Somatic Alteration Burden Involving Non-Cancer Genes Predicts Prognosis in Early-Stage Non-Small Cell Lung Cancer

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
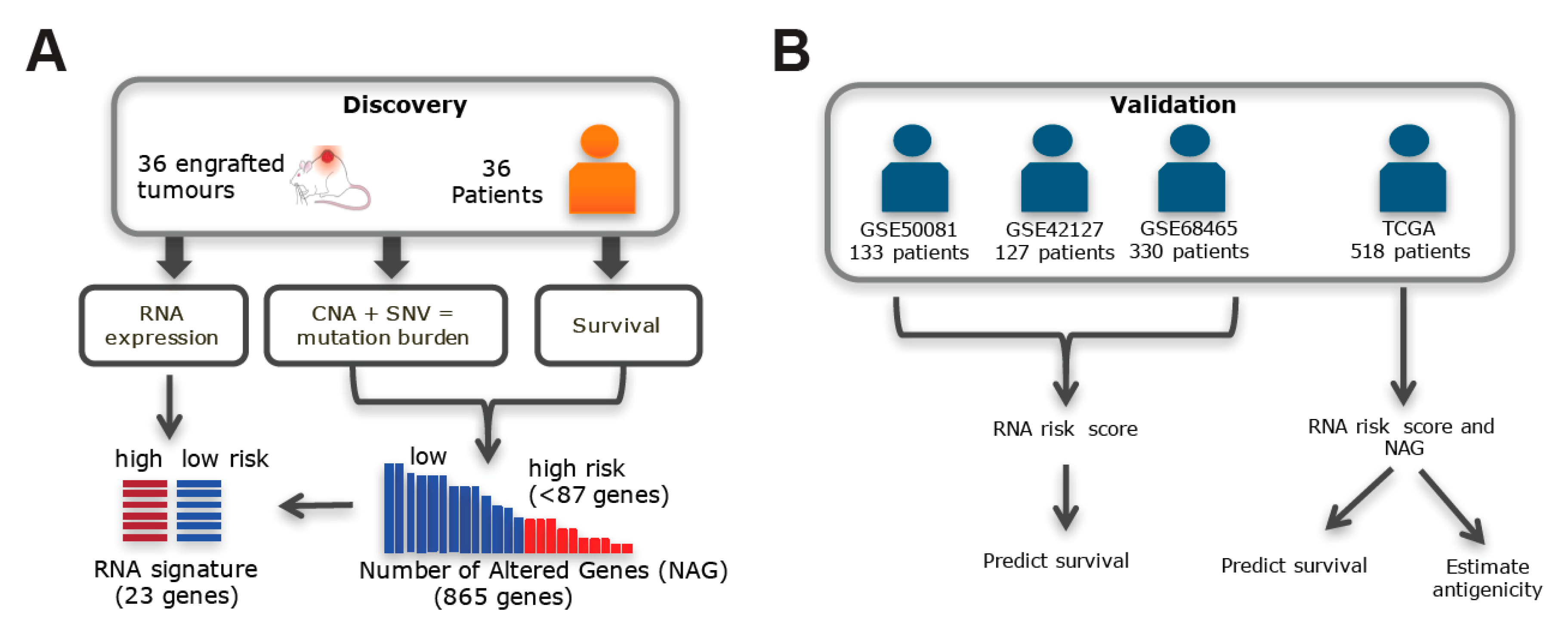
2.1. Somatic Alteration Burden in PDX Is Correlated with Patient Survival
2.2. Validation Using the Gene Expression Signature Associated with Somatic Alteration Burden
2.3. Validation in TCGA Datasets
2.4. Immunogenic Mutations Correlate with the Best Survival and Cytotoxic T-Cell Signature
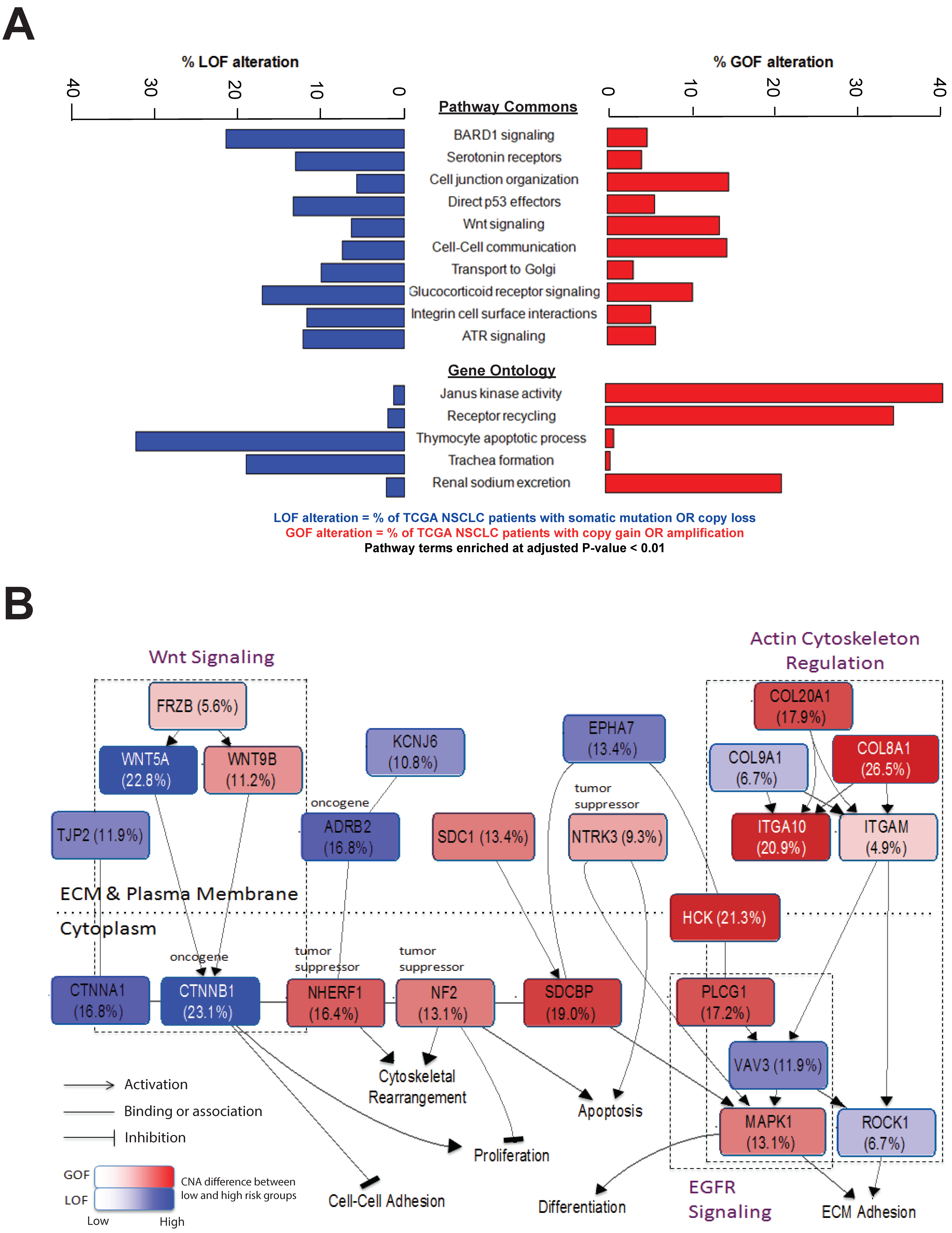
2.5. Prognostic Alterations Enriched in Genes Involved in Extracellular Signaling
3. Discussion
4. Materials and Methods
4.1. Patient and NSCLC PDX for Genomics Profiling
4.2. TCGA Patients
4.3. Stratification by High-Level Mutation Burden
4.4. Stratification by Gene Expression Profiles Corresponding to NAGs
4.5. Estimating Immunogenicity
4.6. Pathway Enrichment
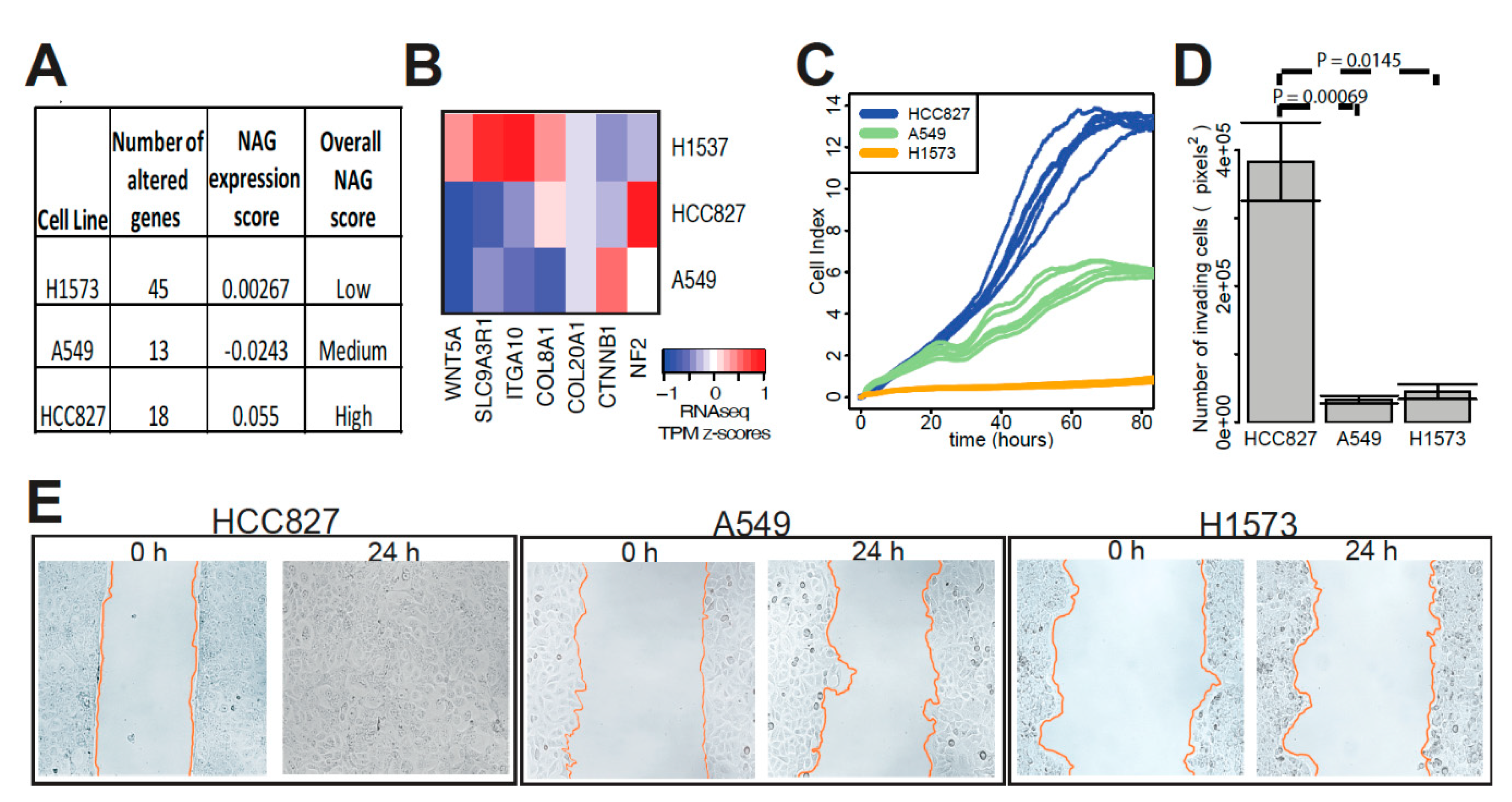
4.7. In Vitro Functional Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma; Shedden, K.; Taylor, J.M.G.; Enkemann, S.A.; Tsao, M.-S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [PubMed]
- Zhu, C.-Q.; Pintilie, M.; John, T.; Strumpf, D.; Shepherd, F.A.; Der, S.D.; Jurisica, I.; Tsao, M.