Gene Expression Analysis of Aggressive Clinical T1 Stage Clear Cell Renal Cell Carcinoma for Identifying Potential Diagnostic and Prognostic Biomarkers

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Baseline Characteristics
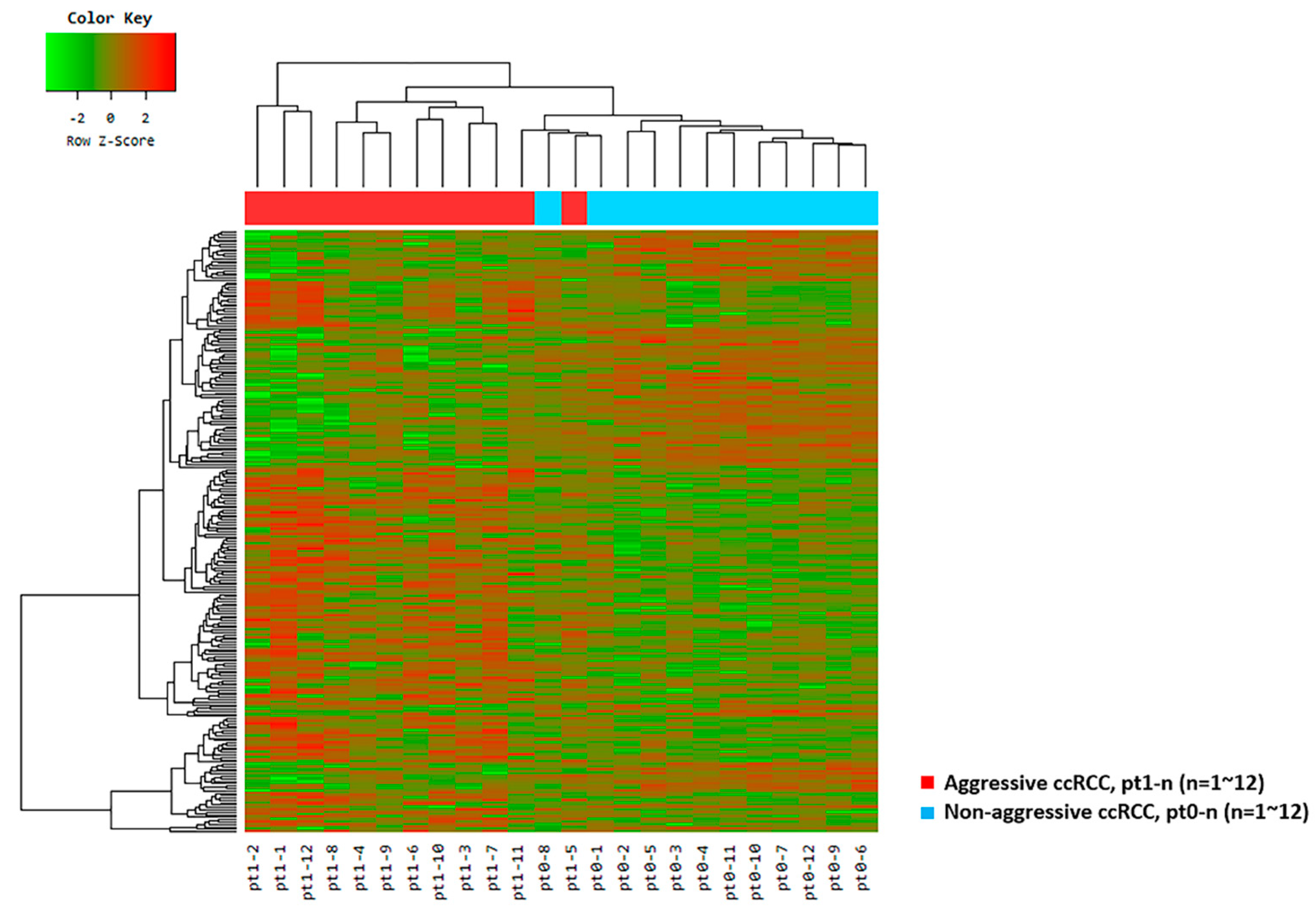
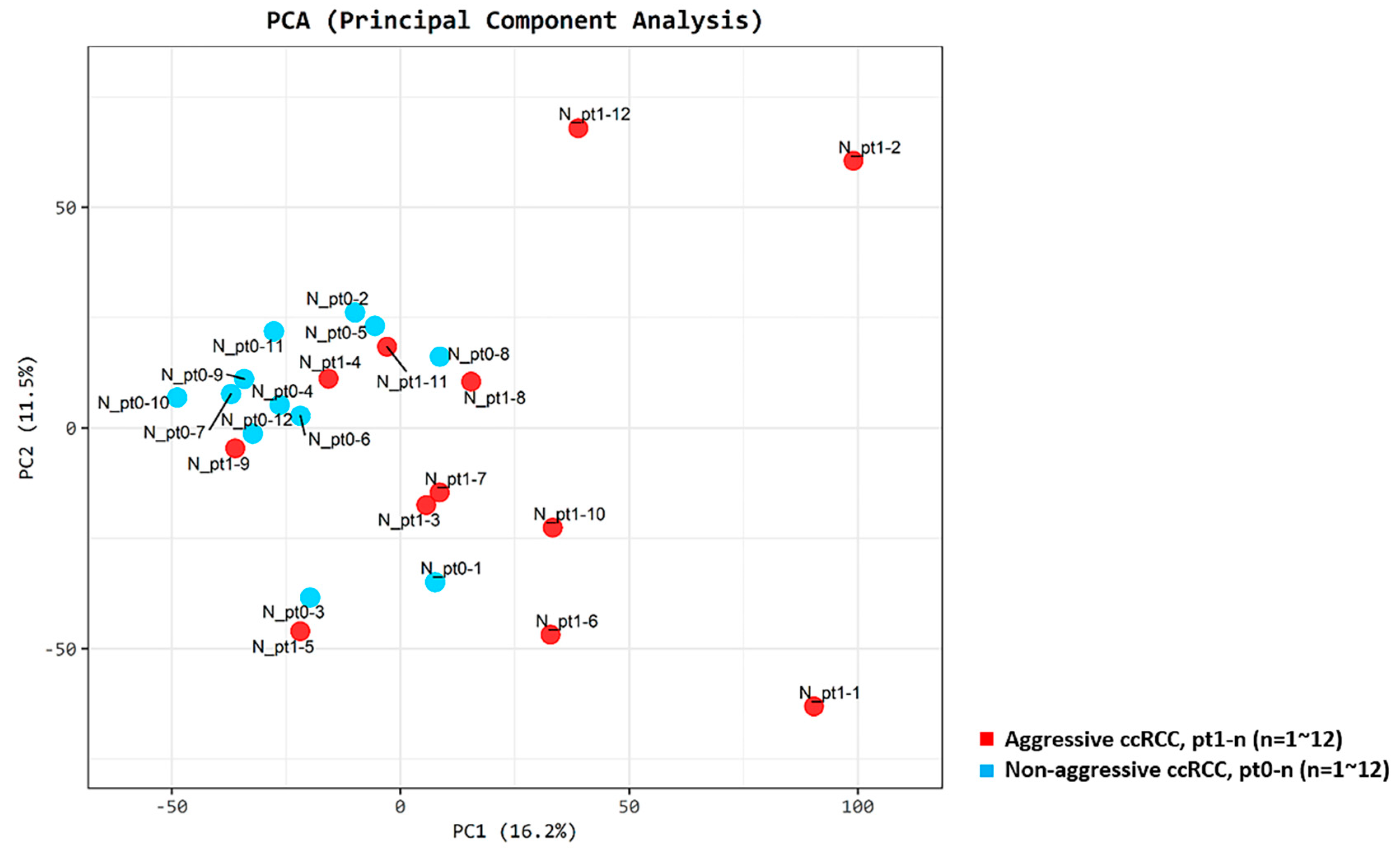
2.2. Results from the RNA-Seq Analysis
2.3. Differentially Expressed Genes
2.4. Association between Oncological Outcomes and Expression of the 10 Newly Selected Genes
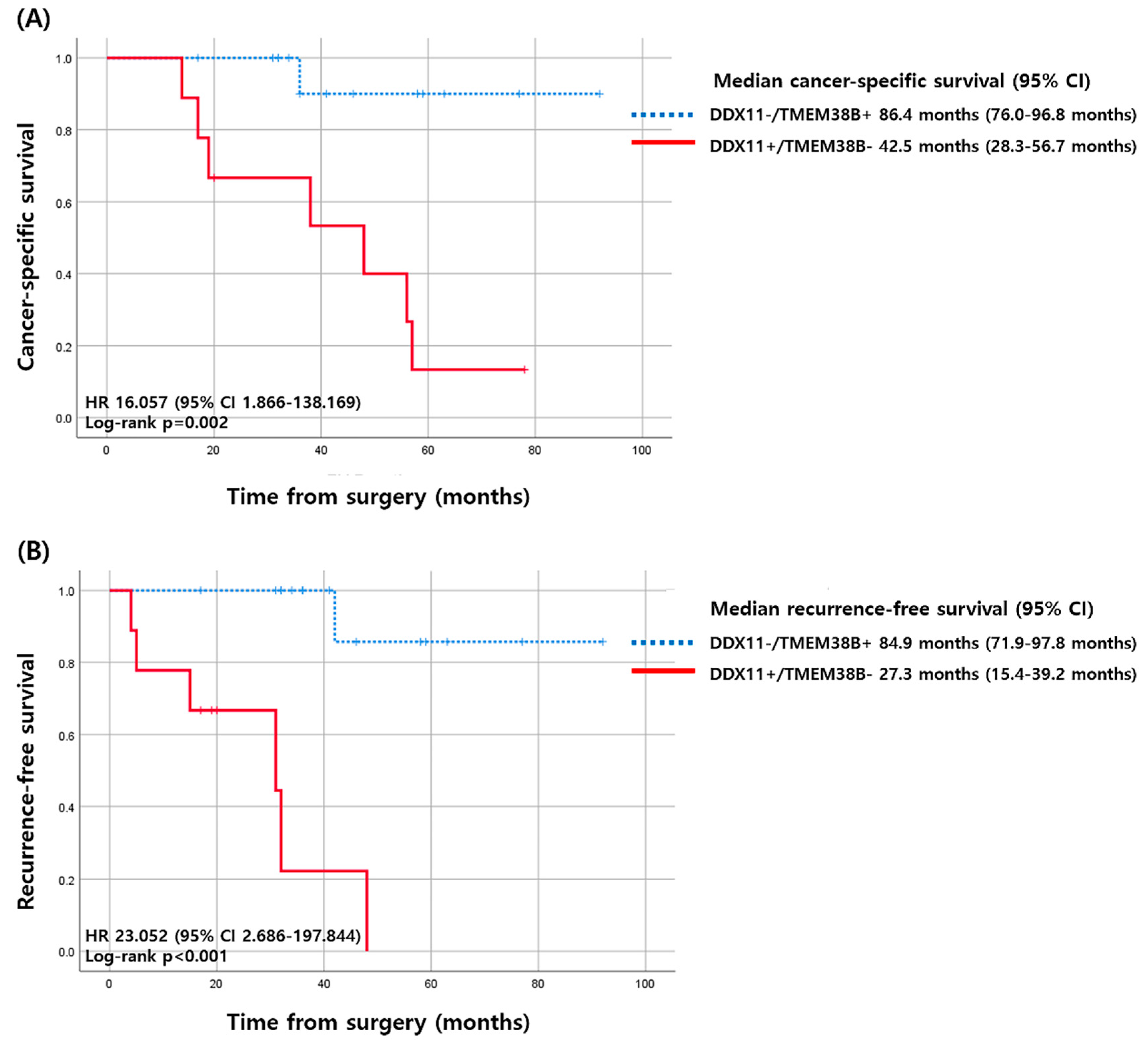
2.5. Survival Analysis of 16 Genes
2.6. GO Analysis and KEGG Analysis of DEGs
2.7. Validation of Target Genes Using Frozen Tissue PCR
3. Discussion
4. Materials and Methods
4.1. Patients and Tissues
4.2. Tissue Preparation
4.3. RNA Extraction and Sequencing
4.4. Analysis of RNA Sequencing—Differentially Expressed Gene (DEG) Selection
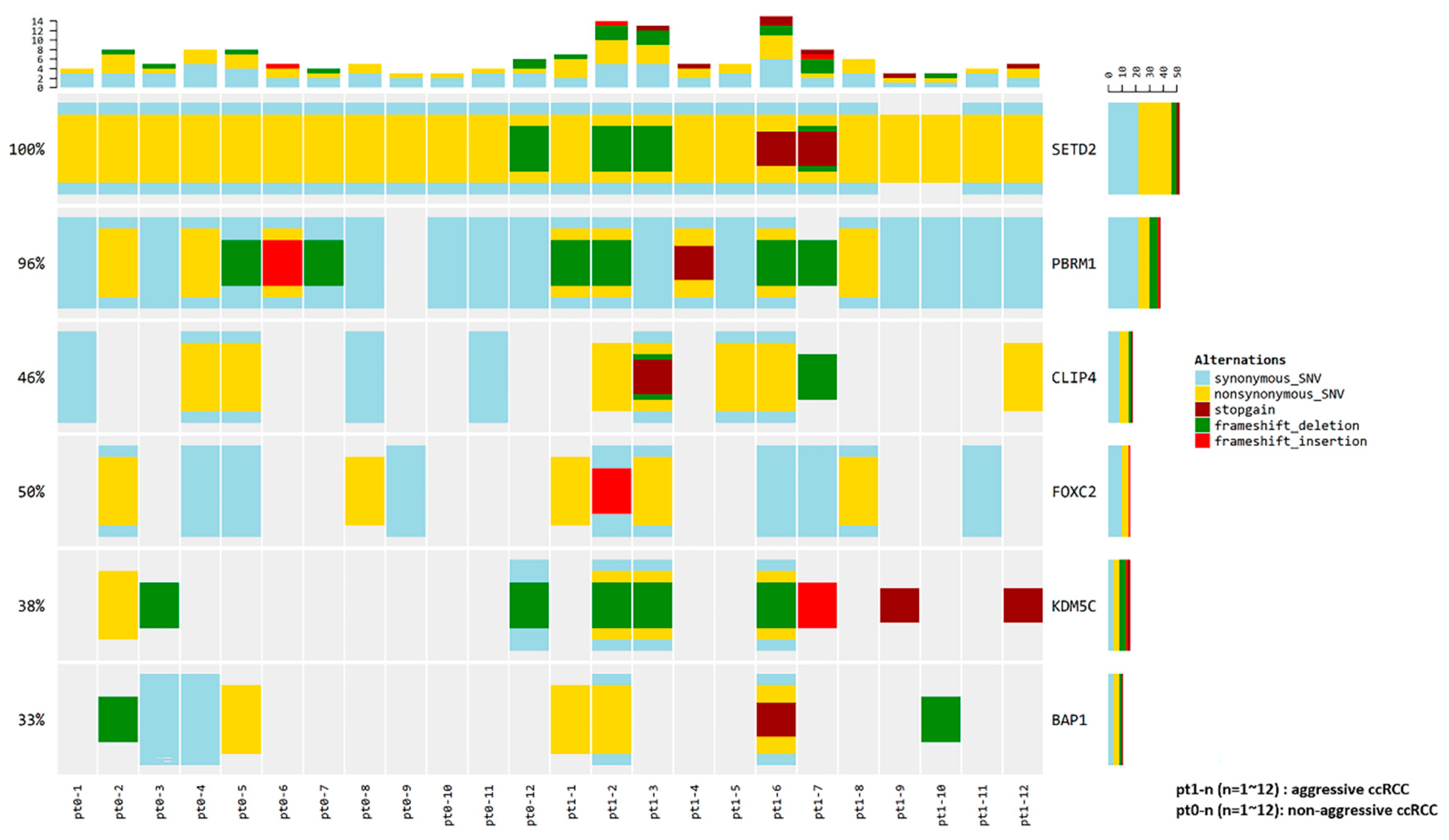
4.5. Variant Calling
4.6. qRT-PCR
4.7. GO Analysis and KEGG Analysis
4.8. Validation of Gene Expression, Survival Analyses, and Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Manley, B.J.; Reznik, E.; Ghanaat, M.; Kashan, M.; Becerra, M.F.; Casuscelli, J.; Tennenbaum, D.; Redzematovic, A.; Carlo, M.I.; Sato, Y.; et al. Characterizing recurrent and lethal small renal masses in clear cell renal cell carcinoma using recurrent somatic mutations. Urol. Oncol. 2019, 37, 12. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. Lancet 2009, 373, 1119. [Google Scholar] [CrossRef]
- Ahn, J.; Han, K.S.; Heo, J.H.; Bang, D.; Kang, Y.H.; Jin, H.A.; Hong, S.J.; Lee, J.H.; Ham, W.S. FOXC2 and CLIP4: A potential biomarker for synchronous metastasis of ≤7-cm clear cell renal cell carcinomas. Oncotarget 2016, 7, 51423. