholar] [CrossRef]
- Petit, B.; Le Meur, Y.; Jaccard, A.; Paraf, F.; Robert, C.L.; Bordessoule, D.; Labrousse, F.; Drouet, M. Influence of host-recipient origin on clinical aspects of posttransplantation lymphoproliferative disorders in kidney transplantation. Transplantation 2002, 73, 265–271. [Google Scholar] [CrossRef]
- Shapiro, R.S.; McClain, K.; Frizzera, G.; Gajl-Peczalska, K.J.; Kersey, J.H.; Blazar, B.R.; Arthur, D.C.; Patton, D.F.; Greenberg, J.S.; Burke, B.; et al. Epstein-Barr virus associated B cell lymphoproliferative disorders following bone marrow transplantation. Blood 1988, 71, 1234–1243. [Google Scholar] [CrossRef] [Green Version]
- Curtis, R.E.; Travis, L.B.; Rowlings, P.A.; Socie, G.; Kingma, D.W.; Banks, P.M.; Jaffe, E.S.; Sale, G.E.; Horowitz, M.M.; Witherspoon, R.P.; et al. Risk of lymphoproliferative disorders after bone marrow transplantation: A multi-institutional study. Blood 1999, 94, 2208–2216. [Google Scholar]
- Gross, T.G.; Steinbuch, M.; DeFor, T.; Shapiro, R.S.; McGlave, P.; Ramsay, N.K.; Wagner, J.E.; Filipovich, A.H. B cell lymphoproliferative disorders following hematopoietic stem cell transplantation: Risk factors, treatment and outcome. Bone Marrow Transplant. 1999, 23, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Sundin, M.; Le Blanc, K.; Ringden, O.; Barkholt, L.; Omazic, B.; Lergin, C.; Levitsky, V.; Remberger, M. The role of HLA mismatch, splenectomy and recipient Epstein-Barr virus seronegativity as risk factors in post-transplant lymphoproliferative disorder following allogeneic hematopoietic stem cell transplantation. Haematologica 2006, 91, 1059–1067. [Google Scholar]
- Ocheni, S.; Kroeger, N.; Zabelina, T.; Sobottka, I.; Ayuk, F.; Wolschke, C.; Muth, A.; Lellek, H.; Petersen, L.; Erttmann, R.; et al. EBV reactivation and post transplant lymphoproliferative disorders following allogeneic SCT. Bone Marrow Transplant. 2008, 42, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Buyck, H.C.; Ball, S.; Junagade, P.; Marsh, J.; Chakrabarti, S. Prior immunosuppressive therapy with antithymocyte globulin increases the risk of EBV-related lymphoproliferative disorder following allo-SCT for acquired aplastic anaemia. Bone Marrow Transplant. 2009, 43, 813–816. [Google Scholar] [CrossRef]
- Hou, H.A.; Yao, M.; Tang, J.L.; Chen, Y.K.; Ko, B.S.; Huang, S.Y.; Tien, H.F.; Chang, H.H.; Lu, M.Y.; Lin, T.T.; et al. Poor outcome in post transplant lymphoproliferative disorder with pulmonary involvement after allogeneic hematopoietic SCT: 13 years’ experience in a single institute. Bone Marrow Transplant. 2009, 43, 315–321. [Google Scholar] [CrossRef]
- Landgren, O.; Gilbert, E.S.; Rizzo, J.D.; Socie, G.; Banks, P.M.; Sobocinski, K.A.; Horowitz, M.M.; Jaffe, E.S.; Kingma, D.W.; Travis, L.B.; et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 4992–5001. [Google Scholar] [CrossRef] [Green Version]
- Johansson, J.E.; Remberger, M.; Lazarevic, V.; Hallbook, H.; Wahlin, A.; Kimby, E.; Juliusson, G.; Omar, H.; Hagglund, H. Allogeneic haematopoietic stem-cell transplantation with reduced intensity conditioning for advanced stage Hodgkin’s lymphoma in Sweden: High incidence of post transplant lymphoproliferative disorder. Bone Marrow Transplant. 2011, 46, 870–875. [Google Scholar] [CrossRef] [Green Version]
- Styczynski, J.; Gil, L.; Tridello, G.; Ljungman, P.; Donnelly, J.P.; van der Velden, W.; Omar, H.; Martino, R.; Halkes, C.; Faraci, M.; et al. Response to rituximab-based therapy and risk factor analysis in Epstein Barr Virus-related lymphoproliferative disorder after hematopoietic stem cell transplant in children and adults: A study from the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Clin. Infect. Dis. 2013, 57, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Uhlin, M.; Wikell, H.; Sundin, M.; Blennow, O.; Maeurer, M.; Ringden, O.; Winiarski, J.; Ljungman, P.; Remberger, M.; Mattsson, J. Risk factors for Epstein-Barr virus-related post-transplant lymphoproliferative disease after allogeneic hematopoietic stem cell transplantation. Haematologica 2014, 99, 346–352. [Google Scholar] [CrossRef]
