Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: Path to Identification and a Proposal for Genetic Screening Guidelines

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
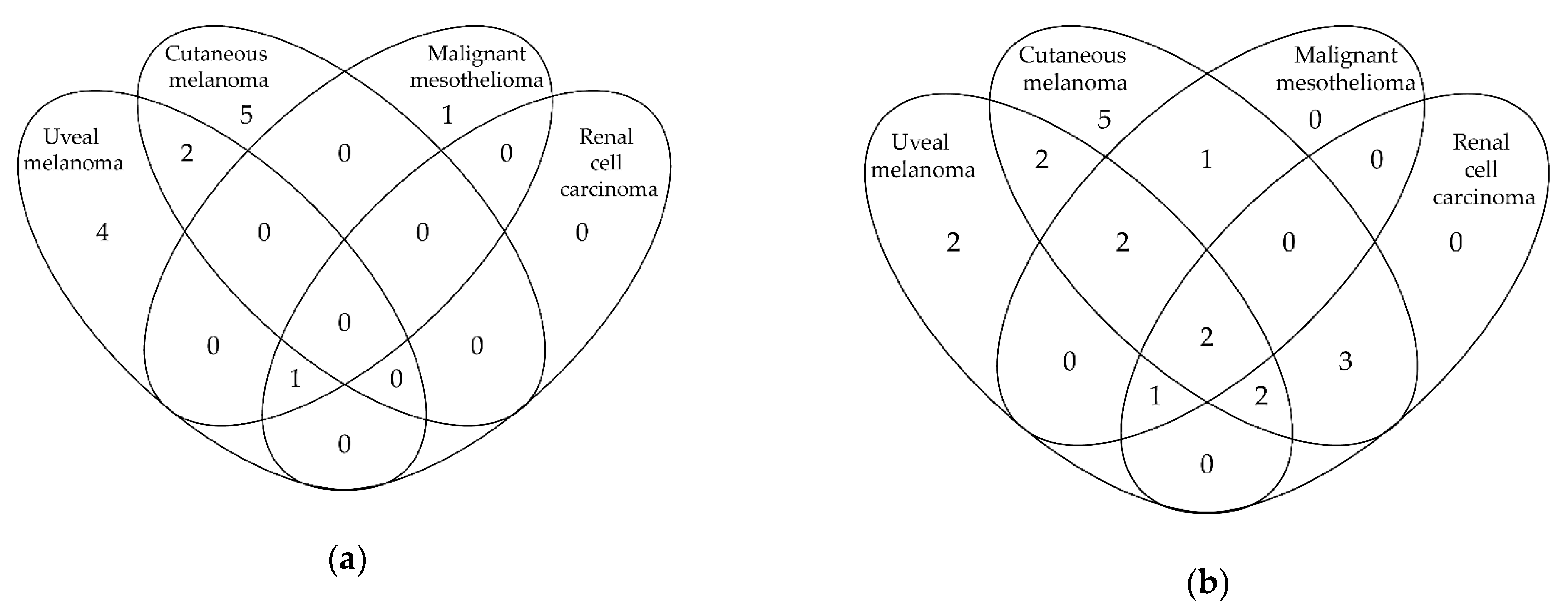
2.1. Identification of BAP1-TPDS Families
2.2. BAP1 Variants in The Netherlands
2.3. Clinical Characteristics of BAP1-TPDS Families in The Netherlands
2.4. BAP1-TPDS Core Malignancies
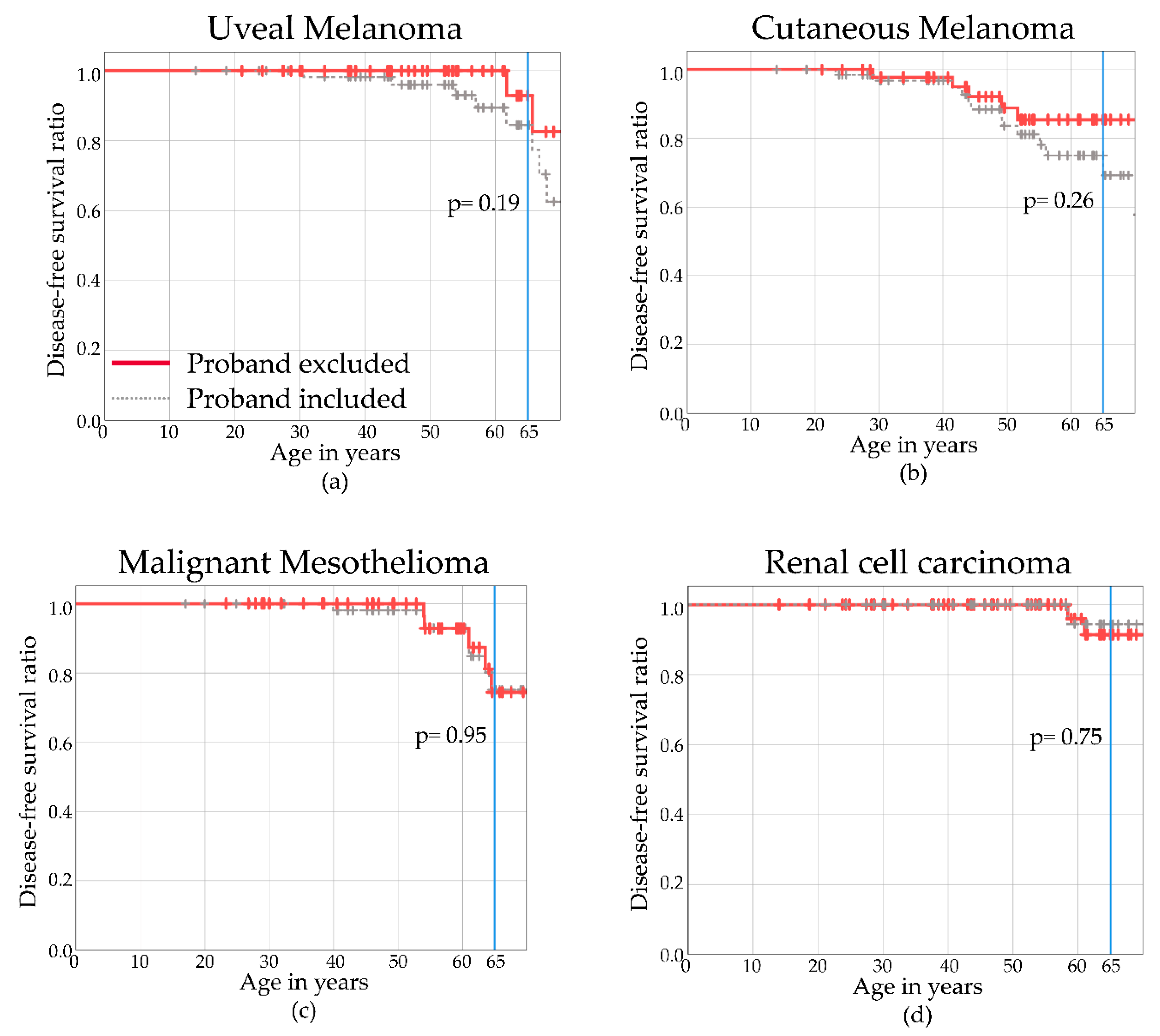
2.4.1. Uveal Melanoma
2.4.2. Cutaneous Melanoma
2.4.3. Malignant Mesothelioma
2.4.4. Renal Cell Carcinoma
2.4.5. BAP1-Inactivated Nevus
2.4.6. Other Malignancies
2.5. Disease-Free Survival
3. Discussion
3.1. BAP1-TPDS-Associated Malignancies
3.1.1. Different Types of Melanomas
3.1.2. Mesothelial Malignancy
3.1.3. Renal Cancer
3.1.4. Non-Melanoma Skin Tumors
3.1.5. Tumor Frequencies Compared to International Cohorts
3.2. Guidelines for Referral for Genetic Analysis
3.3. Proposed Surveillance Guidelines in BAP1-TPDS
4. Materials and Methods
4.1. Clinical Mutational Data
4.2. Clinical Review
4.3. Survival Curves
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pilarski, R.; Rai, K.; Cebulla, C.; Abdel-Rahman, M. BAP1 Tumor Predisposition Syndrome. In GeneReviews((R)); GeneReviews is a Registered Trademark of the University of Washington, Seattle. All Rights Reserved; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, M.H.; Pilarski, R.; Cebulla, C.M.; Massengill, J.B.; Christopher, B.N.; Boru, G.; Hovland, P.; Davidorf, F.H. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J. Med. Genet. 2011, 48, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Njauw, C.N.; Kim, I.; Piris, A.; Gabree, M.; Taylor, M.; Lane, A.M.; DeAngelis, M.M.; Gragoudas, E.; Duncan, L.M.; Tsao, H. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS ONE 2012, 7, e35295. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Ferris, L.K.; Baumann, F.; Napolitano, A.; Lum, C.A.; Flores, E.G.; Gaudino, G.; Powers, A.; Bryant-Greenwood, P.; Krausz, T.; et al. BAP1 cancer syndrome: Malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J. Transl. Med. 2012, 10, 179. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.; Talarchek, J.; Schindeler, K.; Saraiva, E.; Penney, L.S.; Ludman, M.; Testa, J.R. Further evidence for germline BAP1 mutations predisposing to melanoma and malignant mesothelioma. Cancer Genet. 2013, 206, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Fried, I.; Ulz, P.; Stacher, E.; Popper, H.; Murali, R.; Kutzner, H.; Lax, S.; Smolle-Juttner, F.; Geigl, J.B.; et al. Toward an improved definition of the tumor spectrum associated with BAP1 germline mutations. J. Clin. Oncol. 2012, 30, e337–e340. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Hebert, L.; Jacquemin, V.; Gad, S.; Caux-Moncoutier, V.; Dubois-d’Enghien, C.; Richaudeau, B.; Renaudin, X.; Sellers, J.; Nicolas, A.; et al. Germline BAP1 mutations predispose to renal cell carcinomas. Am. J. Hum. Genet. 2013, 92, 974–980. [Google Scholar] [CrossRef]