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.; Goddard, A.; Knezevic, D.; Maddala, T.; Zhou, M.; Aydin, H.; Campbell, S.; Elson, P.; Koscielny, S.; Lopatin, M.; et al. A 16-gene assay to predict recurrence after surgery in localised renal cell carcinoma: Development and validation studies. Lancet Oncol. 2015, 16, 676. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Linehan, W.M. The origin, evolution and route to metastasis of clear cell RCC. Nat. Rev. Nephrol. 2018, 14, 538. [Google Scholar] [CrossRef]
- Mitchell, T.J.; Turajilic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx Renal. Cell 2018, 173, 611. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell 2018, 173, 581. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx Renal. Cell 2018, 173, 595. [Google Scholar] [CrossRef] [Green Version]
- Peña-Llopis, S.; Vega-Rubin-de-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751. [Google Scholar] [CrossRef]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Lee, H.J.; Cho, N.H.; Kim, J.; Jang, W.S.; Heo, J.E.; Ham, W.S. Risk prediction tool for aggressive tumors in clinical T1 stage clear cell renal cell carcinoma using molecular biomarkers. Comput. Struct. Biotechnol. J. 2019, 17, 371. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Wu, C.P.; Yang, K.; Wang, S.P.; Wu, S. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860. [Google Scholar] [CrossRef]
- Kapur, P.; Pena-Llopis, S.; Christie, A.; Zhrebker, L.; Pavía-Jiménez, A.; Rathmell, W.K.; Xie, X.J.; Brugarolas, J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation. Lancet Oncol. 2013, 14, 159. [Google Scholar] [CrossRef] [Green Version]