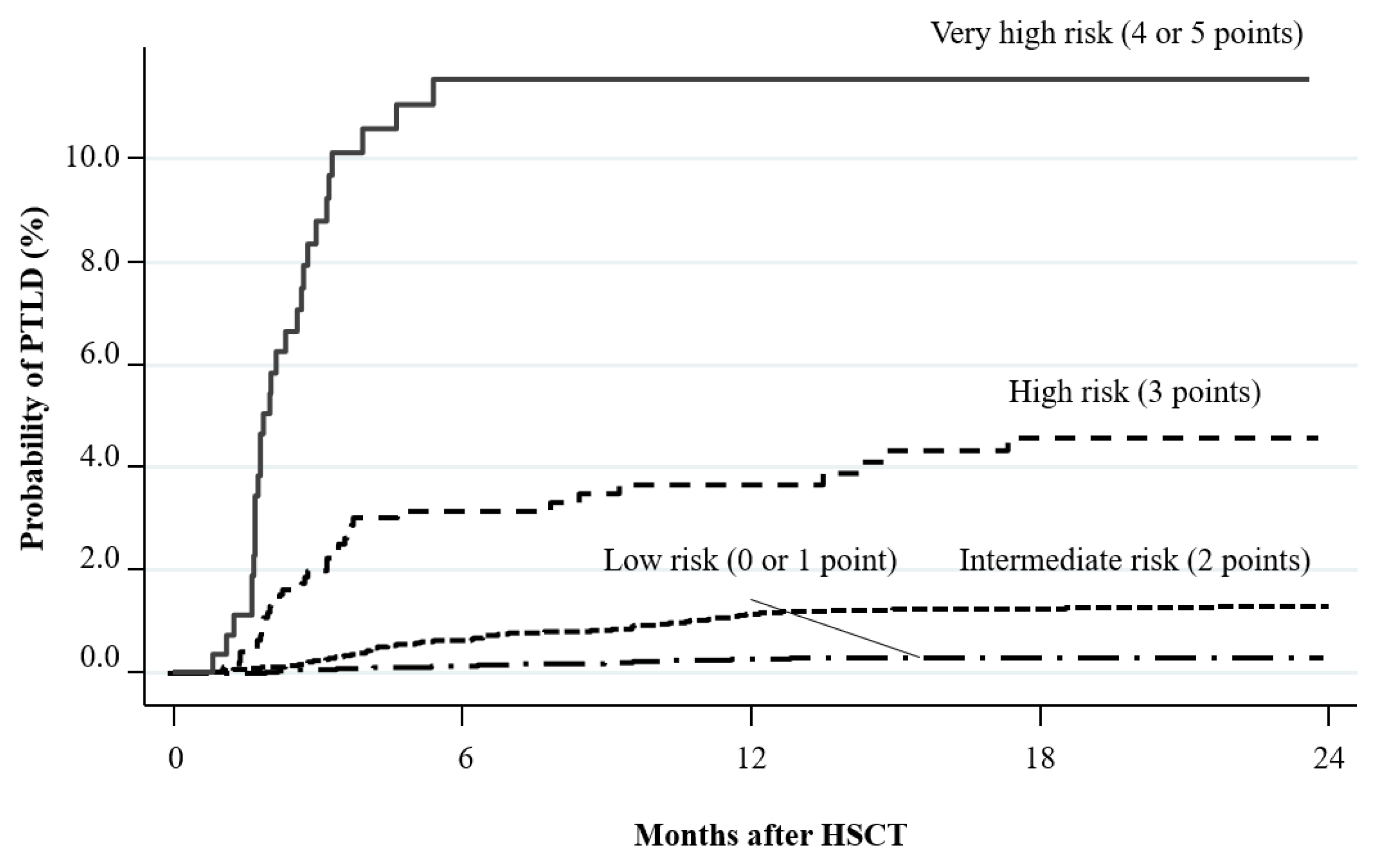
- Fujimoto, A.; Hiramoto, N.; Yamasaki, S.; Inamoto, Y.; Uchida, N.; Maeda, T.; Mori, T.; Kanda, Y.; Kondo, T.; Shiratori, S.; et al. Risk Factors and Predictive Scoring System For Post-Transplant Lymphoproliferative Disorder after Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2019, 25, 1441–1449. [Google Scholar] [CrossRef]
- Barker, J.N.; Martin, P.L.; Coad, J.E.; DeFor, T.; Trigg, M.E.; Kurtzberg, J.; Weisdorf, D.J.; Wagner, J. Low incidence of Epstein-Barr virus-associated posttransplantation lymphoproliferative disorders in 272 unrelated-donor umbilical cord blood transplant recipients. Biol Blood Marrow Transplant. 2001, 7, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Brunstein, C.G.; Weisdorf, D.J.; DeFor, T.; Barker, J.N.; Tolar, J.; van Burik, J.A.; Wagner, J.E. Marked increased risk of Epstein-Barr virus-related complications with the addition of antithymocyte globulin to a nonmyeloablative conditioning prior to unrelated umbilical cord blood transplantation. Blood 2006, 108, 2874–2880. [Google Scholar] [CrossRef]
- Dumas, P.Y.; Ruggeri, A.; Robin, M.; Crotta, A.; Abraham, J.; Forcade, E.; Bay, J.O.; Michallet, M.; Bertrand, Y.; Socie, G.; et al. Incidence and risk factors of EBV reactivation after unrelated cord blood transplantation: A Eurocord and Societe Francaise de Greffe de Moelle-Therapie Cellulaire collaborative study. Bone Marrow Transplant. 2013, 48, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Sanz, J.; Arango, M.; Senent, L.; Jarque, I.; Montesinos, P.; Sempere, A.; Lorenzo, I.; Martin, G.; Moscardo, F.; Mayordomo, E.; et al. EBV-associated post-transplant lymphoproliferative disorder after umbilical cord blood transplantation in adults with hematological diseases. Bone Marrow Transplant. 2014, 49, 397–402. [Google Scholar] [CrossRef]
- Ballen, K.K.; Cutler, C.; Yeap, B.Y.; McAfee, S.L.; Dey, B.R.; Attar, E.C.; Chen, Y.B.; Haspel, R.L.; Liney, D.; Koreth, J.; et al. Donor-derived second hematologic malignancies after cord blood transplantation. Biol Blood Marrow Transplant. 2010, 16, 1025–1031. [Google Scholar] [CrossRef] [Green Version]
- Zutter, M.M.; Martin, P.J.; Sale, G.E.; Shulman, H.M.; Fisher, L.; Thomas, E.D.; Durnam, D.M. Epstein-Barr virus lymphoproliferation after bone marrow transplantation. Blood 1988, 72, 520–529. [Google Scholar] [CrossRef]
- Reddiconto, G.; Chiusolo, P.; Fiorini, A.; Farina, G.; Laurenti, L.; Martini, M.; Marchetti, S.; Fadda, G.; Leone, G.; Sica, S. Assessment of cellular origin and EBV status in a PTLD after double cord blood transplantation. Leukemia 2007, 21, 2552–2554. [Google Scholar] [CrossRef] [Green Version]
- Dierickx, D.; Habermann, T.M. Post-Transplantation Lymphoproliferative Disorders in Adults. N. Engl. J. Med. 2018, 378, 549–562. [Google Scholar] [CrossRef]
- Scheinberg, P.; Nunez, O.; Weinstein, B.; Scheinberg, P.; Biancotto, A.; Wu, C.O.; Young, N.S. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N. Engl. J. Med. 2011, 365, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Hoegh-Petersen, M.; Goodyear, D.; Geddes, M.N.; Liu, S.; Ugarte-Torres, A.; Liu, Y.; Walker, J.T.; Fonseca, K.; Daly, A.; Duggan, P.; et al. High incidence of post transplant lymphoproliferative disorder after antithymocyte globulin-based conditioning and ineffective prediction by day 28 EBV-specific T lymphocyte counts. Bone Marrow Transplant. 2011, 46, 1104–1112. [Google Scholar] [CrossRef]
- Szabolcs, P.; Cairo, M.S. Unrelated umbilical cord blood transplantation and immune reconstitution. Semin. Hematol. 2010, 47, 22–36. [Google Scholar] [CrossRef] [Green Version]
