PTEN Tumor-Suppressor: The Dam of Stemness in Cancer
by
 , , and
, , and
Francesca Luongo
1,†, 
Francesca Colonna
1,†,
Federica Calapà
1,†,
Sara Vitale
1,
Micol E. Fiori
2,* and
Ruggero De Maria
1,3,* 1
Istituto di Patologia Generale, Università Cattolica del Sacro Cuore, Largo Francesco Vito 1, 00168 Rome, Italy
2
Department of Oncology and Molecular Medicine, Istituto Superiore di Sanità, 00161 Rome, Italy
3
Scientific Vice-Direction, Fondazione Policlinico Universitario “A. Gemelli”–I.R.C.C.S., Largo Francesco Vito 1-8, 00168 Rome, Italy
*
Authors to whom correspondence should be addressed.
†
These authors equally contributed.
Cancers 2019, 11(8), 1076; https://doi.org/10.3390/cancers11081076
Submission received: 2 July 2019
/
Revised: 24 July 2019
/
Accepted: 26 July 2019
/
Published: 30 July 2019
(This article belongs to the Special Issue PTEN: A Multifaceted Tumor Suppressor)
Abstract
:PTEN is one of the most frequently inactivated tumor suppressor genes in cancer. Loss or variation in PTEN gene/protein levels is commonly observed in a broad spectrum of human cancers, while germline PTEN mutations cause inherited syndromes that lead to increased risk of tumors. PTEN restrains tumorigenesis through different mechanisms ranging from phosphatase-dependent and independent activities, subcellular localization and protein interaction, modulating a broad array of cellular functions including growth, proliferation, survival, DNA repair, and cell motility. The main target of PTEN phosphatase activity is one of the most significant cell growth and pro-survival signaling pathway in cancer: PI3K/AKT/mTOR. Several shreds of evidence shed light on the critical role of PTEN in normal and cancer stem cells (CSCs) homeostasis, with its loss fostering the CSC compartment in both solid and hematologic malignancies. CSCs are responsible for tumor propagation, metastatic spread, resistance to therapy, and relapse. Thus, understanding how alterations of PTEN levels affect CSC hallmarks could be crucial for the development of successful therapeutic approaches. Here, we discuss the most significant findings on PTEN-mediated control of CSC state. We aim to unravel the role of PTEN in the regulation of key mechanisms specific for CSCs, such as self-renewal, quiescence/cell cycle, Epithelial-to-Mesenchymal-Transition (EMT), with a particular focus on PTEN-based therapy resistance mechanisms and their exploitation for novel therapeutic approaches in cancer treatment.
1. Introduction
First identified in 1997, PTEN (Phosphatase and TENsin homolog deleted on chromosome 10) is among the most commonly mutated tumor suppressor genes in human cancers [1,2,3,4] (Table 1). PTEN is a negative regulator of the phosphatidylinositol-3-kinase (PI3K)/AKT cascade, which controls cell growth, proliferation, survival, and metabolism. The fine regulation of this axis represents a critical node of oncogenic transformation [5].
PTEN is a haploinsufficient tumor-suppressor gene since the loss of 50% of its function compromises tumor suppression [6,7]. Further, even subtle variations of PTEN function can have dramatic effects on cancer predisposition and tumorigenesis, suggesting a strict dosage-dependency of PTEN tumor suppression [8]. The apparent paradox that complete ablation of PTEN can be detrimental to tumor growth, in the absence of other mutations, is due to the activation of p53-dependent senescence that counteracts tumor progression (obligate haploinsufficiency) [9]. Genetic ablation of PTEN leads to the development of different tumor types in mice [10,11,12]. In humans, germline mutations of PTEN cause a group of rare autosomal dominant syndromes (PTEN hamartoma tumor syndromes, PHTS), characterized by increased risk for specific malignancies (breast, thyroid, renal, and endometrial cancers) and neurodevelopmental disorders, such as autism and mental retardation [13,14,15]. In somatic cancers, including glioblastoma (GBM), endometrial, breast and prostate cancer, PTEN function can be compromised by a variety of mechanisms. In addition to genetic inactivation, PTEN expression is tightly controlled both at transcriptional and post-transcriptional level [5]. Epigenetic inhibitory mechanisms involving PTEN promoter hyper-methylation and histone acetylation were reported in several cancers [16,17,18,19,20]. Moreover, multiple transcription factors control PTEN expression and a number of microRNAs (miRNAs, miRs) were identified as PTEN suppressors. Interestingly, post-translational modifications including ubiquitination, phosphorylation, acetylation, oxidation and sumoylation or interaction with other proteins affecting the activity and subcellular localization of PTEN are often associated with tumorigenesis [5,21,22,23]. Furthermore, severe PTEN deficiency is associated with advanced tumor stage (70% in glioblastomas and 60% in advanced prostate cancers) and resistance to therapy, especially in targeted therapies for the receptor tyrosine kinases (RTKs) pathway (e.g., trastuzumab) [24]. Increasing evidence indicates PTEN as a prognostic and predictive biomarker for drug response in several tumors (e.g., prostate, breast, endometrium) [25]. Altogether, the multi-level control of PTEN expression depicts a highly dynamic landscape in which even subtle fluctuations of this protein can induce very significant effects.
PTEN is expressed early during embryonic development and ubiquitously in the adult [11]. The main PTEN catalytic activity is the lipid phosphatase [5,26]. PTEN dephosphorylates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) to phosphatidylinositol-4,5-bisphosphate (PIP2) [26]; PIP3 is generated through the phosphorylation of the 3-position of the PIP2 inositol ring by PI3K, a lipid kinase activated by RTKs and G-protein coupled receptors (GPCRs) [27]. The PI3K complex activates the 3-phosphoinositide-dependent kinase 1 (PDK1) and AKT [24]. Consequently, PTEN antagonizes the PI3K signaling [26,28] and the downstream activation of PDK1/AKT and AKT/mammalian target of rapamycin (mTOR) [27,29,30]. Through the inhibition of the oncogenic and pro-survival PI3K/AKT/mTOR axis, PTEN acts as a tumor suppressor by regulating transcription, translation, cell cycle progression, induction of cell death, stimulation of angiogenesis, and stem cell self-renewal [5]. In addition, this axis is an essential target for anti-cancer agents, especially in tumors displaying elevated activity of the mTOR pathway [31].
PTEN exerts a dual protein-phosphatase activity, targeting both phospho-serine/threonine and phospho-tyrosine substrates [5,32]. Besides the self-regulation (by auto-dephosphorylation), PTEN controls the activity of several target proteins as glycogen synthase kinase-3 (GSK3), BAD, Caspase 9, IκB, focal adhesion kinase 1, cAMP-responsive element-binding protein 1 (CREB1), proto-oncogene tyrosine-protein kinase SRC, insulin receptor substrate 1 (IRS1), modulating pivotal biological functions including inhibition of cell migration, invasion, cycle arrest and survival [5,33,34,35]. Interestingly, PTEN plays also a non-enzymatic role, behaving as a scaffold protein in both the nucleus and the cytoplasm [5]. Although the primary subcellular localization of PTEN is cytoplasmic and membrane-bound, PTEN protein can translocate into the nucleus where it controls critical cellular functions like genomic stability, pre-mRNA alternative splicing, apoptosis, cell cycle arrest and senescence [36,37]. These nuclear activities of PTEN are mainly lipid-phosphatase-independent [38,39]. PTEN nuclear localization occurs preferentially in primary, differentiated, and resting cells rather than cancer cell lines, underlining how the nuclear pool is critical for the tumor suppressive functions of PTEN [40,41,42,43,44]. Consistently, the absence of nuclear PTEN is associated with more aggressive cancers. These observations pave the road for the use of PTEN as a prognostic marker. Moreover, PTEN can also be secreted and transferred to neighboring cells, packaged and exported within exosomes. Upon transfer in target cells PTEN localizes in the cytoplasm and mitochondria, where it can regulate mitochondrial function and energy production, or in the nucleolus, where it inhibits ribosomal biogenesis [45,46,47,48]. Recent studies demonstrated the existence of a translational variant, named PTEN-Long (PTEN-L), characterized by an additional 173aa-domain at the N-terminal containing a secretion signal that enables this membrane-permeable isoform to be secreted and uptaken by recipient cells in a paracrine manner [46]. PTEN-L is expressed in normal tissues and detectable in human serum and plasma whereas lower expression is reported in human breast tumor and mouse models of glioblastoma [49]. Of note, administration of PTEN-L protein to tumor-bearing mice showed an interesting therapeutic potential [46]. Moreover, Liang and collaborators reported an N-terminally extended form, named PTEN-α, localized in the cytoplasm and mitochondria, where it can regulate mitochondrial function and energy production [47]. Further, another variant named PTEN-β, was recently identified in the nucleolus, where it binds to and dephosphorylates nucleolin, thus inhibiting ribosomal DNA transcription and ribosomal biogenesis [48]. Strategies focused at improving PTEN secretion and uptake by target cells may offer a feasible way for tumor treatment [5,50].
In addition to the intrinsic onco-suppressive function of PTEN, several studies underline the pivotal role of PTEN in the modulation of the tumor microenvironment, acting at different levels on cancer cells, stromal compartment, and immune response, thus controlling disease initiation, progression and metastases. PTEN loss in mammary stromal fibroblasts induces an oncogenic secretome that modifies the tumor microenvironment by promoting tumor angiogenesis, extracellular matrix (ECM) deposition, and macrophage recruitment, accelerating malignant transformation [51]. Moreover, a PTEN-null stroma can increase the expansion of mammary epithelial stem cells (MaSC), influencing the mammary stem cell niche [52]. In accordance, the simultaneous ablation of PTEN and Src homology phosphatase 2 (Shp2) in the hepatic microenvironment promotes the earlier expansion of liver tumor-initiating cells (TIC) and enhances tumorigenesis [53]. Further, PTEN-null senescent prostate cancer creates an immunosuppressive tumor microenvironment through cytokines secretion and recruitment of myeloid-derived suppressor cells [54]. Of note, another study revealed the plasticity of PTEN expression in the “co-evolution” between metastatic cells and the stroma: the unique brain metastatic microenvironment induces tumor cells to lose PTEN expression by astrocyte-derived exosomal miRNA targeting PTEN, promoting metastatic outgrowth [55].
Finally, despite the well-defined anti-tumoral function of PTEN in the greatest majority of cancers, in some specific contexts a pro-tumoral role of PTEN has been documented. Specifically, in pre B acute lymphoblastic leukemia (ALL), a cancer of immature B cells, PTEN ablation unexpectedly prevents malignant transformation: loss of one or both alleles of PTEN induces pre-B ALL cells death and is able to clear transplanted recipient mice of leukemia [56]. In line with these data, it was shown that PTEN can acquire tumor-promoting functions by enhancing the stability of gain-of-function p53 mutants in glioma cells, both in vitro and in vivo [57]. Another novel gain-of-function PTEN mutation, frequently observed in gliomas, is the truncation of the C-terminal region (PTENΔ51) that shifts PTEN towards tumor promoting activity [58].
The term Cancer Stem Cells (CSCs) defines a subpopulation of multipotent cells with unique properties of self-renewal and long-term clonal maintenance, responsible for tumor progression, metastatic spread, recurrence and therapeutic resistance [59,60]. This nomenclature is based on the common properties that these cells share with normal stem cells, behaving as a reservoir for cancer. However, the definition of stemness applied to the complex tumor context relies on peculiar parameters that do not strictly stick to classical concepts [61].
Although CSCs do not necessarily coincide with the “cell of origin” of the tumor, they are endowed with and defined by the capacity to sustain serial xenograft formation in mice. This consideration has led to the interchangeable use of different designations, as CSCs and TICs. Given the capacity to foster tumor propagation and spread, these cells have also been referred to as Tumor Propagating Cells (TPCs). For ease of reading, we adopt throughout this review the term CSCs to refer to cancer cells endowed with stem-like phenotypes, tumorigenic potential and therapy resistance properties.
CSCs were first described in acute myeloid leukemia, where the clear hierarchical organization of stem/progenitor/precursor cells reflects the pattern observed in normal hematopoiesis. Subsequently, putative CSCs were identified in multiple solid tumors, including melanoma, brain, breast, colon, lung, and prostate cancers at variable frequencies across different tumor types (2–40%) [62,63]. Several groups proposed the isolation of CSCs based on the expression of specific markers such as CD34, CD38, CD44, CD24, CD90, CD133, aldehyde dehydrogenase 1, Hoechst 33342 dye exclusion [64] and/or by the capacity to grow in selective media [65,66]. However, cancer cell stemness is better defined by functional assays, such as the ability to self-renew and differentiate, giving rise to a tumor reproducing the patient’s pathology and propagating in serial transplantation experiments [67]. Furthermore, CSCs exhibit multiple treatment resistance features, including upregulation of drug-efflux pumps, a superior DNA-repair capacity, enhanced protection against ROS, quiescent state, immune evasion [68].
Recent advances allowed genetic-lineage tracing experiments that enabled the in-situ identification and tracking of stem cells in solid tissues/tumors. These studies revealed discrepancies with the models derived by transplantation-based approaches, possibly because the potential of a given cell type does not necessarily correspond to the real behavior in the original environment. Interestingly, lineage-tracing and other studies have pointed out the concept of ‘cancer stem state’, where transit-amplifying cells can reversibly acquire the stem cell attributes in response to epigenetic and microenvironmental factors [59,68,69,70,71]. This CSC feature, named ‘plasticity’, enhances the complexity of intratumor heterogeneity and urges the understanding of the crosstalk between tumor and non-tumor cells as well as the identification of key players responsible for the transition into the stem cell status, which would facilitate the identification of therapeutic strategies able to eradicate cancer.