- Hakimi, A.A.; Ostrovnaya, I.; Reva, B.; Schultz, N.; Chen, Y.B.; Gonen, M.; Liu, H.; Takeda, S.; Voss, M.H.; Tickoo, S.K.; et al. ccRCC Cancer Genome Atlas (KIRC TCGA) Research Network investigators. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: A report by MSKCC and the KIRC TCGA research network. Clin. Cancer Res. 2013, 19, 3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Guo, R.; Zhang, X.; Liang, Y.; Kong, F.; Wang, J.; Xu, Z. Loss of SETD2, but not H3K36me3, correlates with aggressive clinicopathological features of clear cell renal cell carcinoma patients. Biosci. Trends 2017, 11, 214. [Google Scholar] [CrossRef] [Green Version]
- Joseph, R.W.; Kapur, P.; Serie, D.J.; Parasramka, M.; Ho, T.H.; Cheville, J.C.; Frenkel, E.; Parker, A.S.; Brugarolas, J. Clear cell renal cell carcinoma subtypes identified by BAP1 and PBRM1 expression. J. Urol. 2016, 195, 180. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Bander, N.H.; Nanus, D.M. Renal-cell carcinoma. N. Engl. J. Med. 1996, 335, 865. [Google Scholar] [CrossRef]
- Mani, S.; Todd, M.B.; Katz, K.; Poo, W.J. Prognostic factors for survival in patients with metastatic renal cancer treated with biological response modifiers. J. Urol. 1995, 154, 35. [Google Scholar] [CrossRef]
- Huang, Y.; Murakami, T.; Sano, F.; Kondo, K.; Nakaigawa, N.; Kishida, T.; Kubota, Y.; Nagashima, Y.; Yao, M. Expression of aquaporin 1 in primary renal tumors: A prognostic indicator for clear-cell renal cell carcinoma. Eur. Urol. 2009, 56, 690. [Google Scholar] [CrossRef]
- Bhattacharya, C.; Wang, X.; Becker, D. The DEAD/DEAH box helicase, DDX11, is essential for the survival of advanced melanomas. Mol. Cancer 2012, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, A.; Tsai, C.L.; Jung, S.M.; Chuang, W.C.; Kao, C.; Hsu, A.; Chen, S.H.; Lin, C.Y.; Lee, Y.C.; Lee, Y.S.; et al. BAI1-associated protein 2-like 1 (BAIAP2L1) is a potential biomarker in ovarian cancer. PLoS ONE 2015, 10, e0133081. [Google Scholar] [CrossRef] [Green Version]