-S. Understanding prognostic gene expression signatures in lung cancer. Clin. Lung Cancer 2009, 10, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wang, S.; Xiao, G.; Schiller, J.; Papadimitrakopoulou, V.; Minna, J.; Wistuba, I.I.; Xie, Y. Comprehensive evaluation of published gene expression prognostic signatures for biomarker-based lung cancer clinical studies. Ann. Oncol. 2017, 28, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Domerg, C.; Hainaut, P.; Jänne, P.A.; Pignon, J.-P.; Graziano, S.; Douillard, J.-Y.; Brambilla, E.; Le Chevalier, T.; Seymour, L.; et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J. Clin. Oncol. 2013, 31, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Le Teuff, G.; Lacas, B.; Tsao, M.S.; Graziano, S.; Pignon, J.-P.; Douillard, J.-Y.; Le Chevalier, T.; Seymour, L.; Filipits, M.; et al. Prognostic and Predictive Effect of TP53 Mutations in Patients with Non-Small Cell Lung Cancer from Adjuvant Cisplatin-Based Therapy Randomized Trials: A LACE-Bio Pooled Analysis. J. Thorac. Oncol. 2016, 11, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Zer, A.; Ding, K.; Lee, S.M.; Goss, G.D.; Seymour, L.; Ellis, P.M.; Hackshaw, A.; Bradbury, P.A.; Han, L.; O’Callaghan, C.J.; et al. Pooled Analysis of the Prognostic and Predictive Value of KRAS Mutation Status and Mutation Subtype in Patients with Non-Small Cell Lung Cancer Treated with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2016, 11, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.; Le Teuff, G.; Brambilla, E.; Shepherd, F.A.; Soria, J.