Reportedly, several pathways exerting a pivotal role in the control of normal stem cell self-renewal and differentiation are altered in CSCs, such as Wnt, Hedgehog, and Notch [72,73]. Given the central function of PTEN in the regulation of these and other pathways controlling stem cell maintenance, homeostasis, self-renewal and migration, as well as the crosstalk with tumor microenvironment, the modulation of this tumor suppressor is deeply involved in the biology of CSCs [49,74]. In this review, we will discuss the current advances in PTEN-mediated regulation of CSC hallmarks, with a particular focus on self-renewal, quiescence, EMT/migration, survival, and therapy resistance.
2. PTEN-Mediated Control of CSCs Hallmarks
2.1. PTEN and Self-Renewal
Self-renewal is a specific feature of stem cells, consisting of controlled division (symmetric or asymmetric) able to maintain the undifferentiated state to the progenies, thus indefinitely perpetuating the stem cell pool [119]. In a variety of malignancies, the ability to alter normal self-renewal pathways allows CSCs to drive malignant transformation [120]. PTEN, p53 and several pathways such as Notch, Wnt, and Hedgehog regulate the self-renewal of healthy stem cells. Deregulation of these pathways promotes stem cell expansion and increased spheroid-forming ability in vitro with the consequent acquisition of a high tumorigenic potential [121]. Loss of PTEN in normal stem cells contributes to malignant transformation in several tissues. Conditional deletion of PTEN in adult hematopoietic stem cells [HSCs] triggers leukemia onset in mice, while alterations of PTEN functions in bronchioalveolar and neural stem cells are involved in increasing their expansion and neoplastic potential [122,123,124].
In cancer cells, several data pointed out the role of PTEN in the control of self-renewal, measured in vitro as sphere-forming activity: PTEN mutant cells display high tumor-sphere formation capacity and maintenance upon serial passages in glioma, prostate, and breast cancer models [74,125,126,127]. Moreover, the mutation of one or both alleles of PTEN is associated with the successful generation of proliferating neurosphere cultures from glioblastoma surgical specimens, suggesting a higher capacity to self-renew [128].
In vivo, PTEN deletion promotes the expansion of CSCs and enhances their ability to form tumors capable of serial passaging in NOD/SCID mice. Serial dilution experiments in mice demonstrated that PTEN knockout cells display a higher tumorigenic potential/CSCs content compared to wild type cells, in models of breast, prostate and lung cancers [126,127,129]. Moreover, both in normal and malignant mammary epithelial cells, PTEN knockdown fosters the CSC compartment, through the generation of hyperplasic lesions and contributing to cancer onset in humanized mice [127].
Interestingly, the concomitant loss of PTEN and p53 promotes an undifferentiated status in neural stem cells and increases their proliferation and self-renewal through C-MYC regulation [130]. Consistently, in primary prostate epithelial cells depleted for both PTEN and P53, a higher self-renewal capacity compared to wild type primary cells was reported. Accordingly, CRE-mediated depletion of PTEN and P53 in normal mouse prostate epithelial cells generates highly tumorigenic cells, containing a stable sub-population endowed with self-renewal and multi-lineage differentiation capacities [131,132].
Mechanistically, PTEN loss promotes self-renewal through the activation of PI3K/AKT signaling, which, in turn, modulates several downstream pathways. Indeed, the inhibition of PI3K/AKT/mTOR activity by specific inhibitors leads to a decrease in the CSC population in nasopharyngeal carcinoma [133], glioma [74] pancreatic carcinoma [134], lung cancer [129,135], prostate cancer [126,131] and breast cancer [127]. Besides the involvement of the PI3K/AKT axis, the AKT-mediated phosphorylation of FOXO3a and GSK-3β increases the nuclear β-catenin localization, which potentiates tumorigenicity through the maintenance of stem-like cells populations and clonogenic potential [126,127,136]. Wnt/β-catenin and PTEN/PI3K/AKT pathways also collaborate to promote self-renewal induced by Aryl hydrocarbon receptor and the cytochrome P450 1A1 (CYP1A1) activation [137]. Further, abnormal activation of PI3K/AKT/mTOR pathway increases chemokine receptor type 4 (CXCR4) expression and, in turn, promotes the maintenance of stemness through STAT signaling [135]. STAT3 hyper-activation can also result from the activity of the PI3K/AKT/IL-6 axis: AKT induces IkB degradation, which allows NF-kB to enter the nucleus and activate IL-6 transcription, able to promote self-renewal and tumorigenesis [129,138] (Figure 1). Upstream of PTEN, several mediators of self-renewal have been identified. As discussed, PTEN expression can be modulated at post-transcriptional level by miRNAs. TGF β-mediated deregulation of miR-216a in cancer, as well as upregulation of miR-106b, miR-10b, and miR-21, affects PTEN levels and thus CSC self-renewal [133,136,139,140]. Interestingly, a recent work on glioblastoma primary cells highlighted a new level of PTEN expression control, focusing on the role of the protein arginine methyltransferase-5 (PRMT5) on self-renewal and proliferation of neurospheres. This report shows that PTEN is a target of PRMT5 in the context of primary GBM neurospheres, but not in the differentiated counterparts and that PTEN downregulation, upon methylation by PRMT5, is required for stem cells maintenance through the AKT/ERK axis [141].
2.2. PTEN and Cell Cycle
Another hallmark of CSCs is the ability to reversibly enter a state of slow proliferation and/or quiescence, with cells resting in G0 phase, but ready to promptly reactivate the cell cycle upon external stimuli [69]. Quiescence is an actively regulated status observed also in a subset of adult tissue specific stem cells, where it was defined and characterized. In cancer, quiescent CSCs are the driving force for relapse and therapy-resistance. Among the main determinants of quiescence are p53, Rb, cyclin-dependent kinase inhibitors, paired box protein 7 (PAX7), Notch signalling. Interestingly, quiescent cells are under the control of post-transcriptional mechanisms of gene expression regulation, with miRNAs playing a central role as underlined by the preferential use of distal polyadenylation sites, generating long 3′UTRs exposed to miRNA binding. Relying on post-transcriptional regulation allows a rapid switch toward active cycling upon proper stimuli. This is further accomplished by the maintenance of key genes in a heterochromatic status that is permissive for transcription, with RNA Polymerase II (RNA PolII) stuck on specific promoters, ready to activate the elongation phase and switch on proliferative programs [158].
PTEN plays a crucial role in the regulation of the quiescence and the cell cycle entry of both normal and transformed stem cells (SCs), with PTEN loss often fostering the cancerous phenotype. Yilmaz and colleagues took advantage of a mouse model for leukemia to show that PTEN deletion leads to the exhaustion of the normal stem compartment in favor of leukemic cancer stem cell (L-CSC) expansion and acute leukemia development [122,144]. Interestingly, treatment with rapamycin is sufficient to restore the normal HSCs pool, suggesting the involvement of mTOR protein downstream of PTEN and envisaging the chance to selectively target L-CSCs without consequence for the normal HSCs [122]. The same mechanism occurs in BCR-ABL chronic myeloid leukemia (CML) where an increased percentage of dividing HSCs was found in PTEN deficient mice with overt leukemia, thus indicating that PTEN governs the transition between the quiescent and activated state of HSCs to maintain their physiologic pool. Consistently, PTEN expression induces cell cycle arrest in BCR-ABL expressing leukemia cells [142,144,159,160]. Mechanistic insights came from muscle stem cells, where PTEN loss induces AKT phosphorylation and by that FOXO1 cytoplasmic translocation and inhibition of NOTCH pathway, resulting in quiescent stem cells exhaustion [161].
Of note, mouse models harboring the brain-specific deletion of PTEN provided evidence on the role of PTEN in restricting neural stem cell proliferation and size [162]. Specifically, loss of PTEN in mouse brain determines enhanced G0 cell cycle exit and self-renewal capacity [163]. An interesting study defined the transforming capacity of PTEN loss in neural stem cells (NSCs). Mouse NSCs derived from both WT and PTEN−/− ESCs were assayed for tumorigenic potential, both in vitro and in vivo, clarifying that PTEN loss acts through PAX7 activation to induce transcriptional and metabolic changes leading to neoplastic transformation [124]. Moreover, the activation of PTEN/mTOR/STAT3 pathway in breast cancer cells is required for the viability and maintenance of a subpopulation enriched in CSCs, defined as the side population (SP). These findings suggest that the SP compartment contains a significant number of quiescent cells, providing a possible explanation for drug resistance [145,146,147]. Similar evidence aroused from studies on glioblastoma CD133+ stem-like SP population, comprising the majority of cells in G0/G1 phase [148,149].
The role of PTEN in cell cycle control of CSCs is not limited to quiescence regulation. Peng and colleagues showed that PI3K/AKT pathway is involved in cyclin D1 activation, maintaining cells at G1 stage. In PTEN deficient acute myeloid leukemia (AML) mice, a high number of cyclin D1-expressing cells populated the bone marrow. Furthermore, after rapamycin administration, L-CSCs were depleted and normal HSCs restored in PTEN-deficient AML mice, providing evidence of PI3K/mTOR pathway involvement [142]. Of note, in contrast to other hematologic malignancies, in PTEN-null T-cell acute lymphatic leukemia (T-ALL) L-CSCs are not in a quiescent state, but rather are actively cycling. This feature is consistent with the overexpression of C-MYC that fuels the cell cycle entry of L-CSCs. C-MYC overexpression is a common feature of PTEN-null T-ALL and is due to Tcrα/δ-c-Myc translocation but also results from PTEN deletion and consequent enhancement mTOR activity [143] (Figure 1).
2.3. PTEN and Cell Survival
Cell survival is a key mechanism for tumor persistence and represents a critical node for the therapeutic outcome. In addition to PI3K/mTOR/AKT axis hyperactivation, PTEN loss influences other pathways involved in cell survival. Li and colleagues showed that PTEN negatively regulates PTEN/HIF-1α angiogenic pathway in glioblastoma. In particular, they observed that under stress conditions such as starvation or chemotherapeutic drug treatment, suppression of PTEN by miR-17 activates HIF-1α, thus promoting survival, migration, and angiogenesis. Furthermore, in GBM cells, HIF-1α overexpression contributes to the generation of tumor stem-like cells endowed with increased capacity to form colonies and neurospheres and marked drug-resistance [150] (Figure 1).
PTEN loss can induce cellular senescence (PTEN-loss-Induced Cellular Senescence, PICS). Although the partial loss of PTEN determines an increases in proliferation, its biallelic inactivation, prevalent in advanced stages of cancers [4,5,164,165], leads to cellular senescence, a protective mechanism to restrict tumorigenesis that is dependent on p53 and may follow mTORC1-mediated increased translation and stabilization [164,166]. Abou-Kehir and colleagues, using a PTEN/P53 null prostate cancer mouse model, explained the oncogenic effect of these double mutations with the absence of TP53-dependent cellular senescence after PTEN loss [131]. Moreover, the function of PTEN in senescence could be mediated by other players. For instance, an interesting study evaluated head and neck squamous-cell carcinoma (HNSCC) development under PTEN and TGFβ loss of signaling. In this model system, PTEN deletion is not sufficient for epithelial cells transformation and HNSCC onset, due to the induction of AKT- and p53-dependent senescence. In this context, mutation of TGFβR1 triggers the evasion of senescence and expansion of cancer stem cells. Indeed, TGFβR1/PTEN double conditional knockout (2cKO) mice show significantly increased malignant lesions with enhanced stem cell features and reduced apoptosis [167].
2.4. PTEN and EMT-Metastasis
Neoplastic cells can reversibly enter the CSC status through the activation of the Epithelial-to-Mesenchymal-Transition (EMT), an epigenetic program which drives cells to lose many of their epithelial attributes and switch to mesenchymal characteristics [168]. Plasticity of CSCs sustains the fluctuation between franc epithelial and mesenchymal programs, generating a gradient of intermediate conditions that determine intra-tumor heterogeneity. CSCs endowed with mesenchymal features gain the ability to disseminate to other tissues and establish new metastatic foci. Conversely, once the metastatic niche is colonized, CSCs revert to a more epithelial status and are able to propagate and generate secondary tumors. This complex transition is controlled by cell intrinsic mechanisms as well as by microenvironmental stimuli. PTEN loss or downregulation correlates with the acquisition of EMT traits. In lung cancer, hypoxic microenvironment determines an unbalance in the phospho-PTEN (pPTEN)/PTEN ratio that is associated with EMT [169]. Further, TGFβ-induced phosphorylation of PTEN C-terminal domain determines a reduced phosphatase activity that mediates EMT induction in lung cancer cells [170].
