Mutation Enrichment and Transcriptomic Activation Signatures of 419 Molecular Pathways in Cancer

 ,
,
Abstract
:1. Introduction
2. Results
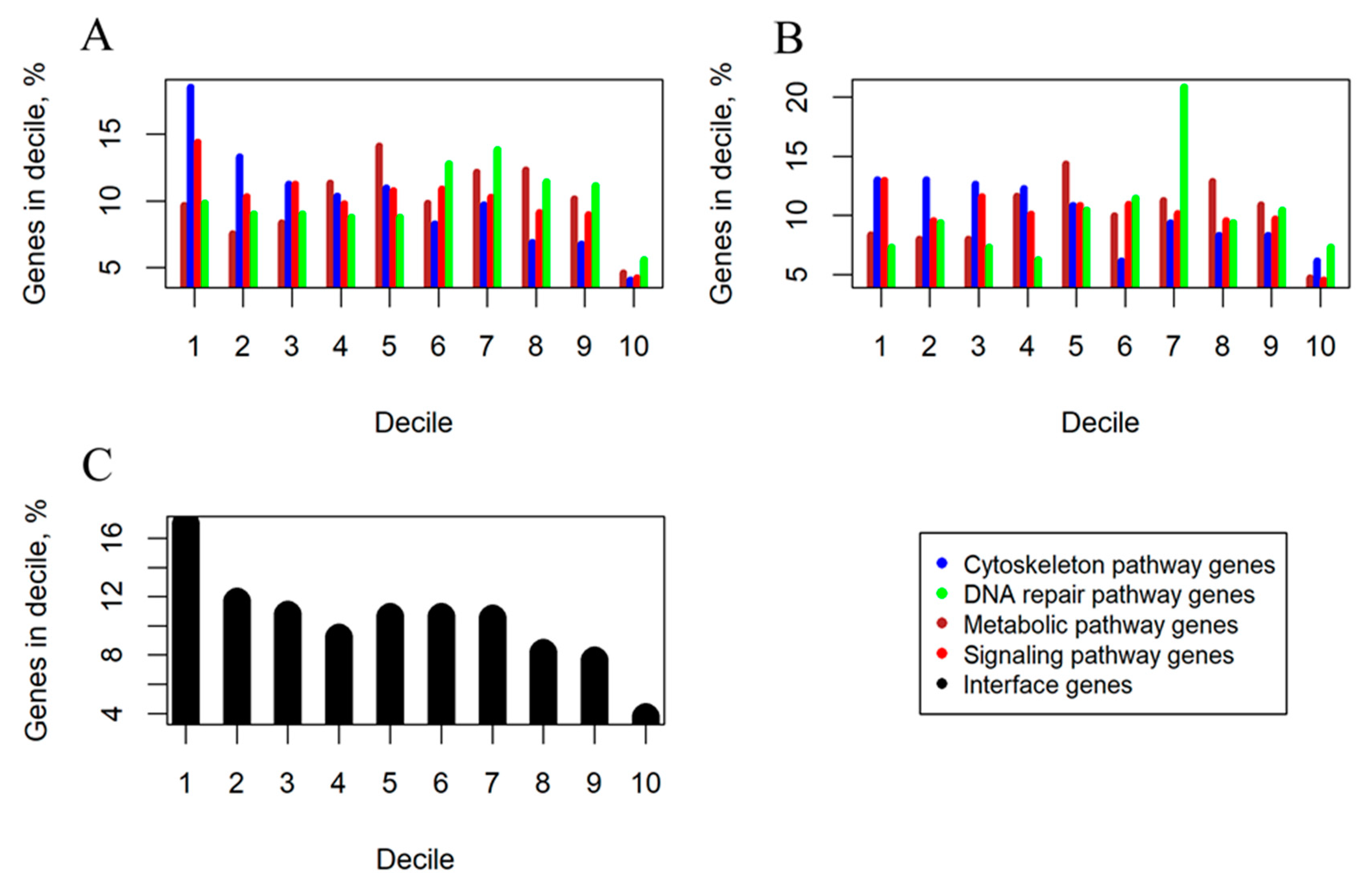
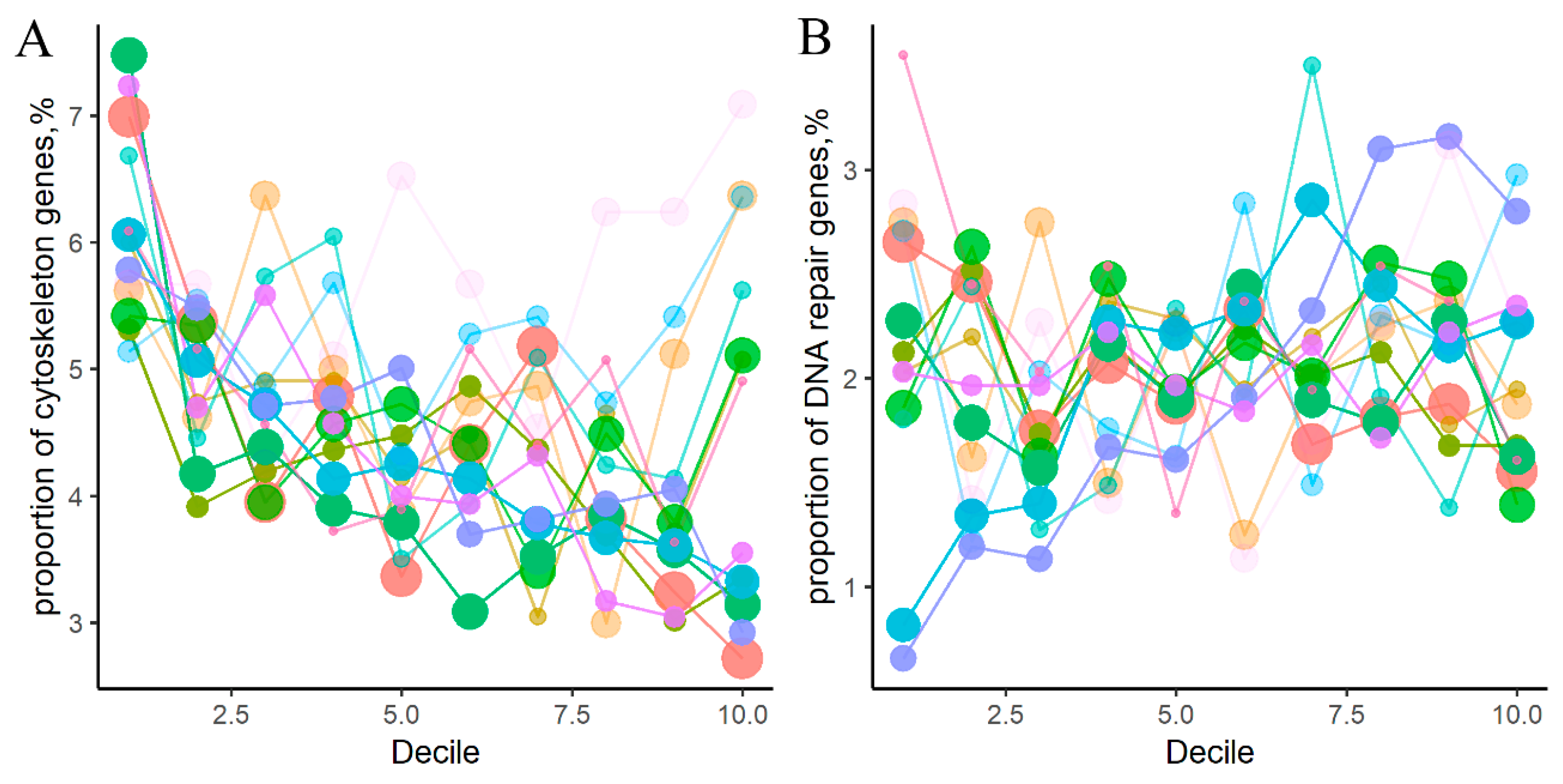
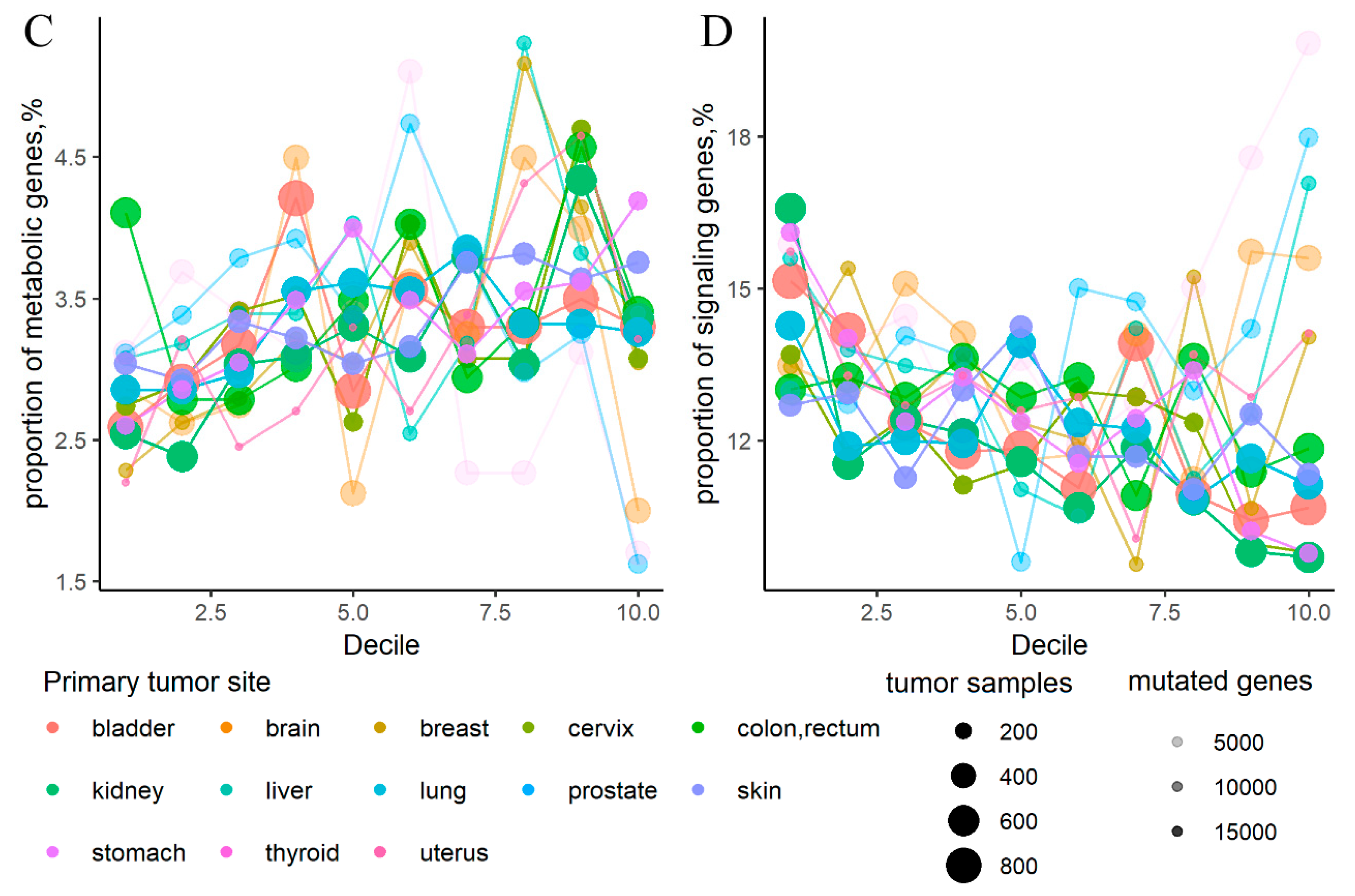
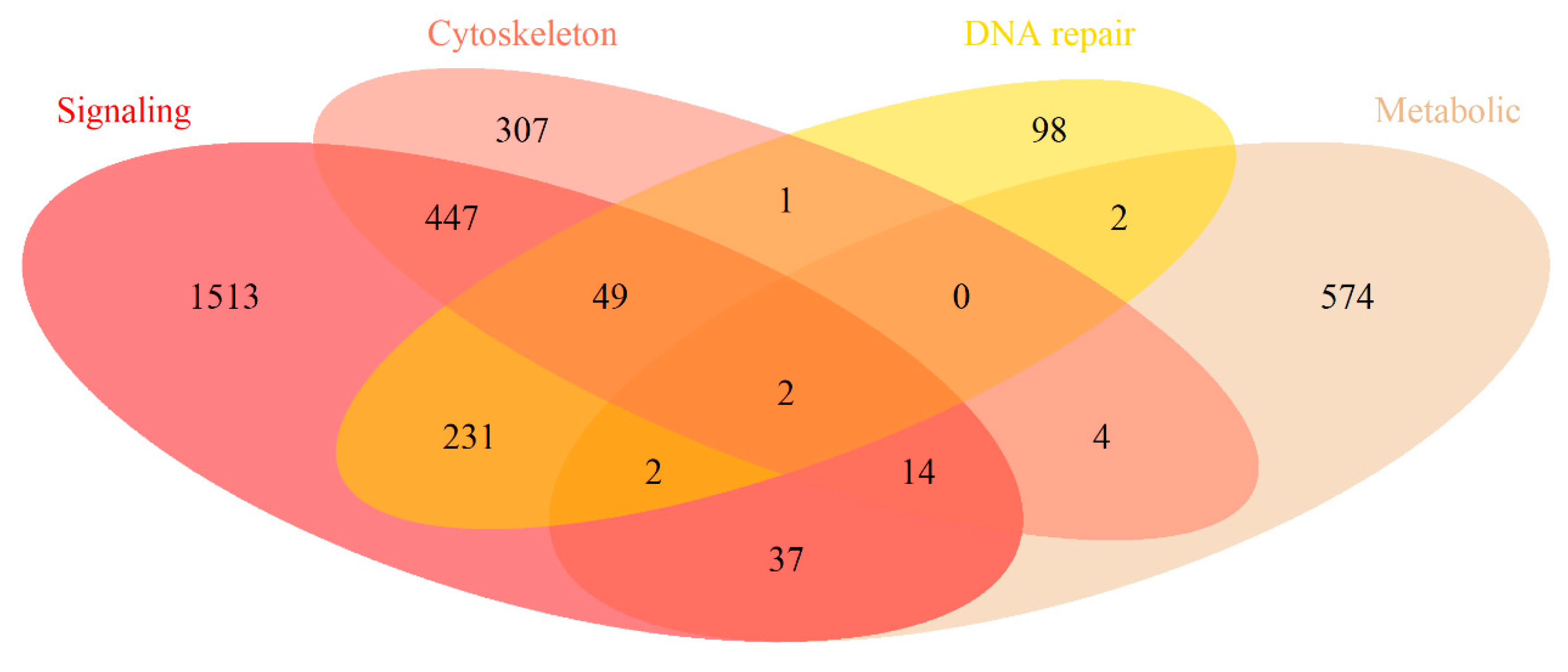
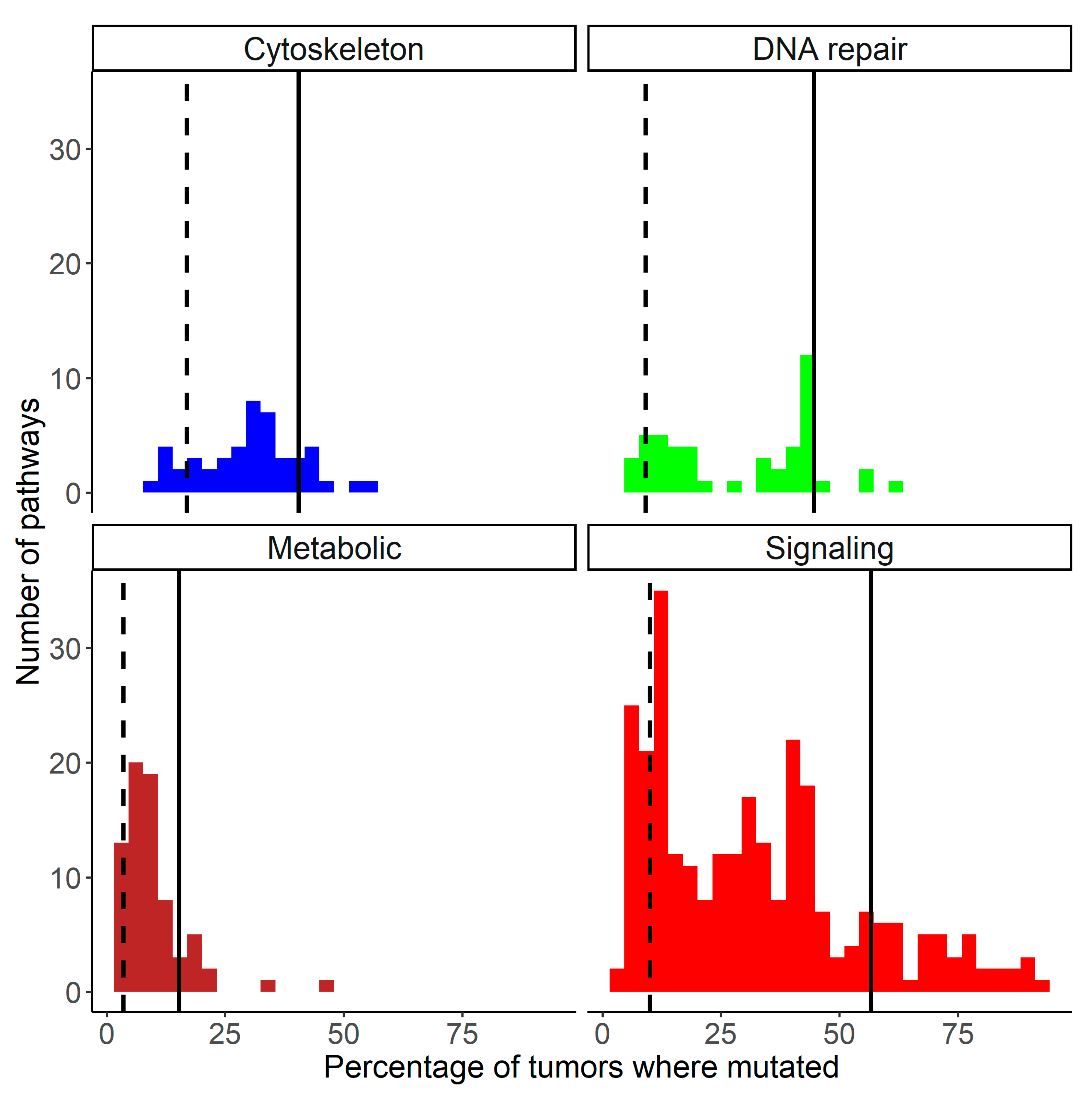
2.1. Cancer Mutation Frequencies of Molecular Pathway Genes
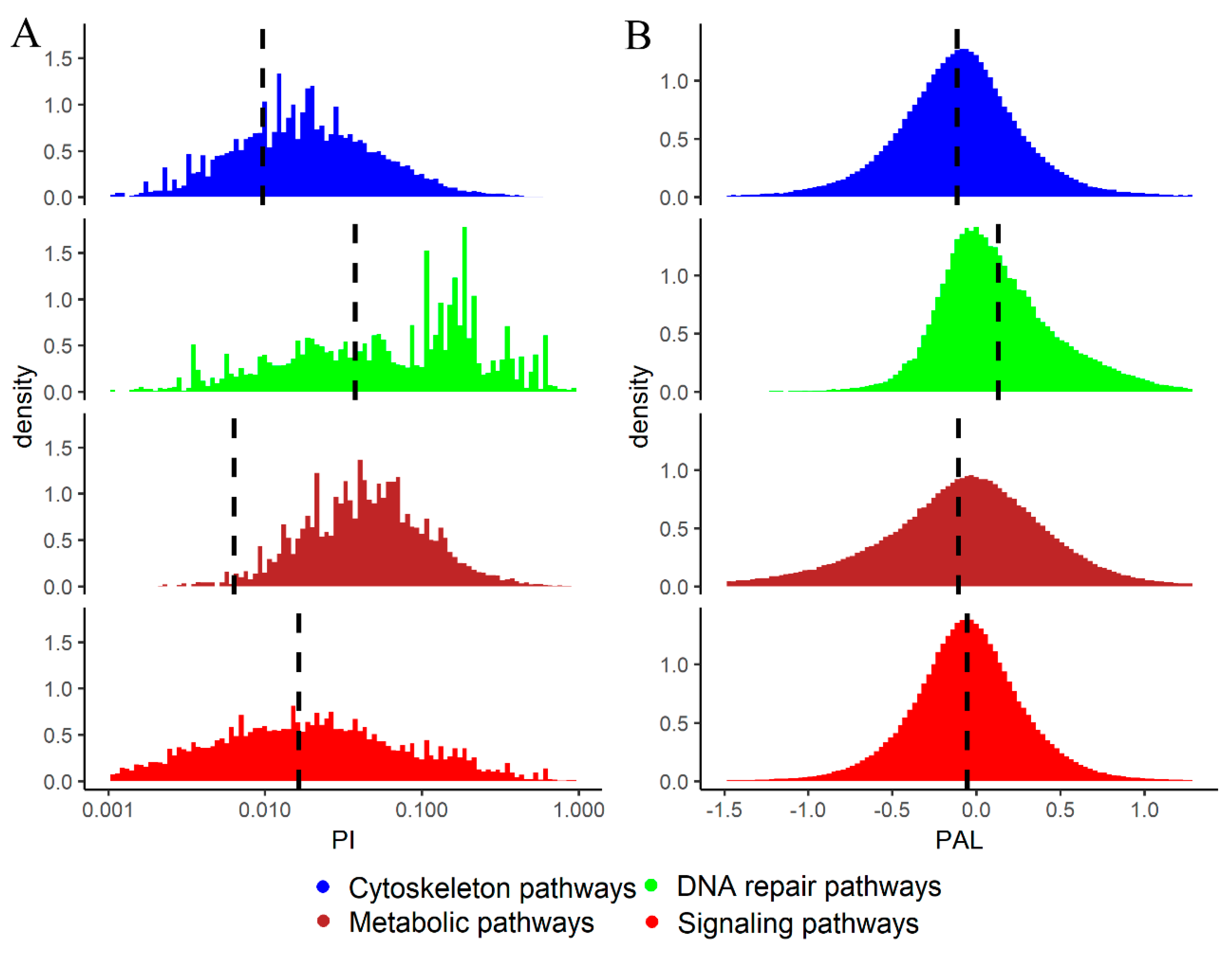
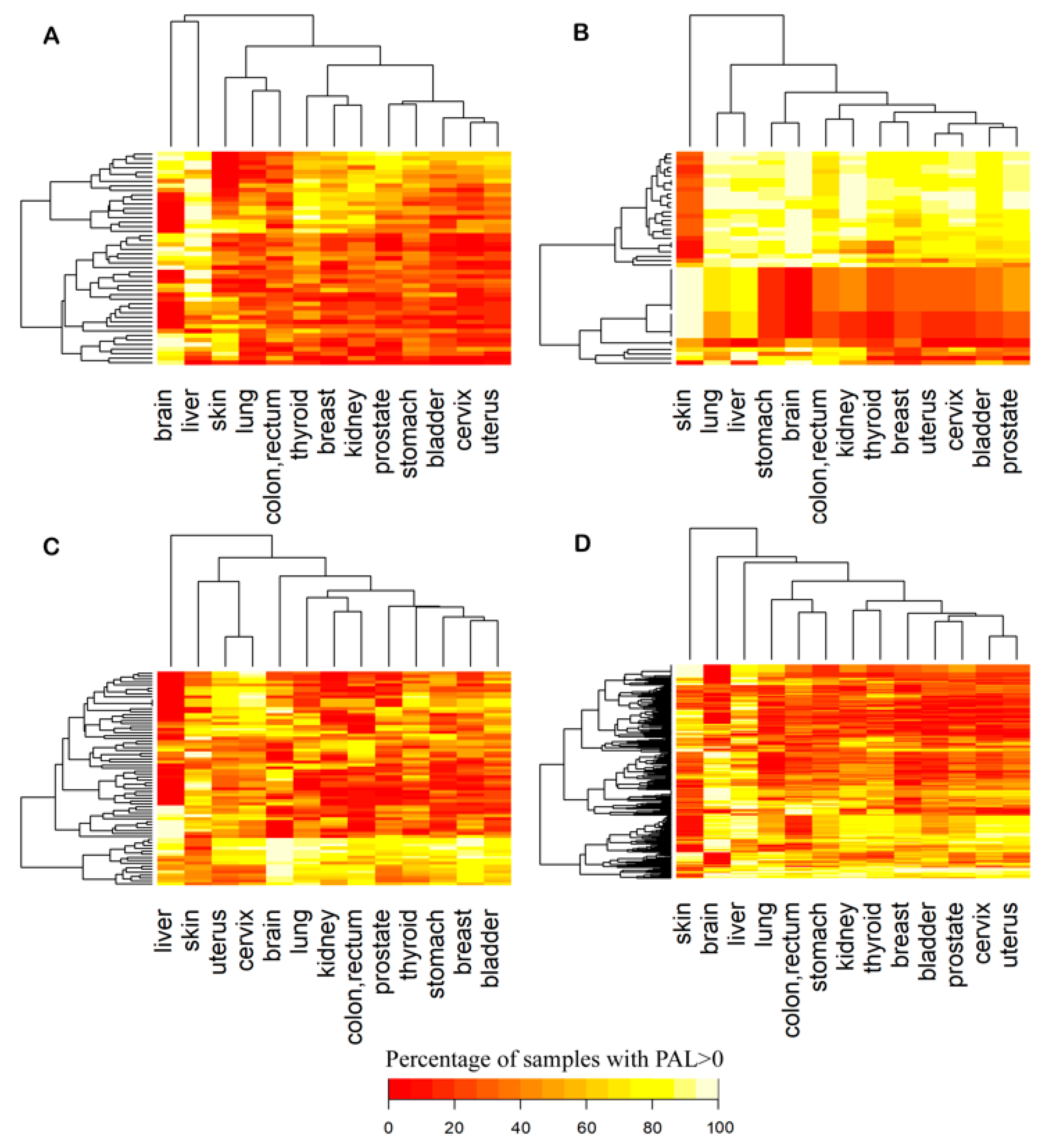
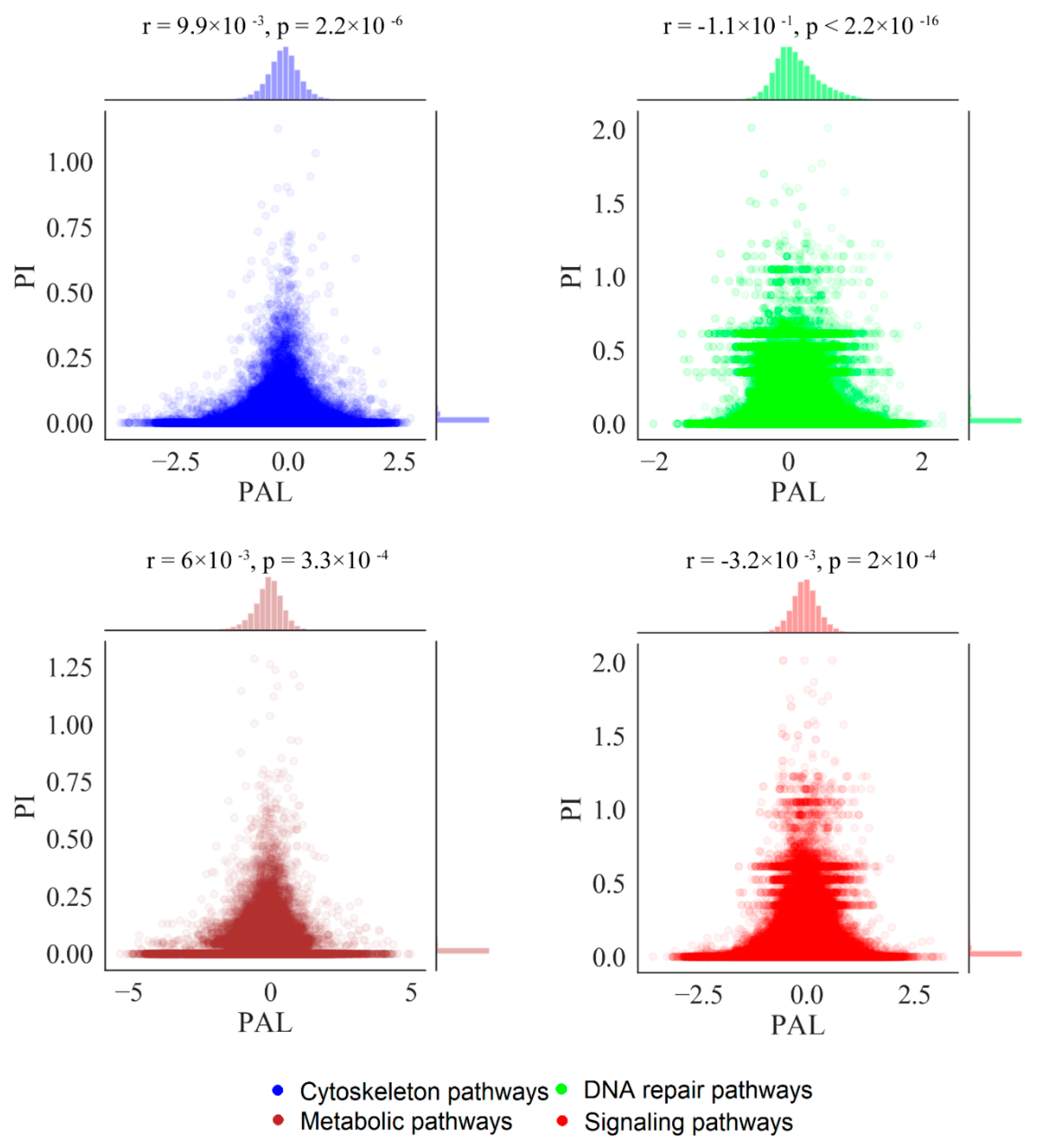
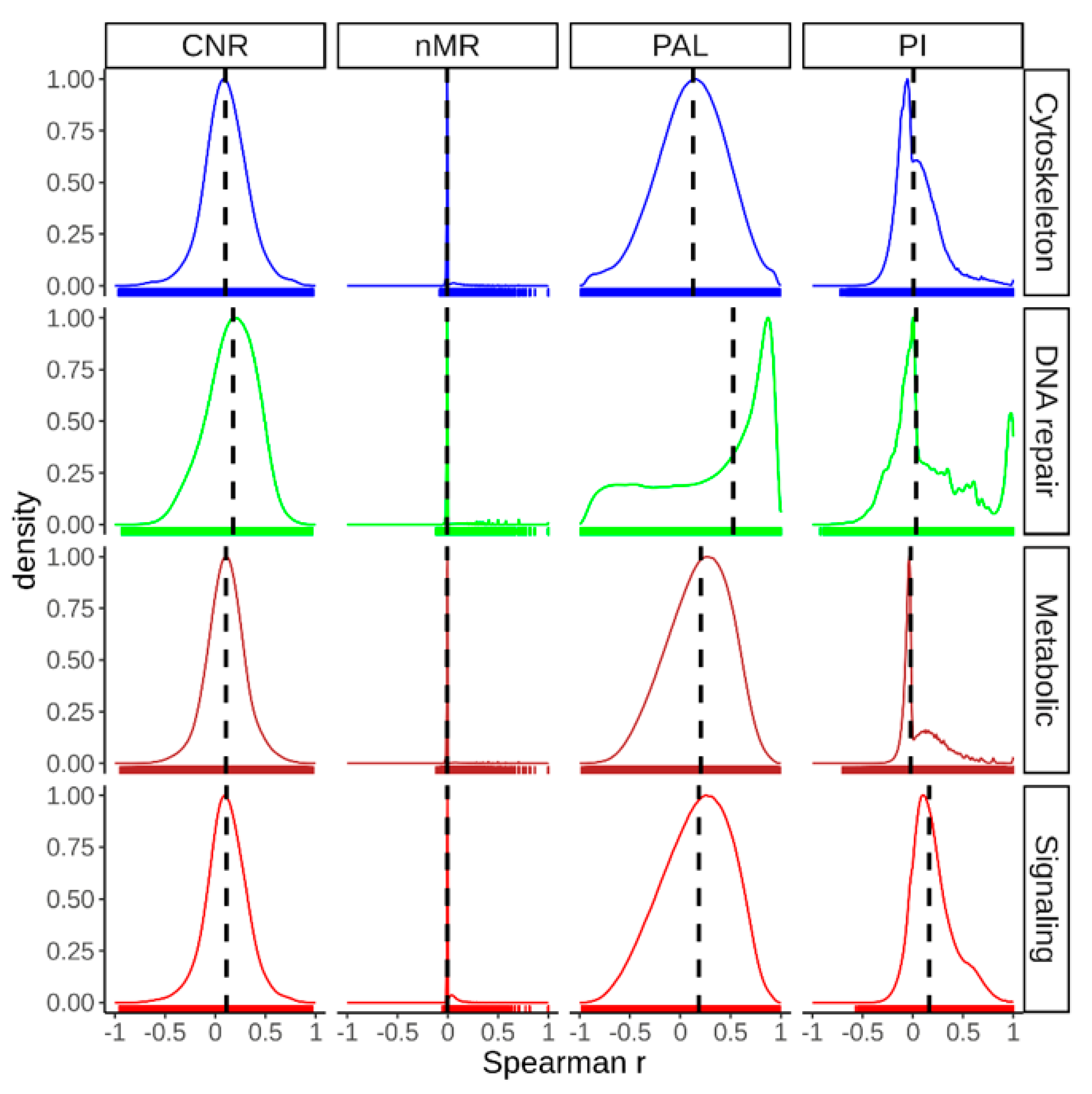
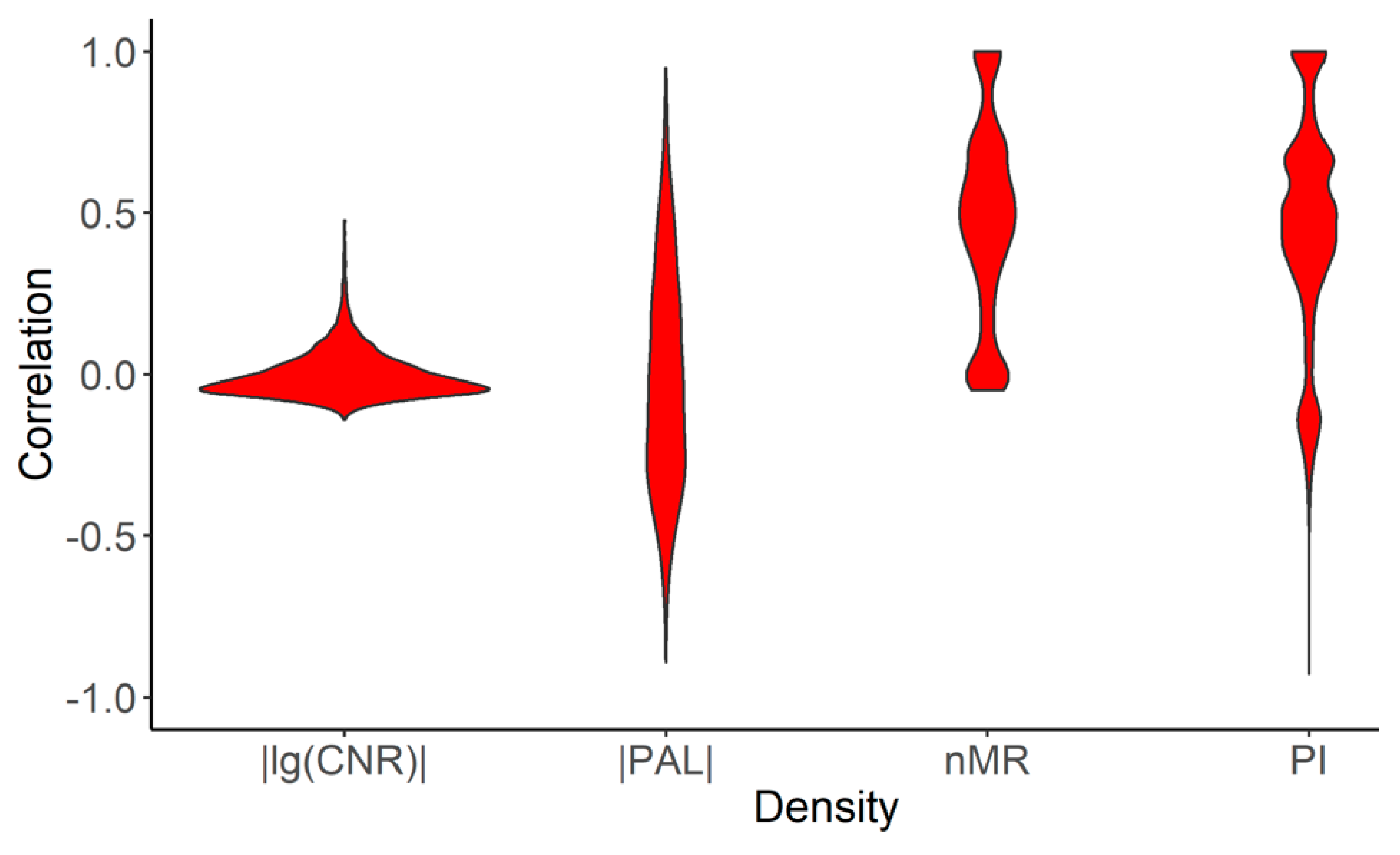
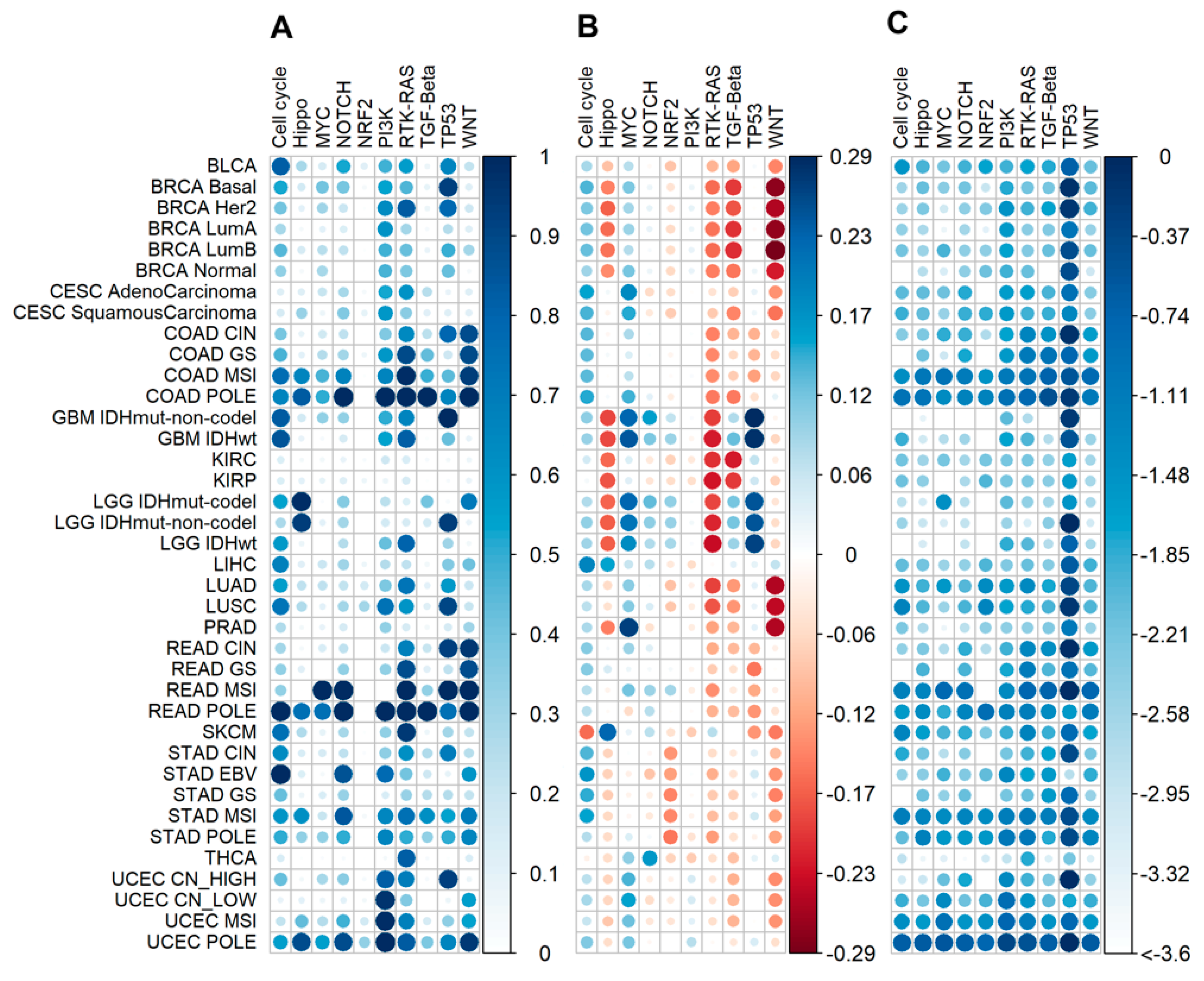
2.2. Mutation Enrichment and Pathway Activation Levels
3. Discussion
4. Materials and Methods
4.1. Mutation Data
4.2. Gene Expression Data
4.3. Pathway Databases
4.4. Calculation of Mutation Frequency Metrics
4.5. Calculation of Pathway Activation Level
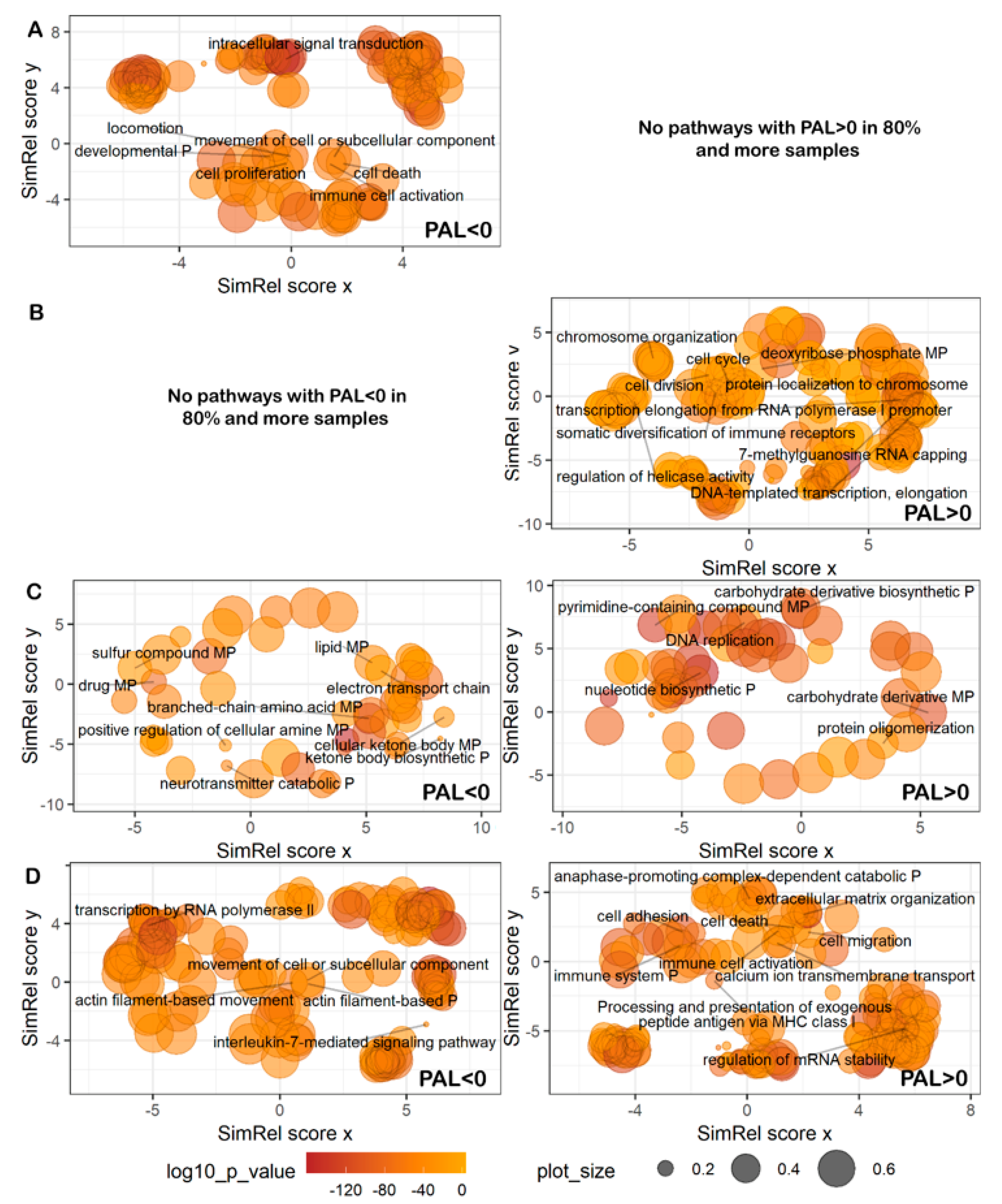
4.6. Gene Ontology Analysis
4.7. Comparison with the Study “Oncogenic Signaling Pathways in The Cancer Genome Atlas”
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sonnenschein, C.; Soto, A.M. Theories of carcinogenesis: An emerging perspective. Semin. Cancer Biol. 2008, 18, 372–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vineis, P.; Schatzkin, A.; Potter, J.D. Models