-C.; Kratzke, R.; Graziano, S.; Douillard, J.-Y.; Rosell, R.; Reiman, A.; et al. LACE-Bio: Validation of Predictive and/or Prognostic Immunohistochemistry/Histochemistry-based Biomarkers in Resected Non-small-cell Lung Cancer. Clin. Lung Cancer 2019, 20, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Xiao, G.; Behrens, C.; Schiller, J.; Allen, J.; Chow, C.-W.; Suraokar, M.; Corvalan, A.; Mao, J.; White, M.A.; et al. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clin. Cancer Res. 2013, 19, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Hai, J.; Zhu, C.-Q.; Wang, T.; Organ, S.L.; Shepherd, F.A.; Tsao, M.-S. TRIM14 is a Putative Tumor Suppressor and Regulator of Innate Immune Response in Non-Small Cell Lung Cancer. Sci. Rep. 2017, 7, 39692. [Google Scholar] [CrossRef] [Green Version]
- Devarakonda, S.; Rotolo, F.; Tsao, M.-S.; Lanc, I.; Brambilla, E.; Masood, A.; Olaussen, K.A.; Fulton, R.; Sakashita, S.; McLeer-Florin, A.; et al. Tumor Mutation Burden as a Biomarker in Resected Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2995–3006. [Google Scholar] [CrossRef]