- Farley, M.N.; Schmidt, L.S.; Mester, J.L.; Pena-Llopis, S.; Pavia-Jimenez, A.; Christie, A.; Vocke, C.D.; Ricketts, C.J.; Peterson, J.; Middelton, L.; et al. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol. Cancer Res. 2013, 11, 1061–1071. [Google Scholar] [CrossRef]
- Pilarski, R.; Cebulla, C.M.; Massengill, J.B.; Rai, K.; Rich, T.; Strong, L.; McGillivray, B.; Asrat, M.J.; Davidorf, F.H.; Abdel-Rahman, M.H. Expanding the clinical phenotype of hereditary BAP1 cancer predisposition syndrome, reporting three new cases. Genes Chromosom. Cancer 2014, 53, 177–182. [Google Scholar] [CrossRef]
- Walpole, S.; Pritchard, A.L.; Cebulla, C.M.; Pilarski, R.; Stautberg, M.; Davidorf, F.H.; de la Fouchardiere, A.; Cabaret, O.; Golmard, L.; Stoppa-Lyonnet, D.; et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J. Natl. Cancer Inst. 2018, 110, 1328–1341. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Abedalthagafi, M.; Vaubel, R.A.; Merrill, P.H.; Nayyar, N.; Gill, C.M.; Brewster, R.; Bi, W.L.; Agarwalla, P.K.; Thorner, A.R.; et al. Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas. Neuro. Oncol. 2017, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- de la Fouchardiere, A.; Cabaret, O.; Savin, L.; Combemale, P.; Schvartz, H.; Penet, C.; Bonadona, V.; Soufir, N.; Bressac-de Paillerets, B. Germline BAP1 mutations predispose also to multiple basal cell carcinomas. Clin. Genet. 2015, 88, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Rawson, R.V.; Watson, G.F.; Maher, A.M.; McCarthy, S.W.; Thompson, J.F.; Scolyer, R.A. Germline BAP1 mutations also predispose to cutaneous squamous cell carcinoma. Pathology 2017, 49, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.J.; Rush, P.S.; Tsao, H.; Duncan, L.M. BRCA1-Associated Protein (BAP1) inactivated melanocytic tumors. J. Cutan. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 2011, 43, 1018–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garfield, E.M.; Walton, K.E.; Quan, V.L.; VandenBoom, T.; Zhang, B.; Kong, B.Y.; Isales, M.C.; Panah, E.; Kim, G.; Gerami, P. Histomorphologic spectrum of germline-related and sporadic BAP1-inactivated melanocytic tumors. J. Am. Acad. Dermatol. 2018, 79, 525–534. [Google Scholar] [CrossRef] [PubMed]
- de la Fouchardiere, A.; Cabaret, O.; Petre, J.; Aydin, S.; Leroy, A.; de Potter, P.; Pissaloux, D.; Haddad, V.; Bressac-de Paillerets, B.; Janin, N. Primary leptomeningeal melanoma is part of the BAP1-related cancer syndrome. Acta Neuropathol. 2015, 129, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Wadt, K.; Choi, J.; Chung, J.Y.; Kiilgaard, J.; Heegaard, S.; Drzewiecki, K.T.; Trent, J.M.; Hewitt, S.M.; Hayward, N.K.; Gerdes, A.M.; et al. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma. Res. 2012, 25, 815–818. [Google Scholar] [CrossRef]
- Rai, K.; Pilarski, R.; Cebulla, C.M.; Abdel-Rahman, M.H. Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin. Genet. 2016, 89, 285–294. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019. [Google Scholar] [CrossRef]