- Soultati, A.; Stares, M.; Swanton, C.; Larkin, J.; Turajilic, S. How should clinicians address intratumour heterogeneity in clear renal cell carcinoma? Curr. Opin. Urol. 2015, 25, 358. [Google Scholar] [CrossRef]
- Wach, S.; Taubert, H.; Weigelt, K.; Hase, N.; Köhn, M.; Misiak, D.; Hüttelmaier, S.; Stöhr, C.G.; Kahlmeyer, A.; Haller, F.; et al. RNA Sequencing of Collecting Duct Renal Cell Carcinoma Suggests an Interaction between miRNA and Target Genes and a Predominance of Deregulated Solute Carrier Genes. Cancers 2019, 12, 64. [Google Scholar] [CrossRef] [Green Version]
- Ingimarsson, J.P.; Sigurdsson, M.I.; Hardarson, S.; Petursdottir, V.; Jonsson, E.; Einarsson, G.V.; Gudbjartsson, T. The impact of tumour size on the probability of synchronous metastasis and survival in renal cell carcinoma patients: A population-based study. BMC Urol. 2014, 14, 72. [Google Scholar] [CrossRef] [Green Version]
- Eble, J.N.; Sauter, G.; Epstein, J.; Sesterhenn, I. World Health Organization Classification of Tumors. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs; IARC Press: Lyon, France, 2004. [Google Scholar]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 2010, 17, 1471. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, S.A.; Lasky, L.C.; Limas, C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am. J. Surg. Pathol. 1982, 6, 655. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast-spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 10–11. [Google Scholar]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2018, 19, 649. [Google Scholar] [CrossRef]
- Lanczky, A.; Nagy., A.; Bottai, G.; Munkácsy, G.; Szabó, A.; Santarpia, L.; Győrffy, B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat. 2016, 160, 439. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aggressiveness | Patient ID | Sex | Age (yr) | Tumor Size (cm) | Fuhrman Grade | Invasion (Perinephric/Sinus Fat/Vascular) | Survival Time a (m) | Outcome b | Recurrence Site | Synchronous Metastatic Site |