Several factors regulate EMT process, migration, and invasion acting upstream of PTEN. It was reported that knockdown of oncogenic BMI-1 (B-lymphoma Moloney murine leukemia virus insertion region-1) and S100A4 (S100 calcium binding protein A4), involved in EMT and invasion ability, decreases phosho-AKT (pAKT) and increases PTEN expression in pancreatic and head-neck CSCs respectively, highlighting the link of PI3K/AKT pathway to EMT process [134,151]. Furthermore, miR-20a, miR-200c, and miR-221/222 regulate PI3K/AKT pathway targeting PTEN, thus modulating self-renewal capabilities, invasion, and metastasis cascade through EMT process in ovarian and breast CSCs [152,153]. Oncogenic miR-17 promotes cell motility and invasion through PTEN suppression and subsequent HIF1α and VEGF up-regulation in glioblastoma [150]. In prostate cancer patients with elevated Gleason score, high Monoamine oxidase (MAOA) expression correlates with low PTEN expression. Wu and colleagues observed that MAOA overexpression increases HIF1α stabilization, activates the EMT-promoting genes AKT/FOXO1 and VEGF-A, thus enhancing TWIST1 expression. Subsequently, Liao and colleagues took advantage of a prostate-specific PTEN/MAOA knockout mouse model and observed a decreased incidence of invasive prostate adenocarcinoma development associated with reduced AKT activity, attenuated CSC markers and reduced clonogenic capacity [157,171].
Recent work has elucidated the role of the long-non-coding RNA Glucose Aroused for EMT Activation (lncRNA-GAEA) on PTEN enzymatic activity. In particular, lncRNA-GEAE, induced by high glucose levels, could drive poly-ubiquitination of PTEN, thus inhibiting its lipid phosphatase while activating the protein phosphatase activity. This switch determines the dephosphorylation and consequent accumulation of EMT master regulators, such as TWIST, SNAIL, and YAP1 [154].
Moreover, Mulholland and colleagues highlighted how alteration of RAS/MAPK signaling potentiates PTEN/PI3K/AKT axis effects, cooperating to promote EMT and drive primary tumor and metastatic prostate cancer progression. In a context of PTEN loss and Ras activation, the authors observed an enhanced expression of EMT markers in stem/progenitor cells in vitro, and metastasis development in orthotopic transplantation models [155].
A recent report showed that Connexin43 (Cx43) regulates PTEN protein phosphatase activity, reducing migration and invasion abilities of glioblastoma stem cells (GSCs). Mechanistically, Cx43 inhibits c-Src activity and upregulates PTEN expression, which contributes to FAK inactivation by dephosphorylation [172].
Collectively, these findings sustain the role of PTEN in the control of several cancer stem cells hallmarks such as self-renewal, quiescence, motility, and invasiveness, all features consistent with the positive association between PTEN loss and poor prognosis and metastasis (Figure 1). In particular, PTEN has a determinant role in the reversible acquisition of specific CSC traits, as quiescence and EMT, thus controlling plasticity and by that the capacity to overcome stressful conditions as hypoxia and nutrient deprivation or resist to therapeutic insults.
3. PTEN Implication in Therapy Resistance
The CSC features mentioned above, together with enhanced DNA repair and efflux pump activities, confer intrinsic resistance to drugs on this cell population. The PI3K/AKT/PTEN/mTOR pathway plays a role in multiple mechanisms of chemo- and radio-resistance, suggesting that targeting of this pathway could be a promising strategy to restore drug sensitivity in CSCs and improve patients’ survival [173]. Here we will discuss PTEN-based resistance mechanisms and possible strategies to modulate PTEN-dependent pathways with the attempt to overcome resistance (Figure 2).
3.1. Breast Cancer
Despite the success of trastuzumab-based therapy in HER2+ breast cancer treatment, the occurrence of resistance remains significant and strongly affects the overall efficacy of the current therapeutic regimens [174,175]. Indeed, about 15% of women treated with trastuzumab face metastatic disease within the first year [176,177] and the majority of patients develop drug resistance after one to two year of treatment [178,179]. Trastuzumab-based therapy targets multiple pathways downstream of HER2 including PI3K and MAPK [180,181]. The first hints proving the role of PTEN in trastuzumab resistance came in 2004 when Nagata and colleagues observed activation of PTEN upon trastuzumab treatment and showed that PTEN is required for the anti-tumor efficacy of trastuzumab, since its inhibition is sufficient to induce resistance [182]. Accordingly, an elegant approach exploiting a short hairpin RNA (shRNA) library demonstrated that PTEN is the mediator of resistance. The authors showed that PTEN loss, together with the more frequent mutation in PIK3CA, can be used as a biomarker with a substantial predictive value for therapy sensitivity and patients’ outcome [183].
As discussed above, PTEN knockdown determines the enrichment of normal and malignant mammary stem cells and triggers a significant increase in AKT phosphorylation levels, leading to GSK3β-dependent β-catenin activation and fueling malignant transformation. Consistently, treatment with perifosine (AKT inhibitor) or Ly294002 (PI3K inhibitor), alone or in combination with chemotherapy, reduces mammary stem cell population and tumor growth in breast cancer xenografts [127].
Recent evidence proposes NOTCH-1 as the main actor in PTEN-mediated resistance to trastuzumab. Baker and colleagues reported the presence of NOTCH-1 modulation together with PTEN down-regulation in HER2+ breast cancer resistant cells. They showed that NOTCH-1 induced the inhibition of PTEN through the activation of ERK1/2, thus promoting survival and self-renewal of trastuzumab-resistant breast CSCs [176]. In accordance with these data, recent studies highlighted that continued use of trastuzumab in HER2+ breast cancer, with loss of PTEN function, induces the epithelial-mesenchymal transition (EMT) and transforms HER2+ to a more aggressive triple negative breast cancer cell type, endowed with increased proliferation and metastatic potential in vivo [178,179].
3.2. Leukemia
Hematopoietic stem cells (HSCs) are the first and best-characterized adult stem cells. Normal hematopoiesis requires specific interactions between the bone marrow niche and HSCs, and perturbation of the microenvironment components may contribute to the generation of leukemic stem cells. L-CSCs are refractory to therapy and eventually sustain resistance and relapse. As already mentioned, alterations in the PTEN pathway are commonly associated with leukemogenesis [144,184]. Interestingly, the inactivation of PTEN leads to increased AKT phosphorylation and mTOR activation, which can be reversed with the specific mTOR inhibitor, rapamycin [185]. In PTEN-null mice, rapamycin is able to restore the normal HSC self-renewal capacity, suggesting that PTEN-null HSCs exhaustion can result from increased mTOR activation [122,160].
Schubbert and colleagues proposed a combination therapy to selectively eliminate L-CSCs, targeting both the PI3K and C-MYC pathway (rapamycin and JQ1 inhibitor) [143]. Of note, replacing rapamycin, with NVP-BEZ235, a dual inhibitor of PI3K and mTOR, resulted in cell cycle arrest and reduced L-CSCs compartment [186]. Interestingly, the identification of a novel regulatory circuit, responsible for leukemia ‘stemness’ maintenance, paved the road for a new approach to hit L-CSCs. Specifically, in a background of several driver mutations, including PTEN loss, the SPI1/PU.1 transcription factor was identified as a master regulator of L-CSCs stemness properties [187]. Treatment of PTEN-null T-ALL leukemic mice with DB1976 (a compound that disrupts the interactions between SPI1 and its targets), alone or in combination with rapamycin, significantly reduces the number of L-CSCs [188,189].
3.3. Liver Cancer
Hepatocellular carcinoma (HCC) accounts for about 70–85% of liver cancers, and it is one of the most common malignancies worldwide [190]. Reportedly, the upregulation of Cyclooxygenase-2 (COX-2), an inducible enzyme frequently found in inflammatory tissue, correlates with angiogenesis, invasion, relapse, chemo-resistance, and tumorigenesis in HCC [191], ultimately promoting hepatoma cell growth by inhibiting apoptosis through AKT activation [192,193]. Inhibition of AKT signaling leads to decreased hepatic CSCs biomarkers, CD133, and CD44 [194,195]. Celecoxib, a selective COX-2 inhibitor, reduces AKT phosphorylation and induces growth inhibition and apoptosis in HCC cells, stimulating peroxisome proliferator-activated receptor gamma (PPARy) as well as PTEN. Treatment with rosiglitazone, a PPARγ agonist, determines an increase in endogenous and celecoxib-induced PTEN protein levels in hepatoma cells, which in turn contributes to the inhibition of hepatic cancer stemness and growth through the reduction of AKT activation and CD133 and CD44 expression levels [196].
Furthermore, PTEN mutant mice are enriched in liver CSCs (CD133+ CD45-) [197]. Previous studies demonstrated the role of TGFβ in hepatic CSCs generation: TGFβ-exposed cells show elevated levels of phosphorylated AKT and suppression of PTEN expression [198]. Among PTEN-targeting microRNAs, miR-216a is upregulated in HCC, and its specific inhibition can rescue PTEN expression and suppress CSC self-renewal capacity [136].
An interesting study showed that lupeol, a phytochemical compound, decreases liver cancer stem population through PTEN upregulation. In vivo, lupeol treatment suppresses tumor growth and synergistically cooperates with standard chemotherapy acting on both the PTEN-AKT pathway and the ATP-binding cassette super-family G member 2 (ABCG2) [199].
3.4. Brain Cancer
Glioblastoma multiforme (GBM) is the most malignant form of glioma with a 5-year survival rate of 5% [150]. Besides surgery, the current therapeutic regimen is based on radiotherapy and temozolomide (TMZ) administration. PTEN loss is extremely frequent in GBM (40%) and correlates with TMZ resistance [200]. Consistently, treatment with TMZ doubles the fraction of stem-like SP cells, thus enhancing their tumorigenic potential. Inhibition of PI3K/AKT pathway with a specific inhibitor (LY294002) has been proposed to reduce multi-drug resistance mediated by ABCG2 in cancer stem-like cells and improve GBM chemosensitivity [74].
As already mentioned, the increased expression of miR-17 and consequent down-regulation of PTEN fuel the tumorigenic features in glioma stem cells (GSCs). Of note, miR-17 overexpression confers drug resistance in docetaxel-treated GSCs by regulating PTEN/HIF-1α pathway [150]. Connexin43 (CX43) is an integral membrane protein, weakly expressed in GSCs [201]. Cell-penetrating peptides containing the CX43 residues 266–283 (TAT-Cx43266-283) can mimic the effects of CX43 [202]. Interestingly, by inhibiting c-Src and increasing PTEN, these peptides reduce FAK (focal adhesion kinase) activity and lead to inhibition of migration and invasion in GSCs [172].
Medulloblastoma (MB) is an aggressive pediatric cerebellar tumor [203], displaying a perivascular niche enriched in CD133+ stem cells, involved in radiotherapy resistance. Specifically, a single dose of radiation induces p53-dependent apoptosis in the tumor bulk and transient cell cycle arrest in resistant stem cells, which survive due to PTEN malfunction and activation of the PI3K/AKT/mTOR pathway. Interestingly, the elucidation of PTEN involvement in this mechanism of resistance suggests the use of AKT inhibitors in the clinic to increase the efficacy of radiotherapy [204].
About 50% of Sonic hedgehog (SHH)-MB are characterized by PTEN loss, which correlates with worse overall survival [205]. Singh and colleagues validated the therapeutic efficacy of PI3K inhibitors (BKM-120) in the treatment of SHH-MB stem cells, suggesting a promising combination of PI3K inhibitors and chemotherapy (cisplatinum and TMZ) for the treatment of this disease [206].
3.5. Prostate Cancer
Conventional therapies for prostate cancer include surgery, hormonal-, radio- and chemo-therapy [207]. Despite the overall initial efficacy of these approaches, a large percentage of patients eventually relapse. The eradication of CSCs should be the primary goal to avoid resistance and relapse. PTEN is one of the most frequently mutated genes in prostate cancer and is often responsible for therapy resistance [208]. Therefore, the analysis of PTEN mutations might predict therapeutic response, while targeting of PTEN and related proteins holds promise for therapeutic efficacy [209]. Interestingly, in 2010 Dubrovska and colleagues showed how a combinatorial approach, based on conventional chemotherapy and NVP-BEZ235, a dual PI3K/mTOR inhibitor, leads to significant tumor regression targeting both bulk tumor cells and prostate cancer progenitors (PCPs), respectively.
Furthermore, in a recent study, Pandolfi and coworkers demonstrated that the indol-3-carbinol (I3C), a derivate of cruciferous vegetables, has potent anti-tumor properties. They provided evidence on the mechanism of action of this novel compound, showing that it strongly inhibits the ubiquitin E3 ligase WWP1, which acts downstream of MYC pathway and mediates the PTEN inactivation. Of note, I3C treatment can restore PTEN activity, leading to a potent suppression of tumorigenesis driven by the PI3K-AKT pathway in prostate tumor spheroids, organoids, and xenografts. These findings open a new perspective of a “reactivation” approach to tumor therapy [210].
3.6. Colon Cancer
The development of targeted therapies has brought to the clinic several molecules able to interfere with specific pathways. In colorectal cancer, the combined treatment of Trametinib and Everolimus, a MAPK/ERK pathway inhibitor and mTOR inhibitor, respectively, results in significant inhibition of tumor growth [211]. The identification of novel potential targets or therapeutic tools is essential to hit CSCs and overcome their innate resistance to conventional therapies. PTEN levels are usually very low in CSCs as compared to the more differentiated cells. We observed that bone morphogenetic protein 4 (BMP4) delivery and thymosine β4 targeting display anti-tumor activity in colorectal (CR) cancer by inducing CR-CSC differentiation and inhibition of PI3K/AKT pathway through the contribution of PTEN upregulation [212,213]. Moreover, recent evidence pointed out how miR-106b, which inversely correlates with PTEN levels, induces radio-resistance in colorectal cancer cells by enhancing tumor-initiating cell capacity [139].