- Gupta, M.P.; Lane, A.M.; DeAngelis, M.M.; Mayne, K.; Crabtree, M.; Gragoudas, E.S.; Kim, I.K. Clinical Characteristics of Uveal Melanoma in Patients with Germline BAP1 Mutations. JAMA Ophthalmol. 2015, 133, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Turunen, J.A.; Markkinen, S.; Wilska, R.; Saarinen, S.; Raivio, V.; Tall, M.; Lehesjoki, A.E.; Kivela, T.T. BAP1 Germline Mutations in Finnish Patients with Uveal Melanoma. Ophthalmology 2016, 123, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Repo, P.; Jarvinen, R.S.; Jantti, J.E.; Markkinen, S.; Tall, M.; Raivio, V.; Turunen, J.A.; Kivela, T.T. Population-based analysis of BAP1 germline variations in patients with uveal melanoma. Hum. Mol. Genet. 2019, 28, 2415–2426. [Google Scholar] [CrossRef]
- O’Shea, S.J.; Robles-Espinoza, C.D.; McLellan, L.; Harrigan, J.; Jacq, X.; Hewinson, J.; Iyer, V.; Merchant, W.; Elliott, F.; Harland, M.; et al. A population-based analysis of germline BAP1 mutations in melanoma. Hum. Mol. Genet. 2017, 26, 717–728. [Google Scholar] [CrossRef]
- Rusch, A.; Ziltener, G.; Nackaerts, K.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Prevalence of BRCA-1 associated protein 1 germline mutation in sporadic malignant pleural mesothelioma cases. Lung Cancer 2015, 87, 77–79. [Google Scholar] [CrossRef]
- Betti, M.; Casalone, E.; Ferrante, D.; Romanelli, A.; Grosso, F.; Guarrera, S.; Righi, L.; Vatrano, S.; Pelosi, G.; Libener, R.; et al. Inference on germline BAP1 mutations and asbestos exposure from the analysis of familial and sporadic mesothelioma in a high-risk area. Genes Chromosom. Cancer 2015, 54, 51–62. [Google Scholar] [CrossRef]
- Sneddon, S.; Leon, J.S.; Dick, I.M.; Cadby, G.; Olsen, N.; Brims, F.; Allcock, R.J.; Moses, E.K.; Melton, P.E.; de Klerk, N.; et al. Absence of germline mutations in BAP1 in sporadic cases of malignant mesothelioma. Gene 2015, 563, 103–105. [Google Scholar] [CrossRef] [Green Version]
- Panou, V.; Gadiraju, M.; Wolin, A.; Weipert, C.M.; Skarda, E.; Husain, A.N.; Patel, J.D.; Rose, B.; Zhang, S.R.; Weatherly, M.; et al. Frequency of Germline Mutations in Cancer Susceptibility Genes in Malignant Mesothelioma. J. Clin. Oncol. 2018, Jco2018785204. [Google Scholar]
- Hassan, R.; Morrow, B.; Thomas, A.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Gadiraju, M.; Panou, V.; Gao, S.; Mian, I.; et al. Inherited predisposition to malignant mesothelioma and overall survival following platinum chemotherapy. Proc. Natl. Acad. Sci. USA 2019, 116, 9008–9013. [Google Scholar] [CrossRef] [Green Version]
- Potjer, T.P.; Bollen, S.; Grimbergen, A.; van Doorn, R.; Gruis, N.A.; van Asperen, C.J.; Hes, F.J.; van der Stoep, N. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int. J. Cancer 2019, 144, 2453–2464. [Google Scholar] [CrossRef] [PubMed]
- Ohar, J.A.; Cheung, M.; Talarchek, J.; Howard, S.E.; Howard, T.D.; Hesdorffer, M.; Peng, H.; Rauscher, F.J.; Testa, J.R. Germline BAP1 Mutational Landscape of Asbestos-Exposed Malignant Mesothelioma Patients with Family History of Cancer. Cancer Res. 2016, 76, 206–215. [Google Scholar] [CrossRef]
- Mensenkamp, A.R.; (Radboud University, Nijmegen, The Netherlands); Sijmons, R.H.; (University of Groningen, Groningen, The Netherlands); van der Hout, A.; (University of Groningen, Groningen, The Netherlands). Personal communication, 2019.