- Kanakry, J.A.; Kasamon, Y.L.; Bolanos-Meade, J.; Borrello, I.M.; Brodsky, R.A.; Fuchs, E.J.; Ghosh, N.; Gladstone, D.E.; Gocke, C.D.; Huff, C.A.; et al. Absence of post-transplantation lymphoproliferative disorder after allogeneic blood or marrow transplantation using post-transplantation cyclophosphamide as graft-versus-host disease prophylaxis. Biol. Blood Marrow Transplant. 2013, 19, 1514–1517. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.P.; Zhang, C.L.; Mo, X.D.; Zhang, X.H.; Chen, H.; Han, W.; Chen, Y.H.; Wang, Y.; Yan, C.H.; Wang, J.Z.; et al. Epstein-Barr Virus-Related Post-Transplantation Lymphoproliferative Disorder after Unmanipulated Human Leukocyte Antigen Haploidentical Hematopoietic Stem Cell Transplantation: Incidence, Risk Factors, Treatment, and Clinical Outcomes. Biol. Blood Marrow Transplant. 2015, 21, 2185–2191. [Google Scholar] [CrossRef] [Green Version]
- Solomon, S.R.; Sizemore, C.A.; Sanacore, M.; Zhang, X.; Brown, S.; Holland, H.K.; Morris, L.E.; Bashey, A. Haploidentical transplantation using T cell replete peripheral blood stem cells and myeloablative conditioning in patients with high-risk hematologic malignancies who lack conventional donors is well tolerated and produces excellent relapse-free survival: Results of a prospective phase II trial. Biol. Blood Marrow Transplant. 2012, 18, 1859–1866. [Google Scholar] [CrossRef] [Green Version]
- Bashey, A.; Zhang, X.; Sizemore, C.A.; Manion, K.; Brown, S.; Holland, H.K.; Morris, L.E.; Solomon, S.R. T-cell-replete HLA-haploidentical hematopoietic transplantation for hematologic malignancies using post-transplantation cyclophosphamide results in outcomes equivalent to those of contemporaneous HLA-matched related and unrelated donor transplantation. J. Clin. Oncol. 2013, 31, 1310–1316. [Google Scholar] [CrossRef]
- Raiola, A.M.; Dominietto, A.; Ghiso, A.; Di Grazia, C.; Lamparelli, T.; Gualandi, F.; Bregante, S.; Van Lint, M.T.; Geroldi, S.; Luchetti, S.; et al. Unmanipulated haploidentical bone marrow transplantation and posttransplantation cyclophosphamide for hematologic malignancies after myeloablative conditioning. Biol. Blood Marrow Transplant. 2013, 19, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Retiere, C.; Willem, C.; Guillaume, T.; Vie, H.; Gautreau-Rolland, L.; Scotet, E.; Saulquin, X.; Gagne, K.; Bene, M.C.; Imbert, B.M.; et al. Impact on early outcomes and immune reconstitution of high-dose post-transplant cyclophosphamide vs anti-thymocyte globulin after reduced intensity conditioning peripheral blood stem cell allogeneic transplantation. Oncotarget 2018, 9, 11451–11464. [Google Scholar] [CrossRef] [Green Version]
- Walker, R.C.; Marshall, W.F.; Strickler, J.G.; Wiesner, R.H.; Velosa, J.A.; Habermann, T.M.; McGregor, C.G.; Paya, C.V. Pretransplantation assessment of the risk of lymphoproliferative disorder. Clin. Infect. Dis 1995, 20, 1346–1353. [Google Scholar] [CrossRef]
- Cohen, J.M.; Cooper, N.; Chakrabarti, S.; Thomson, K.; Samarasinghe, S.; Cubitt, D.; Lloyd, C.; Woolfrey, A.; Veys, P.; Amrolia, P.J. EBV-related disease following haematopoietic stem cell transplantation with reduced intensity conditioning. Leuk. Lymphoma 2007, 48, 256–269. [Google Scholar] [CrossRef]
- Ogonek, J.; Kralj Juric, M.; Ghimire, S.; Varanasi, P.R.; Holler, E.; Greinix, H.; Weissinger, E. Immune Reconstitution after Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 2016, 7, 507. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, J.L.; Cooke, K.R.; Teshima, T. The pathophysiology of acute graft-versus-host disease. Int. J. Hematol. 2003, 78, 181–187. [Google Scholar] [CrossRef]
- Zallio, F.; Primon, V.; Tamiazzo, S.; Pini, M.; Baraldi, A.; Corsetti, M.T.; Gotta, F.; Bertassello, C.; Salvi, F.; Rocchetti, A.; et al. Epstein-Barr virus reactivation in allogeneic stem cell transplantation is highly related to cytomegalovirus reactivation. Clin. Transplant. 2013, 27, E491–E497. [Google Scholar] [CrossRef]