3.7. Lung Cancer
Lung cancer is the leading cause of cancer-related death worldwide. Non-small cell lung cancer (NSCLC) is frequently characterized by an aberrant AKT activation arising from loss of PTEN or PIK3CA/AKT1 activating mutations. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors (EGFR-TKi) represent the treatment of choice for EGFR-mutated NSCLC, but the occurrence of resistance has sharply limited their efficacy. In about 4% of cases, PTEN loss promotes EGFR-TKi resistance by inducing AKT activation and EGFR reactivation [214]. Several studies pointed out the involvement of PTEN and downstream pathways in CSCs survival and resistance mechanisms. For instance, the activation of PI3K/AKT pathway can promote human bronchial epithelial cells (BEAS-2B) tumorigenic potential, sustained by a subpopulation of CSCs. Furthermore, the treatment of NSCLC cells with specific AKT inhibitors (MK2206 or LY294002) strongly impairs AKT phosphorylation, and thus, the spheroid-forming capability of CSCs [129]. Moreover, PI3K/PTEN/AKT/mTOR pathway influences the maintenance of NSCLC stem cells through CXCR4 modulation. The expression of this chemokine receptor is associated with PTEN downregulation and pAKT upregulation in gefitinib-resistant A549 cell line (A549/GR) [215]. Treatment of A549/GR with the PI3K inhibitor LY294002 suppresses CXCR4 expression and spheroids formation, while restoring wild type PTEN reduces pAKT levels and CXCR4 expression. Conversely, in the presence of mutant PTEN, rapamycin treatment suppresses CXCR4 and CD133 expression, underlining the involvement of mTOR pathway downstream of PTEN [135]. Furthermore, in a model of murine lung carcinogenesis, the modulation of both NOTCH1 and PTEN/PI3K/AKT signaling through miR-494-3p correlated with lung CSCs (LCSCs) maintenance, cancer progression, and metastasis. Interestingly, miR-494-3p upregulation correlates with worse survival rates in a cohort of lung cancer patients, suggesting that this miRNA could be a new therapeutic target [216]. Notably, recent evidence suggests that miR-mediated regulation of PTEN is involved in resistance to anti-EGFR targeted inhibitor (erlotinib). Despite its ability to target EGFR, erlotinib treatment fails to inhibit PI3K and AKT activation in LCSCs. Han and colleagues reported a higher miR-23a expression in LCSCs that correlates with low PTEN expression. Of note, miR-23a inhibition via antisense oligonucleotides restores LCSCs sensitivity to erlotinib by upregulation of PTEN, suggesting their possible combined approach for NSCLC treatment [217].
4. Concluding Remarks
A wealth of studies has provided convincing evidence on the crucial role of PTEN function and modulation in human cancer susceptibility. Due to the pivotal role of PTEN in several key cellular functions, mutations in this tumor suppressor occur in a wide variety of tumors. The frequent loss/alteration of PTEN expression may be envisioned as a possible Achille’s heel for cancer, offering a common strategy to fight several different malignancies. A deeper understanding of the regulatory networks and the major alterations of PTEN expression and its related PI3K/AKT/mTOR pathway opens new insights for pharmacological targets and prognostic tumor biomarkers. Therapeutic strategies directed to activate or reactivate PTEN function represent a promising perspective for tumor treatment [210]. Moreover, PTEN regulates CSCs development and maintenance, specifically affecting critical features of these cells through downstream signaling pathways such as WNT, NOTCH, PI3K/AKT, MAPK and NF-kB (Figure 1). The plastic interconversion between CSCs and non-CSCs is not hardware-defined, but rather depends on multiple environmental cues. PTEN loss can influence the response to such stimuli at multiple levels, eliciting the transition to the cancer stem cells state by promoting EMT, quiescence, self-renewal and by that governing CSCs plasticity which is crucial for tumor progression, metastasis, and therapy resistance.
Thus, highlighting the complex network of PTEN within the CSCs population and the tumor niche microenvironment, may provide considerable information for the identification of successful therapeutic strategies aiming at overcoming CSC-mediated drug resistance and enhancing the tumor suppressive function of PTEN in cancer.
Author Contributions
F.L., F.C. (Francesca Colonna), F.C. (Federica Calapà), S.V. analyzed the bibliographic data, collected them, and drafted the manuscript. M.E.F. supervised the analysis of bibliographic data, designed the review, and critically revised the manuscript with R.D.M. All authors read and approved the final manuscript.
Funding
This work was supported by Italian Association for Cancer Research (AIRC) (5 × 1000 Clinical Oncology Extension Program 9979) to R.D.M.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Li, D.M.; Sun, H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997, 57, 2124–2129. [Google Scholar] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, A.; Pandolfi, P.P. The multiple roles of PTEN in tumor suppression. Cell 2000, 100, 387–390. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; Van Dyke, T.; et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003, 1, e59. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, A.; Kotsi, P.; Peng, Y.F.; Cordon-Cardo, C.; Elkon, K.B.; Pandolfi, P.P. Impaired Fas response and autoimmunity in Pten+/- mice. Science 1999, 285, 2122–2125. [Google Scholar] [CrossRef]
- Berger, A.H.; Knudson, A.G.; Pandolfi, P.P. A continuum model for tumour suppression. Nature 2011, 476, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; de la Pompa, J.L.; Stambolic, V.; Elia, A.J.; Sasaki, T.; del Barco Barrantes, I.; Ho, A.; Wakeham, A.; Itie, A.; Khoo, W.; et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr. Biol. 1998, 8, 1169–1178. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Podsypanina, K.; Ellenson, L.H.; Nemes, A.; Gu, J.; Tamura, M.; Yamada, K.M.; Cordon-Cardo, C.; Catoretti, G.; Fisher, P.E.; Parsons, R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc. Natl. Acad. Sci. USA 1999, 96, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Ngeow, J.; Sesock, K.; Eng, C. Clinical Implications for Germline PTEN Spectrum Disorders. Endocrinol. Metab. Clin. North. Am. 2017, 46, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Karikomi, M.; Naidu, S.; Rajmohan, R.; Caserta, E.; Chen, H.Z.; Rawahneh, M.; Moffitt, J.; Stephens, J.A.; Fernandez, S.A.; et al. Allele-specific tumor spectrum in pten knockin mice. Proc. Natl. Acad. Sci. USA 2010, 107, 5142–5147. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef]
- Khan, S.; Kumagai, T.; Vora, J.; Bose, N.; Sehgal, I.; Koeffler, P.H.; Bose, S. PTEN promoter is methylated in a proportion of invasive breast cancers. Int. J. Cancer 2004, 112, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef]
- Salvesen, H.B.; MacDonald, N.; Ryan, A.; Jacobs, I.J.; Lynch, E.D.; Akslen, L.A.; Das, S. PTEN methylation is associated with advanced stage and microsatellite instability in endometrial carcinoma. Int. J. Cancer 2001, 91, 22–26. [Google Scholar] [CrossRef]
- Soria, J.C.; Lee, H.Y.; Lee, J.I.; Wang, L.; Issa, J.P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar]
- Zhang, J.; Lee, Y.R.; Dang, F.; Gan, W.; Menon, A.V.; Katon, J.M.; Hsu, C.H.; Asara, J.M.; Tibarewal, P.; Leslie, N.R.; et al. PTEN Methylation by NSD2 Controls Cellular Sensitivity to DNA Damage. Cancer Discov. 2019. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.G.; Wang, J.J.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G.H. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Ciuffreda, L.; Di Sanza, C.; Cesta Incani, U.; Eramo, A.; Desideri, M.; Biagioni, F.; Passeri, D.; Falcone, I.; Sette, G.; Bergamo, P.; et al. The mitogen-activated protein kinase (MAPK) cascade controls phosphatase and tensin homolog (PTEN) expression through multiple mechanisms. J. Mol. Med. 2012, 90, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yu, D. PI(3)king apart PTEN’s role in cancer. Clin. Cancer Res. 2010, 16, 4325–4330. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Leevers, S.J.; Vanhaesebroeck, B.; Waterfield, M.D. Signalling through phosphoinositide 3-kinases: The lipids take centre stage. Curr. Opin. Cell Biol. 1999, 11, 219–225. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.P.; Stolarov, J.P.; Eng, C.; Li, J.; Wang, S.I.; Wigler, M.H.; Parsons, R.; Tonks, N.K. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. USA 1997, 94, 9052–9057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.C.; Piccini, A.; Myers, M.P.; Van Aelst, L.; Tonks, N.K. Functional analysis of the protein phosphatase activity of PTEN. Biochem. J. 2012, 444, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Tibarewal, P.; Zilidis, G.; Spinelli, L.; Schurch, N.; Maccario, H.; Gray, A.; Perera, N.M.; Davidson, L.; Barton, G.J.; Leslie, N.R. PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci. Signal. 2012, 5, ra18. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.M.; Ji, Y.; Zhang, C.; Dong, S.S.; Yang, S.; Xiong, Z.; Ge, M.K.; Yu, Y.; Xia, L.; Guo, M.; et al. Nuclear PTEN safeguards pre-mRNA splicing to link Golgi apparatus for its tumor suppressive role. Nat. Commun. 2018, 9, 2392. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Zhao, M.; Depinho, R.A.; Furnari, F.B.; Cavenee, W.K. Cellular transformation by the MSP58 oncogene is inhibited by its physical interaction with the PTEN tumor suppressor. Proc. Natl. Acad. Sci. USA 2005, 102, 2703–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchon, S.M.; Waite, K.A.; Eng, C. The nuclear affairs of PTEN. J. Cell Sci 2008, 121, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimm, O.; Perren, A.; Weng, L.P.; Marsh, D.J.; Yeh, J.J.; Ziebold, U.; Gil, E.; Hinze, R.; Delbridge, L.; Lees, J.A.; et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am. J. Pathol. 2000, 156, 1693–1700. [Google Scholar] [CrossRef]
- Ginn-Pease, M.E.; Eng, C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0-G1 in MCF-7 cells. Cancer Res. 2003, 63, 282–286. [Google Scholar] [PubMed]
- Sano, T.; Lin, H.; Chen, X.; Langford, L.A.; Koul, D.; Bondy, M.L.; Hess, K.R.; Myers, J.N.; Hong, Y.K.; Yung, W.K.; et al. Differential expression of MMAC/PTEN in glioblastoma multiforme: Relationship to localization and prognosis. Cancer Res. 1999, 59, 1820–1824. [Google Scholar] [PubMed]
- Lachyankar, M.B.; Sultana, N.; Schonhoff, C.M.; Mitra, P.; Poluha, W.; Lambert, S.; Quesenberry, P.J.; Litofsky, N.S.; Recht, L.D.; Nabi, R.; et al. A role for nuclear PTEN in neuronal differentiation. J. Neurosci. 2000, 20, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Zhou, X.P.; Cummings, M.C.; Pavey, S.; Hayward, N.K.; Eng, C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int. J. Cancer 2002, 99, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Putz, U.; Howitt, J.; Doan, A.; Goh, C.P.; Low, L.H.; Silke, J.; Tan, S.S. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal. 2012, 5, ra70. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; He, S.; Yang, J.; Jia, X.; Wang, P.; Chen, X.; Zhang, Z.; Zou, X.; McNutt, M.A.; Shen, W.H.; et al. PTENalpha, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 2014, 19, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Chen, X.; Yin, Q.; Ruan, D.; Zhao, X.; Zhang, C.; McNutt, M.A.; Yin, Y. PTENbeta is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat. Commun. 2017, 8, 14771. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Falcone, I.; Incani, U.C.; Del Curatolo, A.; Conciatori, F.; Matteoni, S.; Vari, S.; Vaccaro, V.; Cognetti, F.; Milella, M. PTEN expression and function in adult cancer stem cells and prospects for therapeutic targeting. Adv. Biol. Regul. 2014, 56, 66–80. [Google Scholar] [CrossRef]
- Lavictoire, S.J.; Gont, A.; Julian, L.M.; Stanford, W.L.; Vlasschaert, C.; Gray, D.A.; Jomaa, D.; Lorimer, I.A.J. Engineering PTEN-L for Cell-Mediated Delivery. Mol. Ther. Methods Clin. Dev. 2018, 9, 12–22. [Google Scholar] [CrossRef]
- Jiang, B.H.; Liu, L.Z. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv. Cancer Res. 2009, 102, 19–65. [Google Scholar] [CrossRef]
- Sizemore, G.M.; Balakrishnan, S.; Hammer, A.M.; Thies, K.A.; Trimboli, A.J.; Wallace, J.A.; Sizemore, S.T.; Kladney, R.D.; Woelke, S.A.; Yu, L.; et al. Stromal PTEN inhibits the expansion of mammary epithelial stem cells through Jagged-1. Oncogene 2017, 36, 2297. [Google Scholar] [CrossRef]