- Vreeswijk, M.P.; van der Klift, H.M. Analysis and interpretation of RNA splicing alterations in genes involved in genetic disorders. Methods Mol. Biol. 2012, 867, 49–63. [Google Scholar] [PubMed]
- Wadt, K.A.; Aoude, L.G.; Johansson, P.; Solinas, A.; Pritchard, A.; Crainic, O.; Andersen, M.T.; Kiilgaard, J.F.; Heegaard, S.; Sunde, L.; et al. A recurrent germline BAP1 mutation and extension of the BAP1 tumor predisposition spectrum to include basal cell carcinoma. Clin. Genet. 2015, 88, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.; Aspesi, A.; Biasi, A.; Casalone, E.; Ferrante, D.; Ogliara, P.; Gironi, L.C.; Giorgione, R.; Farinelli, P.; Grosso, F.; et al. CDKN2A and BAP1 germline mutations predispose to melanoma and mesothelioma. Cancer Lett. 2016, 378, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Van Paassen, B.W.; (Erasmus University Rotterdam, Rotterdam, The Netherlands). Personal communication, 2019.
- Cabaret, O.; Perron, E.; Bressac-de Paillerets, B.; Soufir, N.; de la Fouchardiere, A. Occurrence of BAP1 germline mutations in cutaneous melanocytic tumors with loss of BAP1-expression: A pilot study. Genes Chromosom. Cancer 2017, 56, 691–694. [Google Scholar] [CrossRef]
- Dutch National Cancer Registration 2013-2017, IKNL. Personal communication, 2019.
- Dono, M.; Angelini, G.; Cecconi, M.; Amaro, A.; Esposito, A.I.; Mirisola, V.; Maric, I.; Lanza, F.; Nasciuti, F.; Viaggi, S.; et al. Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in uveal melanoma: detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. Br J. Cancer 2014, 110, 1058–1065. [Google Scholar] [CrossRef] [Green Version]
- Scholz, S.L.; Moller, I.; Reis, H.; Susskind, D.; van de Nes, J.A.P.; Leonardelli, S.; Schilling, B.; Livingstone, E.; Schimming, T.; Paschen, A.; et al. Frequent GNAQ, GNA11, and EIF1AX Mutations in Iris Melanoma. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3464–3470. [Google Scholar] [CrossRef]
- Van Poppelen, N.M.; Vaarwater, J.; Mudhar, H.S.; Sisley, K.; Rennie, I.G.; Rundle, P.; Brands, T.; van den Bosch, Q.C.C.; Mensink, H.W.; de Klein, A.; et al. Genetic Background of Iris Melanomas and Iris Melanocytic Tumors of Uncertain Malignant Potential. Ophthalmology 2018, 125, 904–912. [Google Scholar] [CrossRef]
- Cruz, F., 3rd; Rubin, B.P.; Wilson, D.; Town, A.; Schroeder, A.; Haley, A.; Bainbridge, T.; Heinrich, M.C.; Corless, C.L. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Res. 2003, 63, 5761–5766. [Google Scholar]
- Zuidervaart, W.; van Nieuwpoort, F.; Stark, M.; Dijkman, R.; Packer, L.; Borgstein, A.M.; Pavey, S.; van der Velden, P.; Out, C.; Jager, M.J.; et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br. J. Cancer 2005, 92, 2032–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.R.; Nanji, A.A.; Galor, A.; Karp, C.L. Management of conjunctival malignant melanoma: A review and update. Expert Rev. Ophthalmol. 2014, 9, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Missotten, G.S.; Keijser, S.; De Keizer, R.J.; De Wolff-Rouendaal, D. Conjunctival melanoma in the Netherlands: A nationwide study. Investig. Ophthalmol. Vis. Sci. 2005, 46, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Westekemper, H.; Murali, R.; Mach, M.; Schilling, B.; Wiesner, T.; Schimming, T.; Livingstone, E.; Sucker, A.; Grabellus, F.; et al. Conjunctival melanomas harbor BRAF and NRAS mutations and copy number changes similar to cutaneous and mucosal melanomas. Clin. Cancer Res. 2013, 19, 3143–3152. [Google Scholar] [CrossRef]