|---|---|---|---|---|---|---|---|---|---|---|
| N | Pt0-1 | M | 67 | 1.6 | 3 | N | 41 | ned | ||
| N | Pt0-2 | M | 56 | 6.3 | 3 | Y | 36 | ned | ||
| N | Pt0-3 | M | 76 | 3.5 | 2 | Y | 32 | ned | ||
| N | Pt0-4 | F | 73 | 5.3 | 2 | Y | 92 | ned | ||
| N | Pt0-5 | M | 81 | 6.6 | 3 | Y | 17 | ned | ||
| N | Pt0-6 | M | 72 | 5.4 | 3 | Y | 77 | ned | ||
| N | Pt0-7 | M | 60 | 6.9 | 3 | Y | 59 | ned | ||
| N | Pt0-8 | M | 59 | 6.4 | 3 | Y | 63 | ned | ||
| N | Pt0-9 | M | 61 | 4.7 | 3 | Y | 58 | ned | ||
| N | Pt0-10 | M | 76 | 5.0 | 2 | N | 46 | ned | ||
| N | Pt0-11 | M | 65 | 4.4 | 2 | N | 32 | ned | ||
| N | Pt0-12 | M | 75 | 2.7 | 2 | N | 34 | ned | ||
| Y | Pt1-1 | M | 73 | 5.5 | 3 | N | 19 | cd | Lung | |
| Y | Pt1-2 | M | 73 | 6.9 | 4 | Y | 17 | cd | Lung | |
| Y | Pt1-3 | M | 60 | 5.1 | 3 | Y | 57 | pd/cd | Local recurrence, liver | |
| Y | Pt1-4 | M | 58 | 2.8 | 3 | N | 36 | cd | Bone | |
| Y | Pt1-5 | M | 74 | 1.6 | 2 | N | 48 | pd/cd | Lung, lymph nodes | |
| Y | Pt1-6 | M | 70 | 6.6 | 3 | Y | 38 | pd/cd | Liver, bone, lymph nodes | |
| Y | Pt1-7 | M | 61 | 6.7 | 3 | Y | 14 | pd/cd | Lung, liver | Lung, bone |
| Y | Pt1-8 | M | 54 | 4.8 | 2 | Y | 56 | pd/cd | Local recurrence | |
| Y | Pt1-9 | M | 66 | 4.2 | 2 | N | 20 | ned | Bone | |
| Y | Pt1-10 | M | 60 | 5.2 | 4 | Y | 78 | pd | Lung, bone | |
| Y | Pt1-11 | M | 70 | 3.5 | 2 | N | 58 | pd | Bone | |
| Y | Pt1-12 | M | 44 | 5.5 | 3 | Y | 31 | ned | Lung |
| Upregulated | ||||||
|---|---|---|---|---|---|---|
| Gene Symbol | Gene Title | FPKM (Mean ± SD) | Log2 Fold Change | p-Value a | Adjusted p b | |