- Swerdlow, S.H. T-cell and NK-cell posttransplantation lymphoproliferative disorders. Am. J. Clin. Pathol. 2007, 127, 887–895. [Google Scholar] [CrossRef]
- Leblond, V.; Davi, F.; Charlotte, F.; Dorent, R.; Bitker, M.O.; Sutton, L.; Gandjbakhch, I.; Binet, J.L.; Raphael, M. Posttransplant lymphoproliferative disorders not associated with Epstein-Barr virus: A distinct entity? J. Clin. Oncol. 1998, 16, 2052–2059. [Google Scholar] [CrossRef]
- Nelson, B.P.; Nalesnik, M.A.; Bahler, D.W.; Locker, J.; Fung, J.J.; Swerdlow, S.H. Epstein-Barr virus-negative post-transplant lymphoproliferative disorders: A distinct entity? Am. J. Surg. Pathol. 2000, 24, 375–385. [Google Scholar] [CrossRef]
- Naik, S.; Riches, M.; Hari, P.; Kim, S.; Chen, M.; Bachier, C.; Shaughnessy, P.; Hill, J.; Ljungman, P.; Battiwalla, M.; et al. Survival outcomes of allogeneic hematopoietic cell transplants with EBV-positive or EBV-negative post-transplant lymphoproliferative disorder, A CIBMTR study. Transplant. Infect. Dis. 2019, 21, e13145. [Google Scholar] [CrossRef]
- Reshef, R.; Morgans, A.K.; Pfanzelter, N.R.; Bloom, R.D.; Brozena, S.C.; Ahya, V.N.; Olthoff, K.M.; Tsai, D.E. EBV-Negative Post-Transplant Lymphoproliferative Disorder (PTLD): A Retrospective Case-Control Study of Clinical and Pathological Characteristics, Response to Treatment and Survival. Blood 2008, 112, 2823. [Google Scholar] [CrossRef]
- Fox, C.P.; Burns, D.; Parker, A.N.; Peggs, K.S.; Harvey, C.M.; Natarajan, S.; Marks, D.I.; Jackson, B.; Chakupurakal, G.; Dennis, M.; et al. EBV-associated post-transplant lymphoproliferative disorder following in vivo T-cell-depleted allogeneic transplantation: Clinical features, viral load correlates and prognostic factors in the rituximab era. Bone Marrow Transplant. 2014, 49, 280–286. [Google Scholar] [CrossRef]
- Gottschalk, S.; Rooney, C.M.; Heslop, H.E. Post-transplant lymphoproliferative disorders. Ann. Rev. Med. 2005, 56, 29–44. [Google Scholar] [CrossRef]
- Rasche, L.; Kapp, M.; Einsele, H.; Mielke, S. EBV-induced post transplant lymphoproliferative disorders: A persisting challenge in allogeneic hematopoetic SCT. Bone Marrow Transplant. 2014, 49, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Dierickx, D.; Tousseyn, T.; Requile, A.; Verscuren, R.; Sagaert, X.; Morscio, J.; Wlodarska, I.; Herreman, A.; Kuypers, D.; Van Cleemput, J.; et al. The accuracy of positron emission tomography in the detection of posttransplant lymphoproliferative disorder. Haematologica 2013, 98, 771–775. [Google Scholar] [CrossRef]
- Panagiotidis, E.; Quigley, A.M.; Pencharz, D.; Ardeshna, K.; Syed, R.; Sajjan, R.; Bomanji, J. (18)F-fluorodeoxyglucose positron emission tomography/computed tomography in diagnosis of post-transplant lymphoproliferative disorder. Leuke. Lymphoma 2014, 55, 515–519. [Google Scholar] [CrossRef]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.F.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J. Clin. Oncol. 2014, 32, 3059–3068. [Google Scholar] [CrossRef]
- Barrington, S.F.; Mikhaeel, N.G.; Kostakoglu, L.; Meignan, M.; Hutchings, M.; Mueller, S.P.; Schwartz, L.H.; Zucca, E.; Fisher, R.I.; Trotman, J.; et al. Role of imaging in the staging and response assessment of lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J. Clin. Oncol. 2014, 32, 3048–3058. [Google Scholar] [CrossRef]
- Styczynski, J.; van der Velden, W.; Fox, C.P.; Engelhard, D.; de la Camara, R.; Cordonnier, C.; Ljungman, P. Management of Epstein-Barr Virus infections and post-transplant lymphoproliferative disorders in patients after allogeneic hematopoietic stem cell transplantation: Sixth European Conference on Infections in Leukemia (ECIL-6) guidelines. Haematologica 2016, 101, 803–811. [Google Scholar] [CrossRef]
- van Esser, J.W.; Niesters, H.G.; van der Holt, B.; Meijer, E.; Osterhaus, A.D.; Gratama, J.W.; Verdonck, L.F.; Lowenberg, B.; Cornelissen, J.J. Prevention of Epstein-Barr virus-lymphoproliferative disease by molecular monitoring and preemptive rituximab in high-risk patients after allogeneic stem cell transplantation. Blood 2002, 99, 4364–4369. [Google Scholar] [CrossRef]