- Larsen, A.C.; Dahl, C.; Dahmcke, C.M.; Lade-Keller, J.; Siersma, V.D.; Toft, P.B.; Coupland, S.E.; Prause, J.U.; Guldberg, P.; Heegaard, S. BRAF mutations in conjunctival melanoma: Investigation of incidence, clinicopathological features, prognosis and paired premalignant lesions. Acta Ophthalmol. 2016, 94, 463–470. [Google Scholar] [CrossRef]
- Cheung, M.; Kadariya, Y.; Talarchek, J.; Pei, J.; Ohar, J.A.; Kayaleh, O.R.; Testa, J.R. Germline BAP1 mutation in a family with high incidence of multiple primary cancers and a potential gene-environment interaction. Cancer Lett. 2015, 369, 261–265. [Google Scholar] [CrossRef]
- Bianchi, C.; Bianchi, T. Malignant mesothelioma: Global incidence and relationship with asbestos. Ind. Health 2007, 45, 379–387. [Google Scholar] [CrossRef]
- Franko, A.; Kotnik, N.; Goricar, K.; Kovac, V.; Dodic-Fikfak, M.; Dolzan, V. The Influence of Genetic Variability on the Risk of Developing Malignant Mesothelioma. Radiol. Oncol. 2018, 52, 105–111. [Google Scholar] [CrossRef]
- Kadariya, Y.; Cheung, M.; Xu, J.; Pei, J.; Sementino, E.; Menges, C.W.; Cai, K.Q.; Rauscher, F.J.; Klein-Szanto, A.J.; Testa, J.R. Bap1 Is a Bona Fide Tumor Suppressor: Genetic Evidence from Mouse Models Carrying Heterozygous Germline Bap1 Mutations. Cancer Res. 2016, 76, 2836–2844. [Google Scholar] [CrossRef]
- Xu, J.; Kadariya, Y.; Cheung, M.; Pei, J.; Talarchek, J.; Sementino, E.; Tan, Y.; Menges, C.W.; Cai, K.Q.; Litwin, S.; et al. Germline mutation of Bap1 accelerates development of asbestos-induced malignant mesothelioma. Cancer Res. 2014, 74, 4388–4397. [Google Scholar] [CrossRef]
- Busam, K.J.; Sung, J.; Wiesner, T.; von Deimling, A.; Jungbluth, A. Combined BRAF(V600E)-positive melanocytic lesions with large epithelioid cells lacking BAP1 expression and conventional nevomelanocytes. Am. J. Surg. Pathol. 2013, 37, 193–199. [Google Scholar] [CrossRef]
- Marusic, Z.; Buljan, M.; Busam, K.J. Histomorphologic spectrum of BAP1 negative melanocytic neoplasms in a family with BAP1-associated cancer susceptibility syndrome. J. Cutan. Pathol. 2015, 42, 406–412. [Google Scholar] [CrossRef]
- Ardakani, N.M.; Palmer, D.L.; Wood, B.A. BAP1 deficient malignant melanoma arising from the intradermal component of a congenital melanocytic naevus. Pathology 2015, 47, 707–710. [Google Scholar] [CrossRef]
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Austrilia, 2017. [Google Scholar]
- Erfelijk en Familair Melanoom. Available online: https://www.stoet.nl/wp-content/uploads/2017/04/STOET-Richtlijnenboekje-april2017_DEF.pdf (accessed on 2 June 2019).
- Pastorino, S.; Yoshikawa, Y.; Pass, H.I.; Emi, M.; Nasu, M.; Pagano, I.; Takinishi, Y.; Yamamoto, R.; Minaai, M.; Hashimoto-Tamaoki, T.; et al. A Subset of Mesotheliomas With Improved Survival Occurring in Carriers of BAP1 and Other Germline Mutations. J. Clin. Oncol. 2018, Jco2018790352. [Google Scholar] [CrossRef]
- Motzer, R.J.; Jonasch, E.; Agarwal, N.; Bhayani, S.; Bro, W.P.; Chang, S.S.; Choueiri, T.K.; Costello, B.A.; Derweesh, I.H.; Fishman, M.; et al. Kidney Cancer, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2017, 15, 804–834. [Google Scholar] [CrossRef]
- Aoude, L.G.; Gartside, M.; Johansson, P.; Palmer, J.M.; Symmons, J.; Martin, N.G.; Montgomery, G.W.; Hayward, N.K. Prevalence of Germline BAP1, CDKN2A, and CDK4 Mutations in an Australian Population-Based Sample of Cutaneous Melanoma Cases. Twin Res. Hum. Genet. 2015, 18, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, K.J.; Jaju, P.D.; Tang, J.Y.; Carbone, M.; Leachman, S.; Sarin, K.Y. Familial skin cancer syndromes: Increased melanoma risk. J. Am. Acad. Dermatol. 2016, 74, 423–434. [Google Scholar] [CrossRef]