of carcinogenesis: An overview. Carcinogenesis 2010, 31, 1703–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedessem, B.; Ruphy, S. SMT or TOFT? How the Two Main Theories of Carcinogenesis are Made (Artificially) Incompatible. Acta Biotheor. 2015, 63, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Antontseva, E.V.; Matveeva, M.Y.; Bondar, N.P.; Kashina, E.V.; Leberfarb, E.Y.; Bryzgalov, L.O.; Gervas, P.A.; Ponomareva, A.A.; Cherdyntseva, N.V.; Orlov, Y.L.; et al. Regulatory single nucleotide polymorphisms at the beginning of intron 2 of the human KRAS gene. J. Biosci. 2015, 40, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Kulikov, A.V.; Luchkina, E.A.; Gogvadze, V.; Zhivotovsky, B. Mitophagy: Link to cancer development and therapy. Biochem. Biophys. Res. Commun. 2017, 482, 432–439. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Buzdin, A.; Sorokin, M.; Poddubskaya, E.; Borisov, N. High-Throughput Mutation Data Now Complement Transcriptomic Profiling: Advances in Molecular Pathway Activation Analysis Approach in Cancer Biology. Cancer Inform. 2019, 18, 1176935119838844. [Google Scholar] [CrossRef] [Green Version]