- Luo, X.; Liao, R.; Hanley, K.L.; Zhu, H.H.; Malo, K.N.; Hernandez, C.; Wei, X.; Varki, N.M.; Alderson, N.; Chu, C.; et al. Dual Shp2 and Pten Deficiencies Promote Non-alcoholic Steatohepatitis and Genesis of Liver Tumor-Initiating Cells. Cell Rep. 2016, 17, 2979–2993. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef]
- Shojaee, S.; Chan, L.N.; Buchner, M.; Cazzaniga, V.; Cosgun, K.N.; Geng, H.; Qiu, Y.H.; von Minden, M.D.; Ernst, T.; Hochhaus, A.; et al. PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat. Med. 2016, 22, 379–387. [Google Scholar] [CrossRef]
- Li, Y.; Guessous, F.; Kwon, S.; Kumar, M.; Ibidapo, O.; Fuller, L.; Johnson, E.; Lal, B.; Hussaini, I.; Bao, Y.; et al. PTEN has tumor-promoting properties in the setting of gain-of-function p53 mutations. Cancer Res. 2008, 68, 1723–1731. [Google Scholar] [CrossRef]
- Costa, H.A.; Leitner, M.G.; Sos, M.L.; Mavrantoni, A.; Rychkova, A.; Johnson, J.R.; Newton, B.W.; Yee, M.C.; De La Vega, F.M.; Ford, J.M.; et al. Discovery and functional characterization of a neomorphic PTEN mutation. Proc. Natl. Acad. Sci. USA 2015, 112, 13976–13981. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Manic, G.; Sistigu, A.; Corradi, F.; Musella, M.; De Maria, R.; Vitale, I. Replication stress response in cancer stem cells as a target for chemotherapy. Semin. Cancer Biol. 2018, 53, 31–41. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—what challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Vermeulen, L.; Todaro, M.; de Sousa Mello, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Fiori, M.E.; Villanova, L.; De Maria, R. Cancer stem cells: At the forefront of personalized medicine and immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 1–11. [Google Scholar] [CrossRef]
- Zeuner, A.; Todaro, M.; Stassi, G.; De Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef]
- Koury, J.; Zhong, L.; Hao, J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017, 2925869. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef]
- Pourmand, G.; Ziaee, A.A.; Abedi, A.R.; Mehrsai, A.; Alavi, H.A.; Ahmadi, A.; Saadati, H.R. Role of PTEN gene in progression of prostate cancer. Urol. J. 2007, 4, 95–100. [Google Scholar]
- Tao, D.L.; Bailey, S.; Beer, T.M.; Foss, E.; Beckett, B.; Fung, A.; Foster, B.R.; Guimaraes, A.; Cetnar, J.P.; Graff, J.N.; et al. Molecular Testing in Patients With Castration-Resistant Prostate Cancer and Its Impact on Clinical Decision Making. JCO Precis. Oncol. 2017, 1–11. [Google Scholar] [CrossRef]
- Geybels, M.S.; Fang, M.; Wright, J.L.; Qu, X.; Bibikova, M.; Klotzle, B.; Fan, J.B.; Feng, Z.; Ostrander, E.A.; Nelson, P.S.; et al. PTEN loss is associated with prostate cancer recurrence and alterations in tumor DNA methylation profiles. Oncotarget 2017, 8, 84338–84348. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, M.; Cunha, I.W.; Coudry, R.A.; Fonseca, F.P.; Torres, C.H.; Soares, F.A.; Squire, J.A. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br. J. Cancer 2007, 97, 678–685. [Google Scholar] [CrossRef]
- Gray, I.C.; Phillips, S.M.; Lee, S.J.; Neoptolemos, J.P.; Weissenbach, J.; Spurr, N.K. Loss of the chromosomal region 10q23-25 in prostate cancer. Cancer Res. 1995, 55, 4800–4803. [Google Scholar]
- Schmitz, M.; Grignard, G.; Margue, C.; Dippel, W.; Capesius, C.; Mossong, J.; Nathan, M.; Giacchi, S.; Scheiden, R.; Kieffer, N. Complete loss of PTEN expression as a possible early prognostic marker for prostate cancer metastasis. Int. J. Cancer 2007, 120, 1284–1292. [Google Scholar] [CrossRef]
- Dreher, T.; Zentgraf, H.; Abel, U.; Kappeler, A.; Michel, M.S.; Bleyl, U.; Grobholz, R. Reduction of PTEN and p27kip1 expression correlates with tumor grade in prostate cancer. Analysis in radical prostatectomy specimens and needle biopsies. Virchows Arch. 2004, 444, 509–517. [Google Scholar] [CrossRef]
- Kazim, Z.; Wahabi, K.; Perwez, A.; Lal, P.; Rizvi, M.A. PTEN Genetic and Epigenetic Alterations Define Distinct Subgroups in North Indian Breast Cancer Patients. Asian Pac. J. Cancer Prev. APJCP 2019, 20, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Liang, F.; Jia, Z.L.; Song, S.T.; Jiang, Z.F. PTEN mutation, methylation and expression in breast cancer patients. Oncol. Lett. 2013, 6, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.M.; Silva, J.M.; Dominguez, G.; Gonzalez, R.; Navarro, A.; Carretero, L.; Provencio, M.; Espana, P.; Bonilla, F. Allelic loss of the PTEN region (10q23) in breast carcinomas of poor pathophenotype. Breast Cancer Res. Treat. 1999, 57, 237–243. [Google Scholar] [CrossRef]
- Tanic, N.; Milovanovic, Z.; Tanic, N.; Dzodic, R.; Juranic, Z.; Susnjar, S.; Plesinac-Karapandzic, V.; Tatic, S.; Dramicanin, T.; Davidovic, R.; et al. The impact of PTEN tumor suppressor gene on acquiring resistance to tamoxifen treatment in breast cancer patients. Cancer Biol. Ther. 2012, 13, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Panigrahi, A.R.; Pinder, S.E.; Chan, S.Y.; Paish, E.C.; Robertson, J.F.; Ellis, I.O. The role of PTEN and its signalling pathways, including AKT, in breast cancer; an assessment of relationships with other prognostic factors and with outcome. J. Pathol. 2004, 204, 93–100. [Google Scholar] [CrossRef]
- Kato, H.; Kato, S.; Kumabe, T.; Sonoda, Y.; Yoshimoto, T.; Kato, S.; Han, S.Y.; Suzuki, T.; Shibata, H.; Kanamaru, R.; et al. Functional evaluation of p53 and PTEN gene mutations in gliomas. Clin. Cancer Res. 2000, 6, 3937–3943. [Google Scholar]
- Wang, S.I.; Puc, J.; Li, J.; Bruce, J.N.; Cairns, P.; Sidransky, D.; Parsons, R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997, 57, 4183–4186. [Google Scholar]
- Abdullah, J.M.; Farizan, A.; Asmarina, K.; Zainuddin, N.; Ghazali, M.M.; Jaafar, H.; Isa, M.N.; Naing, N.N. Association of loss of heterozygosity and PTEN gene abnormalities with paraclinical, clinical modalities and survival time of glioma patients in Malaysia. Asian J. Surg. 2006, 29, 274–282. [Google Scholar] [CrossRef]
- Kurose, K.; Zhou, X.P.; Araki, T.; Cannistra, S.A.; Maher, E.R.; Eng, C. Frequent loss of PTEN expression is linked to elevated phosphorylated Akt levels, but not associated with p27 and cyclin D1 expression, in primary epithelial ovarian carcinomas. Am. J. Pathol. 2001, 158, 2097–2106. [Google Scholar] [CrossRef]
- Obata, K.; Morland, S.J.; Watson, R.H.; Hitchcock, A.; Chenevix-Trench, G.; Thomas, E.J.; Campbell, I.G. Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res. 1998, 58, 2095–2097. [Google Scholar] [PubMed]
- Saito, M.; Okamoto, A.; Kohno, T.; Takakura, S.; Shinozaki, H.; Isonishi, S.; Yasuhara, T.; Yoshimura, T.; Ohtake, Y.; Ochiai, K.; et al. Allelic imbalance and mutations of the PTEN gene in ovarian cancer. Int. J. Cancer 2000, 85, 160–165. [Google Scholar] [CrossRef]
- Abubaker, J.; Bavi, P.; Al-Haqawi, W.; Jehan, Z.; Munkarah, A.; Uddin, S.; Al-Kuraya, K.S. PIK3CA alterations in Middle Eastern ovarian cancers. Mol. Cancer 2009, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.C.; Santiago, I.; Trinh, A.; Xian, J.; Guo, A.; Sayal, K.; Jimenez-Linan, M.; Deen, S.; Driver, K.; Mack, M.; et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biol. 2014, 15, 526. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, N.; Nagai, H.; Bando, K.; Koyama, M.; Matsumoto, S.; Tajiri, T.; Onda, M.; Fujimoto, J.; Ueki, T.; Konishi, N.; et al. PTEN/MMAC1 mutations in hepatocellular carcinomas: Somatic inactivation of both alleles in tumors. Jpn. J. Cancer Res. 1999, 90, 413–418. [Google Scholar] [CrossRef]
- Rahman, M.A.; Kyriazanos, I.D.; Ono, T.; Yamanoi, A.; Kohno, H.; Tsuchiya, M.; Nagasue, N. Impact of PTEN expression on the outcome of hepatitis C virus-positive cirrhotic hepatocellular carcinoma patients: Possible relationship with COX II and inducible nitric oxide synthase. Int. J. Cancer 2002, 100, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Cong, W.M.; Xian, Z.H.; Wu, M.C. Clinicopathological significance of loss of heterozygosity and microsatellite instability in hepatocellular carcinoma in China. World J. Gastroenterol. 2005, 11, 3034–3039. [Google Scholar] [CrossRef]
- Dong-Dong, L.; Xi-Ran, Z.; Xiang-Rong, C. Expression and significance of new tumor suppressor gene PTEN in primary liver cancer. J. Cell. Mol. Med. 2003, 7, 67–71. [Google Scholar] [CrossRef]
- Marsit, C.J.; Zheng, S.; Aldape, K.; Hinds, P.W.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. PTEN expression in non-small-cell lung cancer: Evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum. Pathol. 2005, 36, 768–776. [Google Scholar] [CrossRef]
- Yoo, S.B.; Xu, X.; Lee, H.J.; Jheon, S.; Lee, C.T.; Choe, G.; Chung, J.H. Loss of PTEN Expression is an Independent Poor Prognostic Factor in Non-small Cell Lung Cancer. J. Pathol. Transl. Med. 2011, 45, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Yanagawa, N.; Leduc, C.; Kohler, D.; Saieg, M.A.; John, T.; Sykes, J.; Yoshimoto, M.; Pintilie, M.; Squire, J.; Shepherd, F.A.; et al. Loss of phosphatase and tensin homolog protein expression is an independent poor prognostic marker in lung adenocarcinoma. J. Thorac. Oncol. 2012, 7, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Kim, M.J.; Jeon, H.S.; Choi, J.E.; Kim, D.S.; Lee, E.B.; Cha, S.I.; Yoon, G.S.; Kim, C.H.; Jung, T.H.; et al. PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer 2010, 69, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Atreya, C.E.; Sangale, Z.; Xu, N.; Matli, M.R.; Tikishvili, E.; Welbourn, W.; Stone, S.; Shokat, K.M.; Warren, R.S. PTEN expression is consistent in colorectal cancer primaries and metastases and associates with patient survival. Cancer Med. 2013, 2, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Dicuonzo, G.; Angeletti, S.; Garcia-Foncillas, J.; Brugarolas, A.; Okrouzhnov, Y.; Santini, D.; Tonini, G.; Lorino, G.; De Cesaris, M.; Baldi, A. Colorectal carcinomas and PTEN/MMAC1 gene mutations. Clin. Cancer Res. 2001, 7, 4049–4053. [Google Scholar] [PubMed]
- Nassif, N.T.; Lobo, G.P.; Wu, X.; Henderson, C.J.; Morrison, C.D.; Eng, C.; Jalaludin, B.; Segelov, E. PTEN mutations are common in sporadic microsatellite stable colorectal cancer. Oncogene 2004, 23, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negri, F.V.; Bozzetti, C.; Lagrasta, C.A.; Crafa, P.; Bonasoni, M.P.; Camisa, R.; Pedrazzi, G.; Ardizzoni, A. PTEN status in advanced colorectal cancer treated with cetuximab. Br. J. Cancer 2010, 102, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Aggerholm, A.; Gronbaek, K.; Guldberg, P.; Hokland, P. Mutational analysis of the tumour suppressor gene MMAC1/PTEN in malignant myeloid disorders. Eur. J. Haematol. 2000, 65, 109–113. [Google Scholar] [CrossRef]
- Liu, T.C.; Lin, P.M.; Chang, J.G.; Lee, J.P.; Chen, T.P.; Lin, S.F. Mutation analysis of PTEN/MMAC1 in acute myeloid leukemia. Am. J. Hematol. 2000, 63, 170–175. [Google Scholar] [CrossRef]
- Mendes, R.D.; Sarmento, L.M.; Cante-Barrett, K.; Zuurbier, L.; Buijs-Gladdines, J.G.; Povoa, V.; Smits, W.K.; Abecasis, M.; Yunes, J.A.; Sonneveld, E.; et al. PTEN microdeletions in T-cell acute lymphoblastic leukemia are caused by illegitimate RAG-mediated recombination events. Blood 2014, 124, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Jenkinson, S.; Kirkwood, A.A.; Goulden, N.; Vora, A.; Linch, D.C.; Gale, R.E. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia 2016, 30, 39–47. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson Gedman, A.; Chen, Q.; Kugel Desmoulin, S.; Ge, Y.; LaFiura, K.; Haska, C.L.; Cherian, C.; Devidas, M.; Linda, S.B.; Taub, J.W.; et al. The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Leukemia 2009, 23, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.; Gatalica, Z.; Knezetic, J.; Reddy, S.; Nathan, C.A.; Javadi, N.; Teknos, T. Molecular profiling of head and neck squamous cell carcinoma. Head Neck 2016, 38 (Suppl. 1), E1625–E1638. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agrawal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.M.; et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef]