- Haugh, A.M.; Njauw, C.N.; Bubley, J.A.; Verzi, A.E.; Zhang, B.; Kudalkar, E.; VandenBoom, T.; Walton, K.; Swick, B.L.; Kumar, R.; et al. Genotypic and Phenotypic Features of BAP1 Cancer Syndrome: A Report of 8 New Families and Review of Cases in the Literature. JAMA Dermatol. 2017, 153, 999–1006. [Google Scholar] [CrossRef]
- Ewens, K.G.; Lalonde, E.; Richards-Yutz, J.; Shields, C.L.; Ganguly, A. Comparison of Germline versus Somatic BAP1 Mutations for Risk of Metastasis in Uveal Melanoma. BMC Cancer 2018, 18, 1172. [Google Scholar] [CrossRef]
- Cebulla, C.M.; Binkley, E.M.; Pilarski, R.; Massengill, J.B.; Rai, K.; Liebner, D.A.; Marino, M.J.; Singh, A.D.; Abdel-Rahman, M.H. Analysis of BAP1 Germline Gene Mutation in Young Uveal Melanoma Patients. Ophthalmic. Genet. 2015, 36, 126–131. [Google Scholar] [CrossRef]
- Abdel-Rahman, M.H.; Pilarski, R.; Ezzat, S.; Sexton, J.; Davidorf, F.H. Cancer family history characterization in an unselected cohort of 121 patients with uveal melanoma. Fam. Cancer 2010, 9, 431–438. [Google Scholar] [CrossRef]
- Rai, K.; Pilarski, R.; Boru, G.; Rehman, M.; Saqr, A.H.; Massengill, J.B.; Singh, A.; Marino, M.J.; Davidorf, F.H.; Cebulla, C.M.; et al. Germline BAP1 alterations in familial uveal melanoma. Genes Chromosomes Cancer 2017, 56, 168–174. [Google Scholar] [CrossRef]
- Betti, M.; Aspesi, A.; Ferrante, D.; Sculco, M.; Righi, L.; Mirabelli, D.; Napoli, F.; Rondon-Lagos, M.; Casalone, E.; Vignolo Lutati, F.; et al. Sensitivity to asbestos is increased in patients with mesothelioma and pathogenic germline variants in BAP1 or other DNA repair genes. Genes Chromosomes Cancer 2018, 57, 573–583. [Google Scholar] [CrossRef]
- Christensen, M.B.; Wadt, K.; Jensen, U.B.; Lautrup, C.K.; Bojesen, A.; Krogh, L.N.; Overeem Hansen, T.V.; Gerdes, A.M. Exploring the hereditary background of renal cancer in Denmark. PLoS ONE 2019, 14, e0215725. [Google Scholar] [CrossRef]
- Gruis, N.A.; Sandkuijl, L.A.; van der Velden, P.A.; Bergman, W.; Frants, R.R. CDKN2 explains part of the clinical phenotype in Dutch familial atypical multiple-mole melanoma (FAMMM) syndrome families. Melanoma. Res. 1995, 5, 169–177. [Google Scholar] [CrossRef]
- Read, J.; Wadt, K.A.; Hayward, N.K. Melanoma genetics. J. Med. Genet. 2016, 53, 1–14. [Google Scholar] [CrossRef]
- Rodrigues, M.; Mobuchon, L.; Houy, A.; Fievet, A.; Gardrat, S.; Barnhill, R.L.; Popova, T.; Servois, V.; Rampanou, A.; Mouton, A.; et al. Outlier response to anti-PD1 in uveal melanoma reveals germline MBD4 mutations in hypermutated tumors. Nat. Commun. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Johansson, P.A.; Stark, A.; Palmer, J.M.; Bigby, K.; Brooks, K.; Rolfe, O.; Pritchard, A.L.; Whitehead, K.; Warrier, S.; Glasson, W.; et al. Prolonged stable disease in a uveal melanoma patient with germline MBD4 nonsense mutation treated with pembrolizumab and ipilimumab. Immunogenetics 2019, 71, 433–436. [Google Scholar] [CrossRef]
- Nielsen, M.; Dogrusoz, M.; Bleeker, J.C.; Kroes, W.G.; van Asperen, C.A.; Marinkovic, M.; Luyten, G.P.; Jager, M.J. The genetic basis of uveal melanoma. J. Fr. Ophtalmol. 2015, 38, 516–521. [Google Scholar] [CrossRef]
- Erfelijk en Familiar Niercelcarcinoom. Available online: https://www.stoet.nl/wp-content/uploads/2017/04/STOET-Richtlijnenboekje-april2017_DEF.pdf (accessed on 2 June 2019).