- Shtam, T.; Naryzhny, S.; Samsonov, R.; Karasik, D.; Mizgirev, I.; Kopylov, A.; Petrenko, E.; Zabrodskaya, Y.; Kamyshinsky, R.; Nikitin, D.; et al. Plasma exosomes stimulate breast cancer metastasis through surface interactions and activation of FAK signaling. Breast Cancer Res. Treat. 2019, 174, 129–141. [Google Scholar] [CrossRef]
- Petrov, I.; Suntsova, M.; Mutorova, O.; Sorokin, M.; Garazha, A.; Ilnitskaya, E.; Spirin, P.; Larin, S.; Kovalchuk, O.; Prassolov, V.; et al. Molecular pathway activation features of pediatric acute myeloid leukemia (AML) and acute lymphoblast leukemia (ALL) cells. Aging 2016, 8, 2936–2947. [Google Scholar] [CrossRef] [Green Version]
- Kurz, S.; Thieme, R.; Amberg, R.; Groth, M.; Jahnke, H.-G.; Pieroh, P.; Horn, L.-C.; Kolb, M.; Huse, K.; Platzer, M.; et al. The anti-tumorigenic activity of A2M-A lesson from the naked mole-rat. PLoS ONE 2017, 12, e0189514. [Google Scholar]
- Borisov, N.M.; Terekhanova, N.V.; Aliper, A.M.; Venkova, L.S.; Smirnov, P.Y.; Roumiantsev, S.; Korzinkin, M.B.; Zhavoronkov, A.A.; Buzdin, A.A. Signaling pathways activation profiles make better markers of cancer than expression of individual genes. Oncotarget 2014, 5, 10198–10205. [Google Scholar] [CrossRef] [PubMed]
- Borisov, N.; Suntsova, M.; Sorokin, M.; Garazha, A.; Kovalchuk, O.; Aliper, A.; Ilnitskaya, E.; Lezhnina, K.; Korzinkin, M.; Tkachev, V.; et al. Data aggregation at the level of molecular pathways improves stability of experimental