| RCC with Aggressive Characteristics | RCC without Aggressive Characteristics | (With Aggressive/without Aggressive Characteristics) | ||||
| MOCOS | Molybdenum cofactor sulfurase | 16.76 ± 15.41 | 1.40 ± 1.29 | +3.77 | 9.29 × 10−6 | 1.66 × 10−2 |
| RGPD8 | RANBP2-like and GRIP domain containing 8 | 41.49 ± 35.62 | 6.51 ± 5.38 | +2.78 | 3.67 × 10−5 | 4.59 × 10−2 |
| BAIAP2L1 | BAI1 associated protein 2 like 1 | 7.05 ± 3.39 | 2.18 ± 1.28 | +1.87 | 2.18 × 10−6 | 7.98 × 10−3 |
| DDX11 | DEAD/H-box helicase 11 | 31.95 ± 13.09 | 14.25 ± 5.18 | +1.32 | 2.55 × 10−6 | 7.98 × 10−3 |
| Downregulated | ||||||
| Gene Symbol | Gene Title | FPKM (Mean ± SD) | Log2 Fold Change | p-Value a | Adjusted p b | |
| RCC with Aggressive Characteristics | RCC without Aggressive Characteristics | (With Aggressive/without Aggressive Characteristics) | ||||
| SLC16A9 | Solute carrier family 16 member 9 | 2.13 ± 2.51 | 11.90 ± 8.48 | −2.41 | 5.91 × 10−6 | 1.48 × 10−2 |
| FRAS1 | Fraser extracellular matrix complex subunit 1 | 3.15 ± 3.80 | 13.66 ± 6.60 | −2.12 | 4.37 × 10−8 | 5.47 × 10−4 |
| NPR3 | Natriuretic peptide receptor 3 | 3.22 ± 3.37 | 12.53 ± 7.84 | −1.88 | 3.61 × 10−5 | 4.59 × 10−2 |
| AQP1 | Aquaporin 1 (Colton blood group) | 11.02 ± 11.86 | 36.80 ± 17.38 | −1.63 | 1.73 × 10−5 | 2.71 × 10−2 |
| TMEM38B | Transmembrane protein 38B | 2.74 ± 1.81 | 8.07 ± 2.70 | −1.47 | 1.21 × 10−6 | 7.57 × 10−3 |
| PRUNE2 | Prune homolog 2 | 19.10 ± 9.42 | 49.39 ± 15.65 | −1.24 | 8.48 × 10−6 | 1.66 × 10−2 |
| Cancer-Specific Death | |||||
|---|---|---|---|---|---|
| FPKM (Mean ± SD) | RCC with Cancer-Specific Death (n = 8) | RCC without Cancer-Specific Death (n = 16) | p-Value a | Multivariate OR (95% CI) | p-Value b |
| MOCOS | 16.4 ± 12.5 | 5.4 ± 12.4 | 0.054 | - | 0.093 |
| RGPD8 | 47.6 ± 38.4 | 12.2 ± 17.5 | 0.036 | 1.048 (1.006–1.093) | 0.026 |