- Garcia-Cadenas, I.; Castillo, N.; Martino, R.; Barba, P.; Esquirol, A.; Novelli, S.; Orti, G.; Garrido, A.; Saavedra, S.; Moreno, C.; et al. Impact of Epstein Barr virus-related complications after high-risk allo-SCT in the era of pre-emptive rituximab. Bone Marrow Transplant. 2015, 50, 579–584. [Google Scholar] [CrossRef] [Green Version]
- van der Velden, W.J.; Mori, T.; Stevens, W.B.; de Haan, A.F.; Stelma, F.F.; Blijlevens, N.M.; Donnelly, J.P. Reduced PTLD-related mortality in patients experiencing EBV infection following allo-SCT after the introduction of a protocol incorporating pre-emptive rituximab. Bone Marrow Transplant. 2013, 48, 1465–1471. [Google Scholar] [CrossRef] [Green Version]
- Dominietto, A.; Tedone, E.; Soracco, M.; Bruno, B.; Raiola, A.M.; Van Lint, M.T.; Geroldi, S.; Lamparelli, T.; Galano, B.; Gualandi, F.; et al. In vivo B-cell depletion with rituximab for alternative donor hemopoietic SCT. Bone Marrow Transplant. 2012, 47, 101–106. [Google Scholar] [CrossRef]
- Liu, Q.; Xuan, L.; Liu, H.; Huang, F.; Zhou, H.; Fan, Z.; Zhao, K.; Wu, M.; Xu, L.; Zhai, X.; et al. Molecular monitoring and stepwise preemptive therapy for Epstein-Barr virus viremia after allogeneic stem cell transplantation. Am. J. Hematol. 2013, 88, 550–555. [Google Scholar] [CrossRef]
- McIver, Z.; Stephens, N.; Grim, A.; Barrett, A.J. Rituximab administration within 6 months of T cell-depleted allogeneic SCT is associated with prolonged life-threatening cytopenias. Biol. Blood Marrow Transplant. 2010, 16, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Petropoulou, A.D.; Porcher, R.; Peffault de Latour, R.; Xhaard, A.; Weisdorf, D.; Ribaud, P.; Rodriguez-Otero, P.; Agbalika, F.; Talbot, A.; Toubert, A.; et al. Increased infection rate after preemptive rituximab treatment for Epstein-Barr virus reactivation after allogeneic hematopoietic stem-cell transplantation. Transplantation 2012, 94, 879–883. [Google Scholar] [CrossRef]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.F.; Rochester, R.J.; et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010, 115, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Raberahona, M.; Wackenheim, C.; Germi, R.; Carre, M.; Bulabois, C.E.; Thiebaut, A.; Lupo, J.; Semenova, T.; Cahn, J.Y.; Morand, P.; et al. Dynamics of Epstein-Barr viral load after hematopoietic stem cell transplantation and effect of preemptive rituximab therapy. Transplant. Infect. Dis. 2016, 18, 889–895. [Google Scholar] [CrossRef]
- Delapierre, B.; Reman, O.; Dina, J.; Breuil, C.; Bellal, M.; Johnson-Ansah, H.; Gac, A.C.; Damaj, G.; Chantepie, S. Low dose Rituximab for pre-emptive treatment of Epstein Barr virus reactivation after allogenic hematopoietic stem cell transplantation. Curr. Res. Transl. Med. 2019, 67, 145–148. [Google Scholar] [CrossRef]
- Heslop, H.E. How I treat EBV lymphoproliferation. Blood 2009, 114, 4002–4008. [Google Scholar] [CrossRef] [Green Version]
- Al Hamed, R.; Bazarbachi, A.H.; Mohty, M. Epstein-Barr virus-related post-transplant lymphoproliferative disease (EBV-PTLD) in the setting of allogeneic stem cell transplantation: A comprehensive review from pathogenesis to forthcoming treatment modalities. Bone Marrow Transplant. 2019. [Google Scholar] [CrossRef]
- Styczynski, J.; Einsele, H.; Gil, L.; Ljungman, P. Outcome of treatment of Epstein-Barr virus-related post-transplant lymphoproliferative disorder in hematopoietic stem cell recipients: A comprehensive review of reported cases. Transplant. Infect. Dis. 2009, 11, 383–392. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Klein, A.K.; Ruthazer, R.; Evens, A.M. Hodgkin lymphoma post-transplant lymphoproliferative disorder: A comparative analysis of clinical characteristics, prognosis, and survival. Am. J. Hematol. 2016, 91, 560–565. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, A.S.; Ruthazer, R.; Paulus, J.K.; Kent, D.M.; Evens, A.M.; Klein, A.K. Survival Analyses and Prognosis of Plasma-Cell Myeloma and Plasmacytoma-Like Posttransplantation Lymphoproliferative Disorders. Clin. Lymphoma Myeloma leuk. 2016, 16, 684–692. [Google Scholar] [CrossRef] [Green Version]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef]