transcriptomic and proteomic data. Cell Cycle 2017, 16, 1810–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolotovskaia, M.A.; Sorokin, M.I.; Roumiantsev, S.A.; Borisov, N.M.; Buzdin, A.A. Pathway Instability Is an Effective New Mutation-Based Type of Cancer Biomarkers. Front. Oncol. 2019, 8, 658. [Google Scholar] [CrossRef] [PubMed]
- von Eichborn, J.; Dunkel, M.; Gohlke, B.O.; Preissner, S.C.; Hoffmann, M.F.; Bauer, J.M.J.; Armstrong, J.D.; Schaefer, M.H.; Andrade-Navarro, M.A.; Le Novere, N.; et al. SynSysNet: Integration of experimental data on synaptic protein–protein interactions with drug-target relations. Nucleic Acids Res. 2012, 41, D834–D840. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Bulashevska, S.; Szakacs, O.; Brors, B.; Eils, R.; Kovacs, G. Pathways of urothelial cancer progression suggested by Bayesian network analysis of allelotyping data. Int. J. Cancer 2004, 110, 850–856. [Google Scholar] [CrossRef]
- Haddad, D.; Socci, N.; Chen, C.-H.; Chen, N.G.; Zhang, Q.; Carpenter, S.G.; Mittra, A.; Szalay, A.A.; Fong, Y. Molecular network, pathway, and functional analysis of time-dependent gene changes associated with pancreatic cancer susceptibility to oncolytic vaccinia virotherapy. Mol. Ther.-Oncolytics 2016, 3, 16008. [Google Scholar] [CrossRef] [Green Version]