- Carlo, M.I.; Mukherjee, S.; Mandelker, D.; Vijai, J.; Kemel, Y.; Zhang, L.; Knezevic, A.; Patil, S.; Ceyhan-Birsoy, O.; Huang, K.C.; et al. Prevalence of Germline Mutations in Cancer Susceptibility Genes in Patients With Advanced Renal Cell Carcinoma. JAMA Oncol. 2018, 4, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef]
- Smith, M.J. Germline and somatic mutations in meningiomas. Cancer Genet. 2015, 208, 107–114. [Google Scholar] [CrossRef]
- Christiaans, I.; Kenter, S.B.; Brink, H.C.; van Os, T.A.; Baas, F.; van den Munckhof, P.; Kidd, A.M.; Hulsebos, T.J. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J. Med. Genet. 2011, 48, 93–97. [Google Scholar] [CrossRef]
- Evans, D.G. Neurofibromatosis type 2: Genetic and clinical features. Ear. Nose Throat. J. 1999, 78, 97–100. [Google Scholar] [CrossRef]
- Evans, D.G.; Baser, M.E.; O’Reilly, B.; Rowe, J.; Gleeson, M.; Saeed, S.; King, A.; Huson, S.M.; Kerr, R.; Thomas, N.; et al. Management of the patient and family with neurofibromatosis 2: A consensus conference statement. Br J. Neurosurg. 2005, 19, 5–12. [Google Scholar] [CrossRef]
- Guerrini-Rousseau, L.; Dufour, C.; Varlet, P.; Masliah-Planchon, J.; Bourdeaut, F.; Guillaud-Bataille, M.; Abbas, R.; Bertozzi, A.I.; Fouyssac, F.; Huybrechts, S.; et al. Germline SUFU mutation carriers and medulloblastoma: Clinical characteristics, cancer risk, and prognosis. Neuro. Oncol. 2018, 20, 1122–1132. [Google Scholar] [CrossRef]
- Cancer risks in BRCA2 mutation carriers. J. Natl. Cancer Inst. 1999, 91, 1310–1316. [CrossRef]
- Star, P.; Goodwin, A.; Kapoor, R.; Conway, R.M.; Long, G.V.; Scolyer, R.A.; Guitera, P. Germline BAP1-positive patients: The dilemmas of cancer surveillance and a proposed interdisciplinary consensus monitoring strategy. Eur. J. Cancer 2018, 92, 48–53. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Family Identifier | Reason for Testing | Tumors Proband (Age) | BAP1-TPDS-Associated Malignancies in Family History (Excl. Proband) |
|---|---|---|---|
| NL-1 | UM and family history of multiple UMs | UM (67) | UM ×3, NMSC ×2 |
| NL-2 | Multiple BINs | BIN × (39 ×2) NMSC (39) | UM, CM ×4, MMe ×7, NMSC ×5 |
| NL-3 | Multiple BINs | BIN ×5 (29 ×5) | CM, NMSC ×4 |
| NL-4 | MMe and family history of MMe | MMe pt (39) | CM, MMe 2×, NMSC 2× |
| NL-5 | UM & meningioma and family history of UM, CM, meningioma, and RCC | UM (66) NMSC (66) Meningioma (44) VS (51) | UM, CM, RCC, Meningioma |
| NL-6 | UM and family history of MMe | UM (30) | CM ×2, MMe, RCC, NMSC ×9 |
| NL-7 | Multiple BINs | BIN ×2 (22 ×2) | CM ×2, NMSC b |
| NL-8 | CM & conjunctival melanoma | CM (49) CoM (44) Lung cancer (54) | CM, RCC, NMSC |
| NL-9 | UM, MMe, and RCC | UM (57)—iris MMe pl (61) RCC (61) NMSC (57) B-cell lymphoma (58) | RCC |
| NL-10 | Multiple BINs | BIN ×2 (20, 26) NMSC (27) | CM a ×2, NMSC ×4 |
| NL-11 | Single BIN | BIN (55) Breast cancer (48) | UM, CM, NMSC ×13 |
| NL-12 | UM & CM | UM (53) CM (56) NMSC (38, 52) | CM, MMe, RCC |
| NL-13 | Multiple BINs | BIN ×4 (15 ×2, 18 ×2) | - |
| NL-14 | Familial CM b | CM ×2 (23, 27) NMSC ×2 (50, 53) | CM ×6, NMSC ×47, metastatic melanoma |
| NL-15 | Multiple BINs | BIN ×2 (21, 25) | UM |
| NL-16 | Familial CM b | CM (65) | CM ×3, RCC |
| NL-17 | Multiple BINs | BIN ×2 (14, 14) | CM, RCC, Meningioma |
| NL-18 | UM & HCC and family history of MMe & HCCs | UM (72) HCC (68) | CMa, MMe ×3, NMSC ×5 |
| NL-19 | CM and family history of UM | CM (44) | UM, NMSC |
| NL-20 | Familial CM b | CM (45) | CM ×3 |
| NL-21 | UM and CM | UM (44) CM (55) | CM a ×3, RCC |
| NL-22 | Familial CM | CM (47) NMSC ×7 (54, ?) Prostate cancer (55) | CM, NMSC ×2 |
| Family Identifier | Region | Germline Variant | Protein Change | Chromosome Position (GRCh37/hg19) | Pathogenicity, ACMG Classification [36] | Previously Published in | Previously Reported in |
|---|---|---|---|---|---|---|---|
| NL-1 | Exon 1 | c.35delC | p.(Pro12fs*) | g.52443860del | Pathogenic | Walpole et al., 2019 [12] | |
| NL-2 | Exon 1 | c.35_37+2delinsAGGG | p.(Pro12fs*) | g.52443856_52443860delinsCCCT | Pathogenic | New variant | |
| NL-3 | Exon 4 | c.178C>T | p.(Arg60*) | g.52442567G>A | Pathogenic | Walpole et al., 2019 [12] | Njauw et al., 2012 [4] Wadt et al., 2015 [36] |
| NL-4 | Exon 4 | c.182delA | p.(Lys61fs*) | g.52442563del | Pathogenic | New variant | |
| NL-5 | Exon 4 | c.188_189delCT | p.(Ser63fs*) | g.52442556_52442557del | Pathogenic | New variant | |
| NL-6 | Exon 6 | c.376_377delAG | p.(Ser126fs*) | g.52441475_52441476del | Pathogenic | New variant | |
| NL-7 | Exon 11 | c.1017_1048del a,b | p.(Gly340fs*) | g.52439194_52439225del | Pathogenic | Walpole et al., 2019 [12] | |
| NL-8 | Exon 12 | c.1153C>T | p.(Arg385*) | g.52438566G>A | Pathogenic | Njauw et al., 2012 [4] Betti et al., 2016 [37] | |