- Gustafsson, A.; Levitsky, V.; Zou, J.Z.; Frisan, T.; Dalianis, T.; Ljungman, P.; Ringden, O.; Winiarski, J.; Ernberg, I.; Masucci, M.G. Epstein-Barr virus (EBV) load in bone marrow transplant recipients at risk to develop posttransplant lymphoproliferative disease: Prophylactic infusion of EBV-specific cytotoxic T cells. Blood 2000, 95, 807–814. [Google Scholar] [CrossRef]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.K.; Sixbey, J.W.; Gan, Y.; Srivastava, D.K.; Bowman, L.C.; Krance, R.A.; Brenner, M.K.; et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood 1998, 92, 1549–1555. [Google Scholar] [CrossRef]
- Doubrovina, E.; Oflaz-Sozmen, B.; Prockop, S.E.; Kernan, N.A.; Abramson, S.; Teruya-Feldstein, J.; Hedvat, C.; Chou, J.F.; Heller, G.; Barker, J.N.; et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012, 119, 2644–2656. [Google Scholar] [CrossRef]
- Jiang, X.; Xu, L.; Zhang, Y.; Huang, F.; Liu, D.; Sun, J.; Song, C.; Liang, X.; Fan, Z.; Zhou, H.; et al. Rituximab-based treatments followed by adoptive cellular immunotherapy for biopsy-proven EBV-associated post-transplant lymphoproliferative disease in recipients of allogeneic hematopoietic stem cell transplantation. Oncoimmunology 2016, 5, e1139274. [Google Scholar] [CrossRef] [Green Version]
- Kazi, S.; Mathur, A.; Wilkie, G.; Cheal, K.; Battle, R.; McGowan, N.; Fraser, N.; Hargreaves, E.; Turner, D.; Campbell, J.D.M.; et al. Long-term follow up after third-party viral-specific cytotoxic lymphocytes for immunosuppression- and Epstein-Barr virus-associated lymphoproliferative disease. Haematologica 2019, 104, e356–e359. [Google Scholar] [CrossRef]
- O’Reilly, R.J.; Prockop, S.; Hasan, A.N.; Koehne, G.; Doubrovina, E. Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transplant. 2016, 51, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Morscio, J.; Finalet Ferreiro, J.; Vander Borght, S.; Bittoun, E.; Gheysens, O.; Dierickx, D.; Verhoef, G.; Wlodarska, I.; Tousseyn, T. Identification of distinct subgroups of EBV-positive post-transplant diffuse large B-cell lymphoma. Mod. Pathol. 2017, 30, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Granato, M.; Romeo, M.A.; Tiano, M.S.; Santarelli, R.; Gonnella, R.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Bortezomib promotes KHSV and EBV lytic cycle by activating JNK and autophagy. Sci. Rep. 2017, 7, 13052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.J.; Iempridee, T.; Wang, X.; Lee, H.C.; Mertz, J.E.; Kenney, S.C.; Lin, H.C.; Baladandayuthapani, V.; Dawson, C.W.; Shah, J.J.; et al. Lenalidomide, Thalidomide, and Pomalidomide Reactivate the Epstein-Barr Virus Lytic Cycle through Phosphoinositide 3-Kinase Signaling and Ikaros Expression. Clin. Cancer Res. 2016, 22, 4901–4912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, A.X.; McPherson, M.C.; Ivison, G.T.; Qu, X.; Rigdon, J.; Esquivel, C.O.; Krams, S.M.; Martinez, O.M. Dual blockade of the PI3K/Akt/mTOR pathway inhibits posttransplant Epstein-Barr virus B cell lymphomas and promotes allograft survival. Am. J. Transplant. 2019, 19, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Veloza, L.; Teixido, C.; Castrejon, N.; Climent, F.; Carrio, A.; Marginet, M.; Soldini, D.; Gonzalez-Farre, B.; Ribera-Cortada, I.; Lopez-Guillermo, A.; et al. Clinicopathological evaluation of the programmed cell death 1 (PD1)/programmed cell death-ligand 1 (PD-L1) axis in post-transplant lymphoproliferative disorders: Association with Epstein-Barr virus, PD-L1 copy number alterations, and outcome. Histopathology 2019, 75, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Schiefer, A.I.; Salzer, E.; Fureder, A.; Szepfalusi, Z.; Muller-Sacherer, T.; Huber, W.D.; Michel-Behnke, I.; Lawitschka, A.; Pichler, H.; Mann, G.; et al. PD-L1 and PD1 expression in post-transplantation lymphoproliferative disease (PTLD) of childhood and adolescence: An inter- and intra-individual descriptive study covering the whole spectrum of PTLD categories. Cancer Med. 2019, 8, 4656–4668. [Google Scholar] [CrossRef] [Green Version]