- Creixell, P.; Reimand, J.; Haider, S.; Wu, G.; Shibata, T.; Vazquez, M.; Mustonen, V.; Gonzalez-Perez, A.; Pearson, J.; Sander, C.; et al. Pathway and network analysis of cancer genomes. Nat. Methods 2015, 12, 615–621. [Google Scholar]
- Zhang, Q.; Burdette, J.E.; Wang, J.-P. Integrative network analysis of TCGA data for ovarian cancer. BMC Syst. Biol. 2014, 8, 1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, N.; Sorokin, M.; Garazha, A.; Buzdin, A. Quantitation of Molecular Pathway Activation Using RNA Sequencing Data. Methods Mol. Biol. 2020, 2063, 189–206. [Google Scholar] [PubMed]
- Zolotovskaia, M.; Sorokin, M.; Garazha, A.; Borisov, N.; Buzdin, A. Molecular Pathway Analysis of Mutation Data for Biomarkers Discovery and Scoring of Target Cancer Drugs. Methods Mol. Biol. 2020, 2063, 207–234. [Google Scholar] [PubMed]
- Dreesen, O.; Brivanlou, A.H. Signaling pathways in cancer and embryonic stem cells. Stem. Cell Rev. 2007, 3, 7–17. [Google Scholar] [CrossRef]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef]
- Petrov, I.; Suntsova, M.; Ilnitskaya, E.; Roumiantsev, S.; Sorokin, M.; Garazha, A.; Spirin, P.; Lebedev, T.; Gaifullin, N.; Larin, S.; et al. Gene expression and molecular pathway activation signatures of MYCN-amplified neuroblastomas. Oncotarget 2017, 8, 83768–83780. [Google Scholar] [CrossRef] [Green Version]