| BAIAP2L1 | 7.5 ± 4.0 | 3.2 ± 2.2 | 0.019 | 1.553 (1.088–2.215) | 0.015 |
| DDX11 | 34.3 ± 13.4 | 17.5 ± 9.3 | 0.002 | 1.130 (1.025–1.245) | 0.014 |
| SLC16A9 | 1.3 ± 2.3 | 9.9 ± 8.2 | 0.001 | 0.585 (0.362–0.946) | 0.029 |
| FRAS1 | 2.4 ± 2.5 | 11.4 ± 7.4 | <0.001 | 0.741 (0.567–0.967) | 0.028 |
| NPR3 | 3.1 ± 3.4 | 10.3 ± 8.0 | 0.006 | - | 0.058 |
| AQP1 | 11.3 ± 12.8 | 30.2 ± 19.7 | 0.023 | 0.926 (0.859–0.997) | 0.042 |
| TMEM38B | 1.9 ± 1.2 | 7.2 ± 2.9 | <0.001 | 0.277 (0.091–0.841) | 0.024 |
| PRUNE2 | 20.1 ± 8.5 | 41.3 ± 20.5 | 0.002 | 0.921 (0.854–0.993) | 0.032 |
| Recurrence | |||||
| FPKM (Mean ± SD) | RCC with Recurrence (n = 7) | RCC without Recurrence (n = 17) | p-Value a | Multivariate OR (95% CI) | p-Value b |
| MOCOS | 9.5 ± 6.6 | 8.9 ± 15.4 | 0.921 | - | 0.917 |
| RGPD8 | 65.5 ± 25.9 | 6.9 ± 6.0 | 0.001 | - | 0.996 |
| BAIAP2L1 | 6.8 ± 3.0 | 3.7 ± 3.4 | 0.048 | - | 0.066 |
| DDX11 | 36.0 ± 10.2 | 17.8 ± 10.6 | 0.001 | 1.140 (1.029–1.264) | 0.012 |
| SLC16A9 | 1.5 ± 2.1 | 9.3 ± 8.3 | 0.002 | - | 0.056 |
| FRAS1 | 3.1 ± 2.8 | 10.6 ± 7.8 | 0.002 | - | 0.051 |
| NPR3 | 2.9 ± 2.5 | 9.9 ± 8.1 | 0.004 | - | 0.073 |
| AQP1 | 12.7 ± 13.0 | 28.5 ± 20.3 | 0.071 | - | 0.090 |
| TMEM38B | 2.5 ± 1.5 | 6.6 ± 3.4 | 0.001 | 0.579 (0.355–0.944) | 0.028 |
| PRUNE2 | 14.5 ± 7.9 | 42.4 ± 17.6 | <0.001 | 0.788 (0.623–0.996) | 0.047 |
| Groups Dichotomized by the Expression of DDX1, TMEM38B and PRUNE2 | Cancer-Specific Death | ||||
|---|---|---|---|---|---|
| RCC with Cancer-Specific Death (n = 8) | RCC without Cancer-Specific Death (n = 16) | p-Value a | Multivariate OR (95% CI) | p-Value b | |
| DDX11+ | 7/8 (87.5%) | 5/16 (31.3%) | 0.027 | - | 0.065 |
| TMEM38B− | 8/8 (100.0%) | 4/16 (25.0%) | 0.001 | - | 0.998 |
| PRUNE2− | 7/8 (87.5%) | 5/16 (31.3%) | 0.027 | - | 0.065 |