- Cesaro, S.; Pegoraro, A.; Tridello, G.; Calore, E.; Pillon, M.; Varotto, S.; Abate, D.; Barzon, L.; Mengoli, C.; Carli, M.; et al. A prospective study on modulation of immunosuppression for Epstein-Barr virus reactivation in pediatric patients who underwent unrelated hematopoietic stem-cell transplantation. Transplantation 2010, 89, 1533–1540. [Google Scholar] [CrossRef]
- Cesaro, S.; Murrone, A.; Mengoli, C.; Pillon, M.; Biasolo, M.A.; Calore, E.; Tridello, G.; Varotto, S.; Alaggio, R.; Zanesco, L.; et al. The real-time polymerase chain reaction-guided modulation of immunosuppression enables the pre-emptive management of Epstein-Barr virus reactivation after allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2005, 128, 224–233. [Google Scholar] [CrossRef]
- Oertel, S.H.; Riess, H. Antiviral treatment of Epstein-Barr virus-associated lymphoproliferations. Recent Results Cancer Res. 2002, 159, 89–95. [Google Scholar] [CrossRef]
- AlDabbagh, M.A.; Gitman, M.R.; Kumar, D.; Humar, A.; Rotstein, C.; Husain, S. The Role of Antiviral Prophylaxis for the Prevention of Epstein-Barr Virus-Associated Posttransplant Lymphoproliferative Disease in Solid Organ Transplant Recipients: A Systematic Review. Am. J. Transplant. 2017, 17, 770–781. [Google Scholar] [CrossRef]
- Lieberman, F.; Yazbeck, V.; Raptis, A.; Felgar, R.; Boyiadzis, M. Primary central nervous system post-transplant lymphoproliferative disorders following allogeneic hematopoietic stem cell transplantation. J. Neuro-Oncol. 2012, 107, 225–232. [Google Scholar] [CrossRef]
- Wroblewska, M.; Gil, L.A.; Komarnicki, M.A. Successful treatment of Epstein-Barr virus-related post-transplant lymphoproliferative disease with central nervous system involvement following allogeneic haematopoietic stem cell transplantation—A case study. Cent. Eur. J. Immunol. 2015, 40, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Dierickx, D.; Tousseyn, T.; Gheysens, O. How I treat posttransplant lymphoproliferative disorders. Blood 2015, 126, 2274–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evens, A.M.; David, K.A.; Helenowski, I.; Nelson, B.; Kaufman, D.; Kircher, S.M.; Gimelfarb, A.; Hattersley, E.; Mauro, L.A.; Jovanovic, B.; et al. Multicenter analysis of 80 solid organ transplantation recipients with post-transplantation lymphoproliferative disease: Outcomes and prognostic factors in the modern era. J. Clin. Oncol. 2010, 28, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Trappe, R.U.; Choquet, S.; Dierickx, D.; Mollee, P.; Zaucha, J.M.; Dreyling, M.H.; Duhrsen, U.; Tarella, C.; Shpilberg, O.; Sender, M.; et al. International prognostic index, type of transplant and response to rituximab are key parameters to tailor treatment in adults with CD20-positive B cell PTLD: Clues from the PTLD-1 trial. Am. J. Transplant. 2015, 15, 1091–1100. [Google Scholar] [CrossRef]
- Dierickx, D.; Tousseyn, T.; Morscio, J.; Fieuws, S.; Verhoef, G. Validation of prognostic scores in post-transplantation lymphoproliferative disorders. J. Clin. Oncol. 2013, 31, 3443–3444. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| 1. | Non-destructive PTLDs | EBV status | ||
| 1.1. | Plasmacytic hyperplasia | Almost 100% positive | ||
| 1.2. | Infectious mononucleosis | |||
| 1.3. | Florid follicular hyperplasia | |||
| 2. | Polymorphic PTLD | >90% positive | ||
| 3. | Monomorphic PTLDs | |||