| NL-9 | Exon 13 | c.1530delT | p.(Ile511fs*) | g.52437631del | Pathogenic | New variant | |
| NL-10 | Exon 13 | c.1621delG | p.(Val541fs*) | g.52437540del | Pathogenic | New variant | |
| NL-11 | Exon 13 | c.1621delG | p.(Val541fs*) | g.52437540del | Pathogenic | New variant | |
| NL-12 | Exon 14 | c.1768C>T | p.(Gln590*) | g.52437276G>A | Pathogenic | Walpole et al., 2019 [12] | |
| NL-13 | Exon 14 | c.1819delA b | p.(Thr607Argfs*) | g.52437225del | Pathogenic | New variant | |
| NL-14 | Exon 15 | c.1936_1937insTT | p.(Tyr646fs*) | g.52436841_52436842insAA | Pathogenic | Walpole et al., 2019 [12] Potjer et al., 2019 [32] |
| Family Identifier | Region | Germline Variant | Protein Change | Chromosome Position (GRCh37/hg19) | Pathogenicity, ACMG Classification [38] | Previously Published in | Previously Reported in |
|---|---|---|---|---|---|---|---|
| NL-15 | Intron 2 | c.67+1G>C | p.? | g.52443729C>G | Likely pathogenic | Repo et al., 2019 [25] | |
| NL-16 | Intron 3 | c.122+1G>T a | p.? | g.52443569C>A | Likely pathogenic | Potjer et al., 2019 a [32] | |
| NL-17 | Intron 3 | c.122+5G>C | p.? | g.52443573G>C | Likely pathogenic b [34] | New variant | |
| NL-18 | Intron 6 | c.437+1G>T | p.? | g.52441414C>A | Likely pathogenic | New variant | |
| NL-19 | Intron 8 | c.660-2A>G | p.? | g.52440394T>C | Pathogenic | Walpole et al., 2019 [12] | |
| NL-20 | Intron 13 | c.1730-1G>A | p.? | g.52437315C>T | Likely pathogenic | Potjer et al., 2019 [32] | |
| NL-21 | Exon 4 | c.200A>G | p.(Asp67Gly) | g.52442545T>C | Likely pathogenic [39] | Walpole et al., 2019 [12] | Cabaret et al., 2017 [40] |
| NL-22 | Exon 13 | c.1387C>G | p.(Leu463Val) | g.52437774G>C | VUS | New variant |
| Item | Referral Indicated If | |
|---|---|---|
| Medical history | ≥2 BAP1-TPDS-associated tumors a,b | |
| Medical history and family history | 1 BAP1-TPDS-associated tumor and first- or second-degree relative with ≥1 BAP1-TPDS-associated tumor(s) a,b | |
| Young age of onset |
| age of onset before 40 years age of onset before 18 years [60] age of onset before 50 years [61] age of onset before 46 years [62] |
| Malignancy | Candidate Susceptibility Genes | References |
|---|---|---|
| Uveal melanoma | MBD4 | [74,75] |
| Cutaneous melanoma | CDKN2A, CDK4, MITF, POT1, ACD, TERF2IP, TERT | [32,72,73] |
| Malignant mesothelioma | CDKN2A, BRCA2, TMEM127, VHL, WT1 | [30,37] |
| Renal cell carcinoma | VHL, FLCN, SDHB, FH, MET, PTEN | [9,77,78,79] |
| Meningioma | NF2, SMARCB1, SMARCE1, SUFU | [80,81,82,83,84] |
| Cholangiocarcinoma | Unknown, possibly BRCA2 | [85] |
| Malignancy | Surveillance as Suggested by Rai et al. [21] | Surveillance as Suggested by Star et al. [86] | ||
|---|---|---|---|---|
| Uveal melanoma | -Dilated eye exams and ophthalmic imaging | -Starting at 11 years annually | -Dilated eye exams, fundus photography, and ocular ultrasound | -Starting at 16 years annually -From 30 years 6 monthly |
| Cutaneous melanoma | -Full body skin exam and self-skin exam | -Starting at 20 years annually | -Full body skin exam and total body photography | -Starting at 18 years 6 monthly |
| Malignant mesothelioma | -Physical examinations | -Annually | -Abdominal and respiratory examination | -Starting at age 30 |
| -Ultrasound exam or MRI -CT or MRI | -Between ages 30–55 biennially -After the age of 55 biennially | |||
| Renal cell carcinoma | -Ultrasound -MRI | -Anually -Biennially | Incorporated in mesothelioma surveillance recommendations | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chau, C.; van Doorn, R.; van Poppelen, N.M.; van der Stoep, N.; Mensenkamp, A.R.; Sijmons, R.H.; van Paassen, B.W.; van den Ouweland, A.M.W.; Naus, N.C.; van der Hout, A.H.; et al. Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: Path to Identification and a Proposal for Genetic Screening Guidelines. Cancers 2019, 11, 1114. https://doi.org/10.3390/cancers11081114
Chau C, van Doorn R, van Poppelen NM, van der Stoep N, Mensenkamp AR, Sijmons RH, van Paassen BW, van den Ouweland AMW, Naus NC, van der Hout AH, et al. Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: Path to Identification and a Proposal for Genetic Screening Guidelines. Cancers. 2019; 11(8):1114. https://doi.org/10.3390/cancers11081114
Chicago/Turabian StyleChau, Cindy, Remco van Doorn, Natasha M. van Poppelen, Nienke van der Stoep, Arjen R. Mensenkamp, Rolf H. Sijmons, Barbara W. van Paassen, Ans M. W. van den Ouweland, Nicole C. Naus, Annemieke H. van der Hout, and et al. 2019. "Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: Path to Identification and a Proposal for Genetic Screening Guidelines" Cancers 11, no. 8: 1114. https://doi.org/10.3390/cancers11081114