| DDX11+ and TMEM38B− | 7/8 (87.5%) | 2/16 (12.5%) | 0.001 | 49.000 (3.765–637.794) | 0.003 |
| DDX11+ and PRUNE2− | 6/8 (75.0%) | 2/16 (12.5%) | 0.005 | 21.000 (2.372–185.930) | 0.006 |
| Recurrence | |||||
| RCC with Recurrence (n = 7) | RCC without Recurrence (n = 17) | p-Value a | Multivariate OR (95% CI) | p-Value b | |
| DDX11+ | 7/7 (100.0%) | 5/17 (29.4%) | 0.005 | - | 0.999 |
| TMEM38B− | 6/7 (85.7%) | 6/11 (54.5%) | 0.069 | 11.000 (1.061–114.086) | 0.045 |
| PRUNE2− | 7/7 (100.0%) | 5/17 (29.4%) | 0.005 | - | 0.999 |
| DDX11+ and TMEM38B− | 6/7 (85.7%) | 3/17 (17.6%) | 0.004 | 28.000 (2.399–326.735) | 0.008 |
| DDX11+ and PRUNE2− | 7/7 (100.0%) | 1/17 (5.9%) | <0.001 | - | 0.998 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.S.; Pierorazio, P.M.; Lee, J.H.; Lee, H.J.; Lim, Y.S.; Jang, W.S.; Kim, J.; Lee, S.H.; Rha, K.H.; Cho, N.H.; et al. Gene Expression Analysis of Aggressive Clinical T1 Stage Clear Cell Renal Cell Carcinoma for Identifying Potential Diagnostic and Prognostic Biomarkers. Cancers 2020, 12, 222. https://doi.org/10.3390/cancers12010222
Park JS, Pierorazio PM, Lee JH, Lee HJ, Lim YS, Jang WS, Kim J, Lee SH, Rha KH, Cho NH, et al. Gene Expression Analysis of Aggressive Clinical T1 Stage Clear Cell Renal Cell Carcinoma for Identifying Potential Diagnostic and Prognostic Biomarkers. Cancers. 2020; 12(1):222. https://doi.org/10.3390/cancers12010222
Chicago/Turabian StylePark, Jee Soo, Phillip M. Pierorazio, Ji Hyun Lee, Hyo Jung Lee, Young Soun Lim, Won Sik Jang, Jongchan Kim, Seung Hwan Lee, Koon Ho Rha, Nam Hoon Cho, and et al. 2020. "Gene Expression Analysis of Aggressive Clinical T1 Stage Clear Cell Renal Cell Carcinoma for Identifying Potential Diagnostic and Prognostic Biomarkers" Cancers 12, no. 1: 222. https://doi.org/10.3390/cancers12010222