- Buzdin, A.A.; Prassolov, V.; Zhavoronkov, A.A.; Borisov, N.M. Bioinformatics Meets Biomedicine: OncoFinder, a Quantitative Approach for Interrogating Molecular Pathways Using Gene Expression Data. Methods Mol. Biol. 2017, 1613, 53–83. [Google Scholar]
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Daye, D.; Wellen, K.E. Metabolic reprogramming in cancer: Unraveling the role of glutamine in tumorigenesis. Semin. Cell Dev. Biol. 2012, 23, 362–369. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Gaude, E.; Hilvo, M.; Frezza, C. The Metabolic Alterations of Cancer Cells. Methods Enzymol. 2014, 542, 1–23. [Google Scholar]
- Xu, X.D.; Shao, S.X.; Jiang, H.P.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Wang, Y.L.; Wang, X.S.; Niu, H.T. Warburg effect or reverse Warburg effect? A review of cancer metabolism. Oncol. Res. Treat. 2015, 38, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Dang, C. V Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welf, E.S.; Haugh, J.M. Signaling pathways that control cell migration: Models and analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, T.C.; El-Osta, A. DNA damage repair and transcription. Cell. Mol. Life Sci. 2004, 61, 2137–2147. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009, 28, 5–14. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Galanos, P.; Pappas, G.; Polyzos, A.; Kotsinas, A.; Svolaki, I.; Giakoumakis, N.N.; Glytsou, C.; Pateras, I.S.; Swain, U.; Souliotis, V.L.; et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol. 2018, 19, 37. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.-Q.; Shi, Y.; Ma, L.-J.; Yang, L.-X.; Wang, X.-Y.; Zhang, S.; Wang, Z.-C.; Duan, M.; Zhang, Z.; Liu, L.-Z.; et al. Spatial and temporal clonal evolution of intrahepatic cholangiocarcinoma. J. Hepatol. 2018, 69, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, M.-O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Qu, Z.; Harata-Lee, Y.; Aung, T.N.; Shen, H.; Adelson, D. Cell Cycle, Energy Metabolism and DNA Repair Pathways in Cancer Cells are Suppressed by Compound Kushen Injection. BMC Cancer 2019, 19, 103. [Google Scholar] [CrossRef] [Green Version]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. (Poznan Poland) 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Wagg, J.; Green, M.L.; Kaiser, D.; Krummenacker, M.; Karp, P.D. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2004, 6, R2. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- QIAGEN-Pathway-Central. Available online: https://www.qiagen.com/us/shop/genes-and-pathways/pathway-central/ (accessed on 19 September 2018).