- Squarize, C.H.; Castilho, R.M.; Abrahao, A.C.; Molinolo, A.; Lingen, M.W.; Gutkind, J.S. PTEN deficiency contributes to the development and progression of head and neck cancer. Neoplasia 2013, 15, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Snietura, M.; Jaworska, M.; Mlynarczyk-Liszka, J.; Goraj-Zajac, A.; Piglowski, W.; Lange, D.; Wozniak, G.; Nowara, E.; Suwinski, R. PTEN as a prognostic and predictive marker in postoperative radiotherapy for squamous cell cancer of the head and neck. PLoS ONE 2012, 7, e33396. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, M.; Lorenz, G.; Kleist, B. Detection of new PTEN/MMAC1 mutations in head and neck squamous cell carcinomas with loss of chromosome 10. Cancer Genet. Cytogenet. 2002, 132, 20–24. [Google Scholar] [CrossRef]
- Okami, K.; Wu, L.; Riggins, G.; Cairns, P.; Goggins, M.; Evron, E.; Halachmi, N.; Ahrendt, S.A.; Reed, A.L.; Hilgers, W.; et al. Analysis of PTEN/MMAC1 alterations in aerodigestive tract tumors. Cancer Res. 1998, 58, 509–511. [Google Scholar] [PubMed]
- Fuchs, E.; Chen, T. A matter of life and death: Self-renewal in stem cells. EMBO Rep. 2013, 14, 39–48. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Wicha, M.S. Selective targeting of cancer stem cells: A new concept in cancer therapeutics. BioDrugs 2007, 21, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagi, S.; Kishimoto, H.; Kawahara, K.; Sasaki, T.; Sasaki, M.; Nishio, M.; Yajima, N.; Hamada, K.; Horie, Y.; Kubo, H.; et al. Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J. Clin. Investig. 2007, 117, 2929–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.; Yuan, G.; Liu, X.; Ren, R.; Li, J.; Zhang, W.; Wu, J.; Xu, X.; Fu, L.; Li, Y.; et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat. Commun. 2015, 6, 10068. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Xin, L.; Morim, A.; Lawson, D.; Witte, O.; Wu, H. Lin-Sca-1+CD49fhigh stem/progenitors are tumor-initiating cells in the Pten-null prostate cancer model. Cancer Res. 2009, 69, 8555–8562. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Kim, S.; Salamone, R.J.; Walker, J.R.; Maira, S.M.; Garcia-Echeverria, C.; Schultz, P.G.; Reddy, V.A. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl. Acad. Sci. USA 2009, 106, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkaya, H.; Paulson, A.; Charafe-Jauffret, E.; Ginestier, C.; Brown, M.; Dutcher, J.; Clouthier, S.G.; Wicha, M.S. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009, 7, e1000121. [Google Scholar] [CrossRef]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S.; et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef]
- Malanga, D.; De Marco, C.; Guerriero, I.; Colelli, F.; Rinaldo, N.; Scrima, M.; Mirante, T.; De Vitis, C.; Zoppoli, P.; Ceccarelli, M.; et al. The Akt1/IL-6/STAT3 pathway regulates growth of lung tumor initiating cells. Oncotarget 2015, 6, 42667–42686. [Google Scholar] [CrossRef]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef]
- Abou-Kheir, W.G.; Hynes, P.G.; Martin, P.L.; Pierce, R.; Kelly, K. Characterizing the contribution of stem/progenitor cells to tumorigenesis in the Pten-/-TP53-/- prostate cancer model. Stem Cells 2010, 28, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Abou-Kheir, W.; Hynes, P.G.; Martin, P.; Yin, J.J.; Liu, Y.N.; Seng, V.; Lake, R.; Spurrier, J.; Kelly, K. Self-renewing Pten-/- TP53-/- protospheres produce metastatic adenocarcinoma cell lines with multipotent progenitor activity. PLoS ONE 2011, 6, e26112. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Yang, G.D.; Huang, T.J.; Li, R.; Chu, Q.Q.; Xu, L.; Wang, M.S.; Cai, M.D.; Zhong, L.; Wei, H.J.; et al. EB-virus latent membrane protein 1 potentiates the stemness of nasopharyngeal carcinoma via preferential activation of PI3K/AKT pathway by a positive feedback loop. Oncogene 2016, 35, 3419–3431. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.C.; Jiao, M.; Wu, T.; Jing, L.; Cui, J.; Guo, H.; Tian, T.; Ruan, Z.P.; Wei, Y.C.; Jiang, L.L.; et al. Polycomb complex protein BMI-1 promotes invasion and metastasis of pancreatic cancer stem cells by activating PI3K/AKT signaling, an ex vivo, in vitro, and in vivo study. Oncotarget 2016, 7, 9586–9599. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.J.; Rho, J.K.; Kim, Y.M.; Jung, J.E.; Jin, Y.B.; Ko, Y.G.; Lee, J.S.; Lee, S.J.; Lee, J.C.; Park, M.J. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene 2013, 32, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.F.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C.; et al. Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Al-Dhfyan, A.; Alhoshani, A.; Korashy, H.M. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and beta-Catenin and Akt activation. Mol. Cancer 2017, 16, 14. [Google Scholar] [CrossRef]
- Salah, M.; Nishimoto, Y.; Kohno, S.; Kondoh, A.; Kitajima, S.; Muranaka, H.; Nishiuchi, T.; Ibrahim, A.; Yoshida, A.; Takahashi, C. An in vitro system to characterize prostate cancer progression identified signaling required for self-renewal. Mol. Carcinog. 2016, 55, 1974–1989. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, Y.; Liu, Y.; Zhou, M.; Lu, Y.; Yuan, L.; Zhang, C.; Hong, M.; Wang, S.; Li, X. MiR-106b induces cell radioresistance via the PTEN/PI3K/AKT pathways and p21 in colorectal cancer. J. Transl. Med. 2015, 13, 252. [Google Scholar] [CrossRef]
- Bahena-Ocampo, I.; Espinosa, M.; Ceballos-Cancino, G.; Lizarraga, F.; Campos-Arroyo, D.; Schwarz, A.; Garcia-Lopez, P.; Maldonado, V.; Melendez-Zajgla, J. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016, 17, 648–658. [Google Scholar] [CrossRef]
- Banasavadi-Siddegowda, Y.K.; Russell, L.; Frair, E.; Karkhanis, V.A.; Relation, T.; Yoo, J.Y.; Zhang, J.; Sif, S.; Imitola, J.; Baiocchi, R.; et al. PRMT5-PTEN molecular pathway regulates senescence and self-renewal of primary glioblastoma neurosphere cells. Oncogene 2017, 36, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Chen, Y.; Li, D.; Li, S. Role of Pten in leukemia stem cells. Oncotarget 2010, 1, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Cardenas, A.; Chen, H.; Garcia, C.; Guo, W.; Bradner, J.; Wu, H. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014, 74, 7048–7059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Zhou, J.; Claypool, K.; Tang, D.G. Side Population Is Enriched in Tumorigenic, Stem-Like Cancer Cells, whereas ABCG2+ and ABCG2− Cancer Cells Are Similarly Tumorigenic. Cancer Res. 2005, 65, 6207–6219. [Google Scholar] [CrossRef]
- Qiang, L.; Yang, Y.; Ma, Y.J.; Chen, F.H.; Zhang, L.B.; Liu, W.; Qi, Q.; Lu, N.; Tao, L.; Wang, X.T.; et al. Isolation and characterization of cancer stem like cells in human glioblastoma cell lines. Cancer Lett. 2009, 279, 13–21. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, B.B. Stress response of glioblastoma cells mediated by miR-17-5p targeting PTEN and the passenger strand miR-17-3p targeting MDM2. Oncotarget 2012, 3, 1653–1668. [Google Scholar] [CrossRef]
- Lo, J.F.; Yu, C.C.; Chiou, S.H.; Huang, C.Y.; Jan, C.I.; Lin, S.C.; Liu, C.J.; Hu, W.Y.; Yu, Y.H. The epithelial-mesenchymal transition mediator S100A4 maintains cancer-initiating cells in head and neck cancers. Cancer Res. 2011, 71, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Dong, Z.; Chen, Y.; Yang, L.; Lai, D. Enrichment of ovarian cancer stem-like cells is associated with epithelial to mesenchymal transition through an miRNA-activated AKT pathway. Cell Prolif. 2013, 46, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lu, Y.; Wang, H.; Han, X.; Mao, J.; Li, J.; Yu, L.; Wang, B.; Fan, S.; Yu, X.; et al. miR-221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed. Pharmacother. 2016, 79, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Li, C.; Wang, S.; Li, Y.; Wen, B.; Zhang, Y.; Liang, K.; Yao, J.; Ye, Y.; Hsiao, H.; et al. LncRNAs-directed PTEN enzymatic switch governs epithelial-mesenchymal transition. Cell Res. 2019, 29, 286–304. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, M.V.; Dahiya, S.; Danielson, L.S.; Segura, M.F.; Vales-Lara, F.M.; Menendez, S.; Popiolek, D.; Mittal, K.; Wei, J.J.; Zavadil, J.; et al. Dual Pten/Tp53 suppression promotes sarcoma progression by activating Notch signaling. Am. J. Pathol. 2013, 182, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.P.; Lin, T.P.; Li, P.C.; Geary, L.A.; Chen, K.; Vaikari, V.P.; Wu, J.B.; Lin, C.H.; Gross, M.E.; Shih, J.C. Loss of MAOA in epithelia inhibits adenocarcinoma development, cell proliferation and cancer stem cells in prostate. Oncogene 2018, 37, 5175–5190. [Google Scholar] [CrossRef]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef]
- Peng, C.; Chen, Y.; Yang, Z.; Zhang, H.; Osterby, L.; Rosmarin, A.G.; Li, S. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood 2010, 115, 626–635. [Google Scholar] [CrossRef]
- Guo, W.; Schubbert, S.; Chen, J.Y.; Valamehr, B.; Mosessian, S.; Shi, H.; Dang, N.H.; Garcia, C.; Theodoro, M.F.; Varella-Garcia, M.; et al. Suppression of leukemia development caused by PTEN loss. Proc. Natl. Acad. Sci. USA 2011, 108, 1409–1414. [Google Scholar] [CrossRef] [Green Version]
- Yue, F.; Bi, P.; Wang, C.; Shan, T.; Nie, Y.; Ratliff, T.L.; Gavin, T.P.; Kuang, S. Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat. Commun. 2017, 8, 14328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groszer, M.; Erickson, R.; Scripture-Adams, D.D.; Lesche, R.; Trumpp, A.; Zack, J.A.; Kornblum, H.I.; Liu, X.; Wu, H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 2001, 294, 2186–2189. [Google Scholar] [CrossRef] [PubMed]
- Groszer, M.; Erickson, R.; Scripture-Adams, D.D.; Dougherty, J.D.; Le Belle, J.; Zack, J.A.; Geschwind, D.H.; Liu, X.; Kornblum, H.I.; Wu, H. PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. Proc. Natl. Acad. Sci. USA 2006, 103, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.G.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Investig. 2010, 120, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Hwang, H.J.; Kang, D.; Park, H.A.; Lee, H.C.; Jeong, D.; Lee, K.; Park, H.J.; Ko, Y.G.; Lee, J.S. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 2019, 38, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Hall, B.; Sun, Z.J.; Molinolo, A.; Chen, W.; Gutkind, J.S.; Waes, C.V.; Kulkarni, A.B. Loss of TGF-beta signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012, 31, 3322–3332. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Kohnoh, T.; Hashimoto, N.; Ando, A.; Sakamoto, K.; Miyazaki, S.; Aoyama, D.; Kusunose, M.; Kimura, M.; Omote, N.; Imaizumi, K.; et al. Hypoxia-induced modulation of PTEN activity and EMT phenotypes in lung cancers. Cancer Cell Int. 2016, 16, 33. [Google Scholar] [CrossRef]
- Aoyama, D.; Hashimoto, N.; Sakamoto, K.; Kohnoh, T.; Kusunose, M.; Kimura, M.; Ogata, R.; Imaizumi, K.; Kawabe, T.; Hasegawa, Y. Involvement of TGFbeta-induced phosphorylation of the PTEN C-terminus on TGFbeta-induced acquisition of malignant phenotypes in lung cancer cells. PLoS ONE 2013, 8, e81133. [Google Scholar] [CrossRef]
- Wu, J.B.; Shao, C.; Li, X.; Li, Q.; Hu, P.; Shi, C.; Li, Y.; Chen, Y.T.; Yin, F.; Liao, C.P.; et al. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J. Clin. Investig. 2014, 124, 2891–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaraiz-Rodriguez, M.; Tabernero, M.D.; Gonzalez-Tablas, M.; Otero, A.; Orfao, A.; Medina, J.M.; Tabernero, A. A Short Region of Connexin43 Reduces Human Glioma Stem Cell Migration, Invasion, and Survival through Src, PTEN, and FAK. Stem Cell Rep. 2017, 9, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.H.; Hao, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Emerging roles of radioresistance in prostate cancer metastasis and radiation therapy. Cancer Metastasis Rev. 2014, 33, 469–496. [Google Scholar] [CrossRef] [PubMed]
- Untch, M.; Fasching, P.A.; Konecny, G.E.; Hasmuller, S.; Lebeau, A.; Kreienberg, R.; Camara, O.; Muller, V.; du Bois, A.; Kuhn, T.; et al. Pathologic complete response after neoadjuvant chemotherapy plus trastuzumab predicts favorable survival in human epidermal growth factor receptor 2-overexpressing breast cancer: Results from the TECHNO trial of the AGO and GBG study groups. J. Clin. Oncol. 2011, 29, 3351–3357. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A. Compensatory pathways in oncogenic kinase signaling and resistance to targeted therapies: Six degrees of separation. Cancer Discov. 2012, 2, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Wyatt, D.; Bocchetta, M.; Li, J.; Filipovic, A.; Green, A.; Peiffer, D.S.; Fuqua, S.; Miele, L.; Albain, K.S.; et al. Notch-1-PTEN-ERK1/2 signaling axis promotes HER2+ breast cancer cell proliferation and stem cell survival. Oncogene 2018, 37, 4489–4504. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Esteva, F.J. Trastuzumab: Triumphs and tribulations. Oncogene 2007, 26, 3637–3643. [Google Scholar] [CrossRef]