| 3.1. | B-cell neoplasms | Both EBV-positive and EBV-negative types exist (EBV-negative in 10–48% of cases, particularly T-cell lymphoma) | ||
| Diffuse large B-cell lymphoma | ||||
| Burkitt lymphoma | ||||
| Plasma cell myeloma | ||||
| Plasmacytoma | ||||
| Other | ||||
| 3.2. | T-cell neoplasms | |||
| Peripheral T-cell lymphoma, NOS | ||||
| Hepatosplenic T-cell lymphoma | ||||
| Other | ||||
| 4. | Classical Hodgkin lymphoma PTLD | >90% positive | ||
| Latency | EBV Proteins | Function of the Proteins | B-Cell Normal Counterpart | Post-Transplant Disease |
|---|---|---|---|---|
| III (growth) | EBER 1–2, EBNA-LP, EBNA 1–2, EBNA 3A–C, LMP 1, LMP 2A–B | Activate B-cells and promote growth and transformation of naïve B-cells | Activated B-lymphoblast | PTLD |
| II (default) | EBER 1–2, EBNA 1, LMP 1–2A | activate B-cells and differentiate naïve B-cells into memory B-cells through germinal center | Germinal center B-cell | (PTLD); Classical Hodgkin lymphoma; T/NK cell lymphoma |
| I (EBNA1 only) | EBER 1–2, EBNA 1 | EBV genomic replication | Dividing memory B-cell | Burkitt lymphoma; Plasmablastic lymphoma |
| 0 (latency) | EBER 1–2 | Lifetime persistence of infection | Resting memory B-cell | Healthy carrier |
| Variable | HSCT | SOT | ||
|---|---|---|---|---|
| Typical cell of origin | Donor origin | Recipient origin | ||
| Frequency | Cord blood | 2.0–4.5% | Multi-visceral, small intestine | >20% |
| Lung | 3–10% | |||
| Bone marrow or peripheral blood | 0.8–4.0% | Heart | 2–8% | |
| Kidney, pancreas, or liver | 1–2% | |||
| Onset time | 6–12 months | 4–5.3 year | ||
| Variable | Category | Risk Factor | References |
|---|---|---|---|
| Established risk factors | |||
| T-cell depletion strategy | In vivo | [51,58,62,72] | |
| Ex vivo | [49,50,58] | ||
| Donor | Unrelated BM/PBSC | [58,60] | |
| HLA-mismatched BM/PBSC | [58,60,61] | ||
| Cord blood | [62] | ||
| Other risk factors | |||
| 1. Patient baseline | Disease | Aplastic anemia, primary immunodeficiency, chronic myeloid leukemia, advanced Hodgkin’s lymphoma | [53,62,70] |
| Age | >50 years old | [58] | |
| Past medial history | Splenectomy | [61] | |
| Number of allogeneic HSCT | Two times or more | [62] | |
| EBV serological mismatch | EBV-negative recipient and EBV-positive donor | [54,61] | |
| 2. Factors before HSCT | Conditioning regimen | Reduced intensity conditioning | [61,79] |
| 3. Factors after HSCT | Acute GVHD development | Grade II–IV | [58,61,62] |
| MSC use | [61] | ||
| CMV reactivation | [80] |
| Category | Risk Factor | Landgren, et al. [58] (CIBMTR/FHCRC) | Uhlin, et al. [61] (Karolinska Univ.) | Fujimoto, et al. [62] (JSHCT Database) |
|---|---|---|---|---|
| T-cell depletion | Selective T-cell depletion | ● | ||
| ATG use | GVHD prophylaxis | ● * | ● | ● † |
| GVHD treatment | ||||
| Donor | HLA mismatch | ● | ● | ● ‡ |
| Unrelated | ||||
| Age | 50 years or older | ● | ||
| EBV status | Recipient −/donor + | ● | ||
| Conditioning regimen | Reduced intensity | ● | ||
| Acute GVHD II-IV | ● | |||
| Splenectomy | ● | |||
| MSC treatment | ● | |||
| Disease | Aplastic anemia | ● |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimoto, A.; Suzuki, R. Epstein-Barr Virus-Associated Post-Transplant Lymphoproliferative Disorders after Hematopoietic Stem Cell Transplantation: Pathogenesis, Risk Factors and Clinical Outcomes. Cancers 2020, 12, 328. https://doi.org/10.3390/cancers12020328
Fujimoto A, Suzuki R. Epstein-Barr Virus-Associated Post-Transplant Lymphoproliferative Disorders after Hematopoietic Stem Cell Transplantation: Pathogenesis, Risk Factors and Clinical Outcomes. Cancers. 2020; 12(2):328. https://doi.org/10.3390/cancers12020328
Chicago/Turabian StyleFujimoto, Ayumi, and Ritsuro Suzuki. 2020. "Epstein-Barr Virus-Associated Post-Transplant Lymphoproliferative Disorders after Hematopoietic Stem Cell Transplantation: Pathogenesis, Risk Factors and Clinical Outcomes" Cancers 12, no. 2: 328. https://doi.org/10.3390/cancers12020328