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Pita-Juárez, Y.; Altschuler, G.; Kariotis, S.; Wei, W.; Koler, K.; Green, C.; Tanzi, R.E.; Hide, W. The Pathway Coexpression Network: Revealing pathway relationships. PLoS Comput. Biol. 2018, 14, e1006042. [Google Scholar] [CrossRef] [Green Version]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Sekacheva, M.; Kim, E.; Zhukov, N.; Wang, Y.; Li, X.; Kar, S.; Hartmann, C.; et al. Molecular pathway activation-New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin. Cancer Biol. 2018, 53, 110–124. [Google Scholar] [CrossRef]
- Poddubskaya, E.V.; Baranova, M.P.; Allina, D.O.; Sekacheva, M.I.; Makovskaia, L.A.; Kamashev, D.E.; Suntsova, M.V.; Barbara, V.S.; Kochergina-Nikitskaya, I.N.; Aleshin, A.A. Personalized prescription of imatinib in recurrent granulosa cell tumor of the ovary: Case report. Mol. Case Stud. 2019, 5, a003434. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Huntley, R.P.; Sawford, T.; Mutowo-Meullenet, P.; Shypitsyna, A.; Bonilla, C.; Martin, M.J.; O’Donovan, C. The GOA database: Gene Ontology annotation updates for 2015. Nucleic Acids Res. 2015, 43, D1057–D1063. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamoorthi, G.; Sivalingam, N. Molecular mechanism of TGF-β signaling pathway in colon carcinogenesis and status of curcumin as chemopreventive strategy. Tumor Biol. 2014, 35, 7295–7305. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Zhang, Z.; Zhou, W.; Zhou, X.; Geng, C.; Chang, L.K.; Tian, X.; Liu, S. WNT10A/β-catenin pathway in tumorigenesis of papillary thyroid carcinoma. Oncol. Rep. 2017, 38, 1287–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzdin, A.A.; Zhavoronkov, A.A.; Korzinkin, M.B.; Venkova, L.S.; Zenin, A.A.; Smirnov, P.Y.; Borisov, N.M. Oncofinder, a new method for the analysis of intracellular signaling pathway activation using transcriptomic data. Front. Genet. 2014, 5, 55. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.; Sarkar, R.R. Comparison of human cell signaling pathway databases—evolution, drawbacks and challenges. Database 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Wittig, U. Analysis and comparison of metabolic pathway databases. Brief. Bioinform. 2001, 2, 126–142. [Google Scholar] [CrossRef] [Green Version]
- McQuerry, J.A.; Jenkins, D.F.; Yost, S.E.; Zhang, Y.; Schmolze, D.; Johnson, W.E.; Yuan, Y.; Bild, A.H. Pathway activity profiling of growth factor receptor network and stemness pathways differentiates metaplastic breast cancer histological subtypes. BMC Cancer 2019, 19, 881. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Jing, C.; Chang, X.; Ding, D.; Han, T.; Yang, J.; Lu, Z.; Hu, X.; Liu, Z.; Wang, J.; et al. Mutational landscape of gastric cancer and clinical application of genomic profiling based on target next-generation sequencing. J. Transl. Med. 2019, 17, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikova, V.; Jourdan, M.; Robert, N.; Requirand, G.; Boireau, S.; Bruyer, A.; Vincent, L.; Cartron, G.; Klein, B.; Elemento, O.; et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics 2019, 9, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S. Is Cancer a Genetic Disease or a Metabolic Disease? EBioMedicine 2015, 2, 478–479. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- PrimePCR Pathways-260 pathway maps|Life Science Research|Bio-Rad. Available online: https://www.bio-rad.com/en-us/prime-pcr-assays/pathway/primepcr-pathways (accessed on 23 November 2019).
- Zhu, Q.; Izumchenko, E.; Aliper, A.M.; Makarev, E.; Paz, K.; Buzdin, A.A.; Zhavoronkov, A.A.; Sidransky, D. Pathway activation strength is a novel independent prognostic biomarker for cetuximab sensitivity in colorectal cancer patients. Hum. Genome Var. 2015, 2, 15009. [Google Scholar] [CrossRef]
- Aliper, A.M.; Korzinkin, M.B.; Kuzmina, N.B.; Zenin, A.A.; Venkova, L.S.; Smirnov, P.Y.; Zhavoronkov, A.A.; Buzdin, A.A.; Borisov, N.M. Mathematical Justification of Expression-Based Pathway Activation Scoring (PAS). Methods Mol. Biol. 2017, 1613, 31–51. [Google Scholar]
- Yates, B.; Braschi, B.; Gray, K.A.; Seal, R.L.; Tweedie, S.; Bruford, E.A. Genenames.org: The HGNC and VGNC resources in 2017. Nucleic Acids Res. 2017, 45, D619–D625. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intersection | Signaling | Metabolic | Cytoskeleton | DNA Repair |
|---|---|---|---|---|
| Signaling | 1 | 0.019 | 0.196 | 0.119 |
| Metabolic | 0.087 | 1 | 0.014 | 0.006 |
| Cytoskeleton | 0.621 | 0.031 | 1 | 0.045 |
| DNA repair | 0.738 | 0.016 | 0.135 | 1 |
| Minimal Number of Mutated Genes | Cytoskelton | DNA Repair | Metabolic | Signaling | Minimal Number of Mutated Genes | Cytoskelton | DNA Repair | Metabolic | Signaling |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 78% | 53% | 58% | 94% | 11 | 17% | 6% | 9% | 46% |
| 2 | 65% | 35% | 41% | 89% | 12 | 16% | 5% | 8% | 42% |
| 3 | 54% | 24% | 31% | 84% | 13 | 15% | 5% | 8% | 39% |
| 4 | 45% | 18% | 24% | 78% | 14 | 14% | 4% | 7% | 37% |
| 5 | 38% | 14% | 19% | 72% | 15 | 13% | 4% | 6% | 35% |
| 6 | 32% | 11% | 16% | 66% | 16 | 12% | 4% | 6% | 33% |
| 7 | 27% | 9% | 14% | 61% | 17 | 11% | 3% | 5% | 31% |
| 8 | 24% | 8% | 12% | 56% | 18 | 10% | 3% | 5% | 29% |
| 9 | 21% | 7% | 11% | 52% | 19 | 10% | 3% | 5% | 28% |
| 10 | 19% | 6% | 10% | 48% | 20 | 9% | 3% | 5% | 26% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zolotovskaia, M.A.; Tkachev, V.S.; Seryakov, A.P.; Kuzmin, D.V.; Kamashev, D.E.; Sorokin, M.I.; Roumiantsev, S.A.; Buzdin, A.A. Mutation Enrichment and Transcriptomic Activation Signatures of 419 Molecular Pathways in Cancer. Cancers 2020, 12, 271. https://doi.org/10.3390/cancers12020271
Zolotovskaia MA, Tkachev VS, Seryakov AP, Kuzmin DV, Kamashev DE, Sorokin MI, Roumiantsev SA, Buzdin AA. Mutation Enrichment and Transcriptomic Activation Signatures of 419 Molecular Pathways in Cancer. Cancers. 2020; 12(2):271. https://doi.org/10.3390/cancers12020271
Chicago/Turabian StyleZolotovskaia, Marianna A., Victor S. Tkachev, Alexander P. Seryakov, Denis V. Kuzmin, Dmitry E. Kamashev, Maxim I. Sorokin, Sergey A. Roumiantsev, and Anton A. Buzdin. 2020. "Mutation Enrichment and Transcriptomic Activation Signatures of 419 Molecular Pathways in Cancer" Cancers 12, no. 2: 271. https://doi.org/10.3390/cancers12020271