- Burnett, J.P.; Korkaya, H.; Ouzounova, M.D.; Jiang, H.; Conley, S.J.; Newman, B.W.; Sun, L.; Connarn, J.N.; Chen, C.S.; Zhang, N.; et al. Trastuzumab resistance induces EMT to transform HER2(+) PTEN(-) to a triple negative breast cancer that requires unique treatment options. Sci. Rep. 2015, 5, 15821. [Google Scholar] [CrossRef]
- Sun, L.; Burnett, J.; Gasparyan, M.; Xu, F.; Jiang, H.; Lin, C.C.; Myers, I.; Korkaya, H.; Liu, Y.; Connarn, J.; et al. Novel cancer stem cell targets during epithelial to mesenchymal transition in PTEN-deficient trastuzumab-resistant breast cancer. Oncotarget 2016, 7, 51408–51422. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Chinratanalab, W.; Ritter, C.A.; King, W.; Seelig, S.; Arteaga, C.L. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002, 62, 4132–4141. [Google Scholar]
- Ebbesen, S.H.; Scaltriti, M.; Bialucha, C.U.; Morse, N.; Kastenhuber, E.R.; Wen, H.Y.; Dow, L.E.; Baselga, J.; Lowe, S.W. Pten loss promotes MAPK pathway dependency in HER2/neu breast carcinomas. Proc. Natl. Acad. Sci. USA 2016, 113, 3030–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Wu, H. PTEN, stem cells, and cancer stem cells. J. Biol. Chem. 2009, 284, 11755–11759. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Grimaldi, C.; Ricci, F.; Tazzari, P.L.; Evangelisti, C.; Ognibene, A.; Battistelli, M.; Falcieri, E.; Melchionda, F.; Pession, A.; et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010, 70, 8097–8107. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, L.; Wu, Y.; Dong, B.; Guo, W.; Wang, M.; Yang, L.; Fan, X.; Tang, Y.; Liu, N.; et al. T-ALL leukemia stem cell ‘stemness’ is epigenetically controlled by the master regulator SPI1. eLife 2018, 7, e38314. [Google Scholar] [CrossRef]
- Antony-Debre, I.; Paul, A.; Leite, J.; Mitchell, K.; Kim, H.M.; Carvajal, L.A.; Todorova, T.I.; Huang, K.; Kumar, A.; Farahat, A.A.; et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J. Clin. Investig. 2017, 127, 4297–4313. [Google Scholar] [CrossRef] [Green Version]
- Munde, M.; Wang, S.; Kumar, A.; Stephens, C.E.; Farahat, A.A.; Boykin, D.W.; Wilson, W.D.; Poon, G.M. Structure-dependent inhibition of the ETS-family transcription factor PU.1 by novel heterocyclic diamidines. Nucleic Acids Res. 2014, 42, 1379–1390. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma: An epidemiologic view. J. Clin. Gastroenterol. 2002, 35, S72–S78. [Google Scholar] [CrossRef]
- Cervello, M.; McCubrey, J.A.; Cusimano, A.; Lampiasi, N.; Azzolina, A.; Montalto, G. Targeted therapy for hepatocellular carcinoma: Novel agents on the horizon. Oncotarget 2012, 3, 236–260. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.A.; Schubert, D.; Sahi, D.; Schoneweiss, M.M.; Moll, I.; Haugg, A.M.; Dienes, H.P.; Breuhahn, K.; Schirmacher, P. Proapoptotic and antiproliferative potential of selective cyclooxygenase-2 inhibitors in human liver tumor cells. Hepatology 2002, 36, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Leng, J.; Han, C.; Demetris, A.J.; Michalopoulos, G.K.; Wu, T. Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: Evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology 2003, 38, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ding, W.; Dang, H.; Jiang, Y.; Rountree, C.B. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011, 54, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, T.H.; Chan, H.H.; Kuo, H.M.; Liu, L.F.; Hu, T.H.; Sun, C.K.; Kung, M.L.; Lin, S.W.; Wang, E.M.; Ma, Y.L.; et al. Celecoxib suppresses hepatoma stemness and progression by up-regulating PTEN. Oncotarget 2014, 5, 1475–1490. [Google Scholar] [CrossRef] [PubMed]
- Rountree, C.B.; Ding, W.; He, L.; Stiles, B. Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem Cells 2009, 27, 290–299. [Google Scholar] [CrossRef]
- Tang, Y.; Kitisin, K.; Jogunoori, W.; Li, C.; Deng, C.X.; Mueller, S.C.; Ressom, H.W.; Rashid, A.; He, A.R.; Mendelson, J.S.; et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2445–2450. [Google Scholar] [CrossRef]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. Lupeol targets liver tumor-initiating cells through phosphatase and tensin homolog modulation. Hepatology 2011, 53, 160–170. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Giaume, C.; Koulakoff, A.; Roux, L.; Holcman, D.; Rouach, N. Astroglial networks: A step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 2010, 11, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanchez, A.; Jaraiz-Rodriguez, M.; Dominguez-Prieto, M.; Herrero-Gonzalez, S.; Medina, J.M.; Tabernero, A. Connexin43 recruits PTEN and Csk to inhibit c-Src activity in glioma cells and astrocytes. Oncotarget 2016, 7, 49819–49833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellino, R.C.; Barwick, B.G.; Schniederjan, M.; Buss, M.C.; Becher, O.; Hambardzumyan, D.; Macdonald, T.J.; Brat, D.J.; Durden, D.L. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PLoS ONE 2010, 5, e10849. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.R.; Joshi, S.; Zulcic, M.; Alcaraz, M.; Garlich, J.R.; Morales, G.A.; Cho, Y.J.; Bao, L.; Levy, M.L.; Newbury, R.; et al. PI-3K Inhibitors Preferentially Target CD15+ Cancer Stem Cell Population in SHH Driven Medulloblastoma. PLoS ONE 2016, 11, e0150836. [Google Scholar] [CrossRef] [PubMed]
- Grubb, R.L., 3rd; Kibel, A.S. Prostate cancer: Screening, diagnosis and management in 2007. Mo. Med. 2007, 104, 408–413. [Google Scholar] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Elliott, J.; Salamone, R.J.; Kim, S.; Aimone, L.J.; Walker, J.R.; Watson, J.; Sauveur-Michel, M.; Garcia-Echeverria, C.; Cho, C.Y.; et al. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clin. Cancer Res. 2010, 16, 5692–5702. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Matteoni, S.; Sacconi, A.; De Luca, T.; Bazzichetto, C.; Corbo, V.; Simbolo, M.; Sperduti, I.; et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci. Rep. 2017, 7, 43013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, Y.; Scopelliti, A.; Cammareri, P.; Todaro, M.; Iovino, F.; Ricci-Vitiani, L.; Gulotta, G.; Dieli, F.; de Maria, R.; Stassi, G. Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology 2011, 140, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Mollinari, C.; di Martino, S.; Biffoni, M.; Pilozzi, E.; Pagliuca, A.; de Stefano, M.C.; Circo, R.; Merlo, D.; De Maria, R.; et al. Thymosin beta4 targeting impairs tumorigenic activity of colon cancer stem cells. FASEB J. 2010, 24, 4291–4301. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.K.; Choi, Y.J.; Lee, J.K.; Ryoo, B.Y.; Na, I.I.; Yang, S.H.; Kim, C.H.; Lee, J.C. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer 2009, 63, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Faversani, A.; Amatori, S.; Augello, C.; Colombo, F.; Porretti, L.; Fanelli, M.; Ferrero, S.; Palleschi, A.; Pelicci, P.G.; Belloni, E.; et al. miR-494-3p is a novel tumor driver of lung carcinogenesis. Oncotarget 2017, 8, 7231–7247. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zhou, X.; Li, S.; Qin, Y.; Chen, Y.; Liu, H. Inhibition of miR-23a increases the sensitivity of lung cancer stem cells to erlotinib through PTEN/PI3K/Akt pathway. Oncol. Rep. 2017, 38, 3064–3070. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mechanisms of PTEN-mediated control of cancer stem cells (CSCs) hallmarks. PTEN deficiency promotes self-renewal through inhibitory phosphorylation of FoxO3a [126,136] and GSK3β, which increases nuclear β-catenin localization [127]. Furthermore, it induces STAT3 activation by NFkB-mediated IL6 transcription [129] and CXCR4 expression [135]. MicroRNAs (miR-10b [140], miR-21 [133], miR-106b [139] and TGFβ-induced miR-216a [136]) and PRMT5-mediated methylation [141] are also involved in the downregulation of PTEN and regulation of self-renewal. Hyperactive AKT acts on cell cycle stimulating proliferation trough PAX7 [124], cyclinD1 [142] or C-MYC [143]. PTEN loss may trigger G0 cell cycle arrest and quiescence of CSCs [122,142,144,145,146,147,148,149]. Activation of mTOR is required for CSC survival and can be associated with the activation of collateral pro-survival pathways such as HIF-1α [150]. S100A4 [151] and BMI1 [134], along with several miRNAs (miR-20a/miR-200c [152], miR-17, miR-221/222 [153]) impair PTEN function, thus promoting EMT and metastatic progression. The lncRNA-GAEA inhibits PTEN lipid phosphatase activity, switching on its protein phosphatase activity and promoting the accumulation of EMT master regulators such as TWIST, SNAIL, and YAP [154]. Moreover, activation of the RAS/MAPK pathway [155], NOTCH1 signaling [156], and MAOA [157] cooperate with PTEN/PI3K/AKT axis to promote the EMT program.
Figure 1.
Mechanisms of PTEN-mediated control of cancer stem cells (CSCs) hallmarks. PTEN deficiency promotes self-renewal through inhibitory phosphorylation of FoxO3a [126,136] and GSK3β, which increases nuclear β-catenin localization [127]. Furthermore, it induces STAT3 activation by NFkB-mediated IL6 transcription [129] and CXCR4 expression [135]. MicroRNAs (miR-10b [140], miR-21 [133], miR-106b [139] and TGFβ-induced miR-216a [136]) and PRMT5-mediated methylation [141] are also involved in the downregulation of PTEN and regulation of self-renewal. Hyperactive AKT acts on cell cycle stimulating proliferation trough PAX7 [124], cyclinD1 [142] or C-MYC [143]. PTEN loss may trigger G0 cell cycle arrest and quiescence of CSCs [122,142,144,145,146,147,148,149]. Activation of mTOR is required for CSC survival and can be associated with the activation of collateral pro-survival pathways such as HIF-1α [150]. S100A4 [151] and BMI1 [134], along with several miRNAs (miR-20a/miR-200c [152], miR-17, miR-221/222 [153]) impair PTEN function, thus promoting EMT and metastatic progression. The lncRNA-GAEA inhibits PTEN lipid phosphatase activity, switching on its protein phosphatase activity and promoting the accumulation of EMT master regulators such as TWIST, SNAIL, and YAP [154]. Moreover, activation of the RAS/MAPK pathway [155], NOTCH1 signaling [156], and MAOA [157] cooperate with PTEN/PI3K/AKT axis to promote the EMT program.

Figure 2.
Targeting PI3K/PTEN/AKT/mTOR pathways for cancer therapy. Schematic depicting of possible strategies/drugs to overcome PTEN-mediated resistance to conventional therapy in several malignancies. Nervous system cancer: specific PI3K/AKT inhibitors, LY294002 and BKM-120, in combination with conventional chemotherapy, inhibit Glioblastoma Multiforme (GBM) and Sonic Hedgehog Medulloblastoma (SHH-MB) CSCs, respectively. Anti-miR-17 and TAT-Cx43266-283, by mimicking the effects of Connexin 43 (CX43), increase PTEN expression and inhibit glioma stem cells tumorigenic features. Liver cancer: treatment with Celecoxib (cyclooxygenase-2 (COX-2) inhibitor) and rosiglitazone reduce AKT phosphorylation and increase PTEN protein levels, thus affecting cell proliferation. Anti-miR-216a and lupeol, a phytochemical compound, decrease the stem population through PTEN modulation. Prostate cancer: PI3K-mTOR dual inhibitor, NVP-BEZ235, in combination with conventional chemotherapy, leads to significant tumor regression in mice, targeting both prostate cancer progenitors (PCPs) and bulk tumor. I3C (indol-3-carbinol), a derivative of cruciferous vegetables, is able to restore PTEN activity leading to suppression of tumorigenesis. Leukemia: mTOR inhibitor rapamicyn, PI3K-mTOR dual inhibitor, NVP-BEZ235, JQ1 inhibitor targeting c-Myc pathway, and DB1976, a compound that disrupts the interactions between SPI1 and its targets, all result in a significant reduction of leukemic stem cells (L-CSCs). Colon cancer: treatment with rapamicyn, in combination with chemotherapy, inhibits tumor growth. Treatment with anti-miR-106b may overcome radio-resistance in colon cancer. BMP4 (Bone Morphogenetic Protein 4) inhibits PI3K/AKT pathway through PTEN up-regulation. Breast cancer: treatment with PI3K/AKT inhibitors, LY294002 and perifosine, alone or in combination with chemotherapy, reduces mammary stem cell population and tumor growth in mice. Lung cancer: treatment with LY294002, MK2206, and rapamicyn inhibits PI3K/AKT/mTOR pathway acting on the maintenance of lung stem cells through chemokine receptor modulation. Anti-miR-494-3p prevents metastasis and tumor progression while anti-miR-23a is able to upregulate PTEN expression, restoring lung cancer stem cells (LCSCs) sensitivity to chemotherapy.
Figure 2.
Targeting PI3K/PTEN/AKT/mTOR pathways for cancer therapy. Schematic depicting of possible strategies/drugs to overcome PTEN-mediated resistance to conventional therapy in several malignancies. Nervous system cancer: specific PI3K/AKT inhibitors, LY294002 and BKM-120, in combination with conventional chemotherapy, inhibit Glioblastoma Multiforme (GBM) and Sonic Hedgehog Medulloblastoma (SHH-MB) CSCs, respectively. Anti-miR-17 and TAT-Cx43266-283, by mimicking the effects of Connexin 43 (CX43), increase PTEN expression and inhibit glioma stem cells tumorigenic features. Liver cancer: treatment with Celecoxib (cyclooxygenase-2 (COX-2) inhibitor) and rosiglitazone reduce AKT phosphorylation and increase PTEN protein levels, thus affecting cell proliferation. Anti-miR-216a and lupeol, a phytochemical compound, decrease the stem population through PTEN modulation. Prostate cancer: PI3K-mTOR dual inhibitor, NVP-BEZ235, in combination with conventional chemotherapy, leads to significant tumor regression in mice, targeting both prostate cancer progenitors (PCPs) and bulk tumor. I3C (indol-3-carbinol), a derivative of cruciferous vegetables, is able to restore PTEN activity leading to suppression of tumorigenesis. Leukemia: mTOR inhibitor rapamicyn, PI3K-mTOR dual inhibitor, NVP-BEZ235, JQ1 inhibitor targeting c-Myc pathway, and DB1976, a compound that disrupts the interactions between SPI1 and its targets, all result in a significant reduction of leukemic stem cells (L-CSCs). Colon cancer: treatment with rapamicyn, in combination with chemotherapy, inhibits tumor growth. Treatment with anti-miR-106b may overcome radio-resistance in colon cancer. BMP4 (Bone Morphogenetic Protein 4) inhibits PI3K/AKT pathway through PTEN up-regulation. Breast cancer: treatment with PI3K/AKT inhibitors, LY294002 and perifosine, alone or in combination with chemotherapy, reduces mammary stem cell population and tumor growth in mice. Lung cancer: treatment with LY294002, MK2206, and rapamicyn inhibits PI3K/AKT/mTOR pathway acting on the maintenance of lung stem cells through chemokine receptor modulation. Anti-miR-494-3p prevents metastasis and tumor progression while anti-miR-23a is able to upregulate PTEN expression, restoring lung cancer stem cells (LCSCs) sensitivity to chemotherapy.


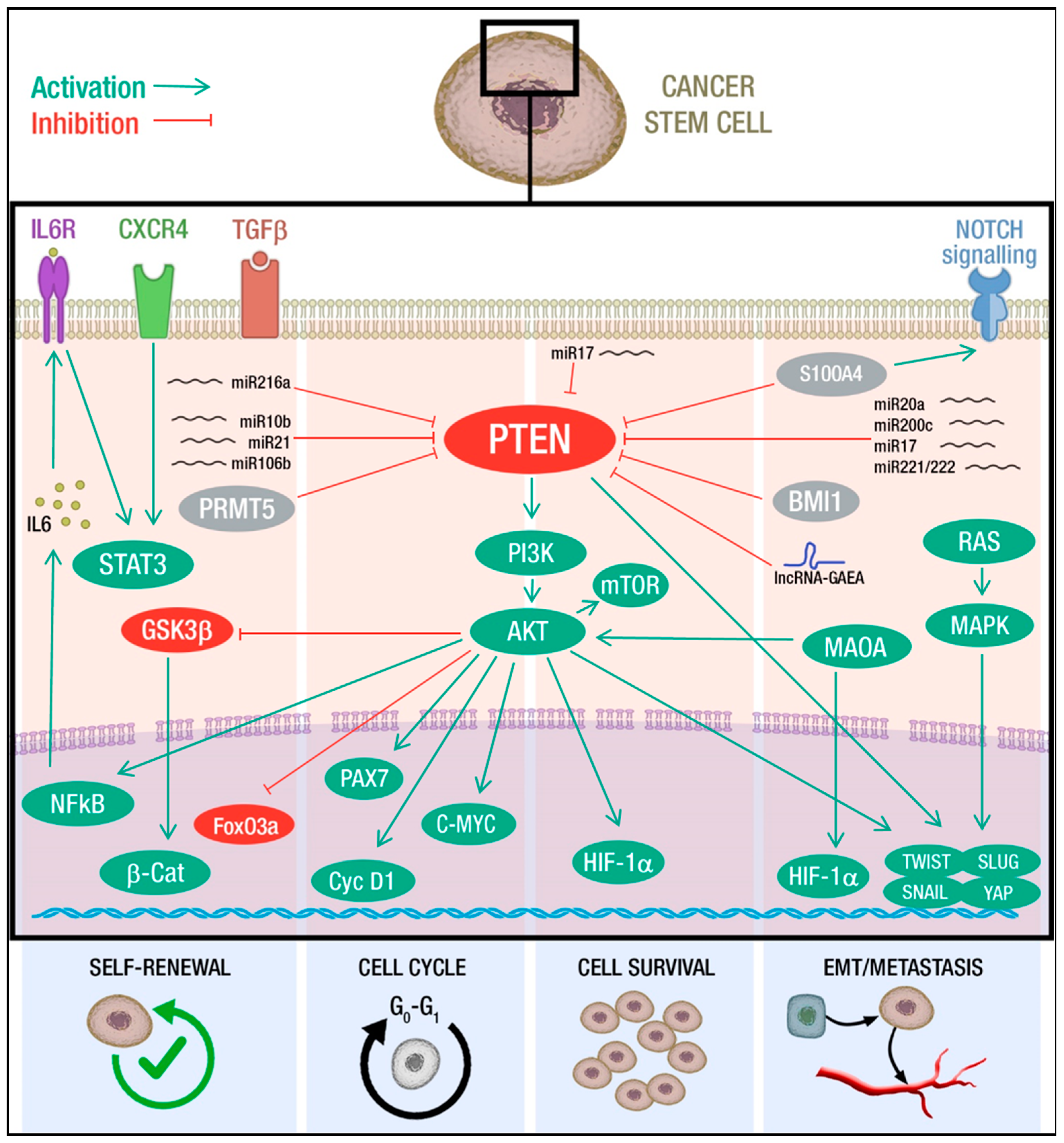{kind=link}
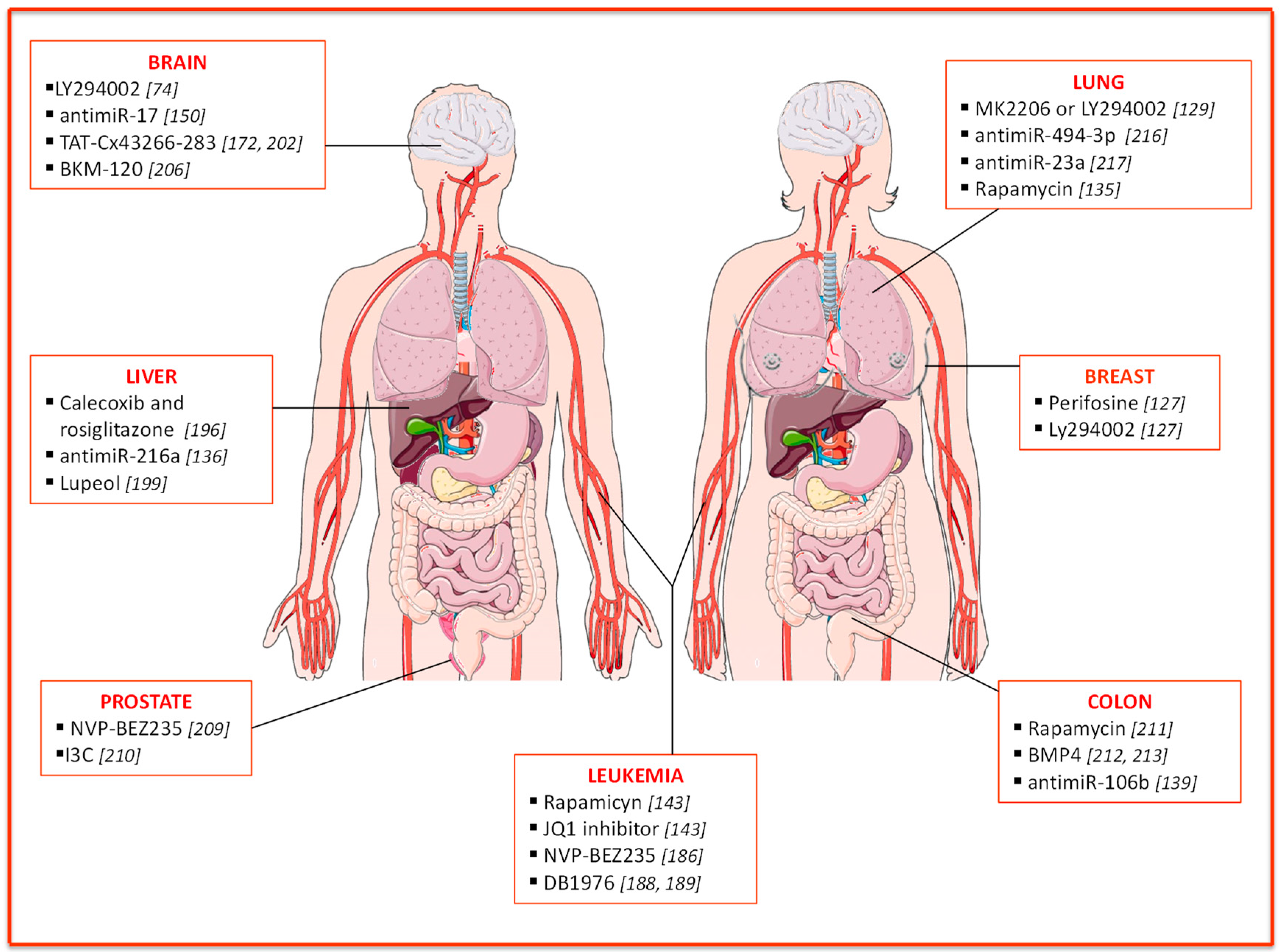{kind=link}
Table 1.
Incidence of PTEN alterations in diverse malignancies. The type of alteration is specified along with the relative % of patients displaying that specific alteration. Studies with more than 30 patients were included in the analysis.
Table 1.
Incidence of PTEN alterations in diverse malignancies. The type of alteration is specified along with the relative % of patients displaying that specific alteration. Studies with more than 30 patients were included in the analysis.
| Site | Malignancy Type | Molecular Mechanism(s) of PTEN Alteration and Incidence (%) | Reference |
|---|---|---|---|
| Prostate | Prostate cancer | Mutation: 12–26% LOH: 10–62% Reduced expression: 27–95% | [75,76,77,78,79,80,81] |
| Breast | Breast cancer | Mutation: <7% LOH: 29–63% Reduced expression: 8–55% | [82,83,84,85,86] |
| Brain | Glioma | Mutation: 12–44% LOH: 32–84% Reduced expression: 69% | [42,87,88,89] |
| Ovary | Ovarian carcinoma | Mutation: <9% LOH: 32–61% Reduced expression: 23–55% | [90,91,92,93,94] |
| Liver | Liver cancer | Mutation: <5% LOH: 27–79% Reduced expression: 30–63% | [95,96,97,98] |
| Lung | Non-small-cell lung cancer | Mutation: <5% LOH: 3–19% Reduced expression: 41–73% | [99,100,101,102] |
| Colorectum | Colorectal cancer | Mutation: 17–20% LOH: 20–30% Reduced expression: 12% | [103,104,105,106] |
| Blood | Myeloid leukemia | Deletion: rare LOH: Absent Reduced expression: 24% | [107,108] |
| Lymphoid leukemia | Deletion: 8–63% LOH: NA Reduced expression: 6–17% | [109,110,111,112] | |
| Head-neck | Head and neck squamous cell carcinoma | Deletion: 2–23% LOH: 41% Reduced expression: 31–60% | [113,114,115,116,117,118] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers 2019, 11, 1076. https://doi.org/10.3390/cancers11081076
AMA Style
Luongo F, Colonna F, Calapà F, Vitale S, Fiori ME, De Maria R. PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers. 2019; 11(8):1076. https://doi.org/10.3390/cancers11081076
Chicago/Turabian StyleLuongo, Francesca, Francesca Colonna, Federica Calapà, Sara Vitale, Micol E. Fiori, and Ruggero De Maria. 2019. "PTEN Tumor-Suppressor: The Dam of Stemness in Cancer" Cancers 11, no. 8: 1076. https://doi.org/10.3390/cancers11081076
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.