- Owada-Ozaki, Y.; Muto, S.; Takagi, H.; Inoue, T.; Watanabe, Y.; Fukuhara, M.; Yamaura, T.; Okabe, N.; Matsumura, Y.; Hasegawa, T.; et al. Prognostic Impact of Tumor Mutation Burden in Patients with Completely Resected Non-Small Cell Lung Cancer: Brief Report. J. Thorac. Oncol. 2018, 13, 1217–1221. [Google Scholar] [CrossRef]
- Yu, H.; Chen, Z.; Ballman, K.; Watson, M.A.; Govindan, R.; Lanc, I.; Beer, D.G.; Bueno, R.; Chirieac, L.; Chui, M.H.; et al. Correlation of PD-L1 expression with tumor mutation burden and gene signatures for prognosis in early stage squamous cell lung carcinoma. J. Thorac. Oncol. 2019, 14, 25–36. [Google Scholar] [CrossRef]
- Dong, Z.-Y.; Zhang, C.; Li, Y.-F.; Su, J.; Xie, Z.; Liu, S.-Y.; Yan, L.-X.; Chen, Z.-H.; Yang, X.-N.; Lin, J.-T.; et al. Genetic and Immune Profiles of Solid Predominant Lung Adenocarcinoma Reveal Potential Immunotherapeutic Strategies. J. Thorac. Oncol. 2018, 13, 85–96. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.-E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Wang, D.; Pham, N.-A.; Tong, J.; Sakashita, S.; Allo, G.; Kim, L.; Yanagawa, N.; Raghavan, V.; Wei, Y.; To, C.; et al. Molecular heterogeneity of non-small cell lung carcinoma patient-derived xenografts closely reflect their primary tumors. Int. J. Cancer 2017, 140, 662–673. [Google Scholar] [CrossRef]
- Wu, L.; Allo, G.; John, T.; Li, M.; Tagawa, T.; Opitz, I.; Anraku, M.; Yun, Z.; Pintilie, M.; Pitcher, B.; et al. Patient-Derived Xenograft Establishment from Human Malignant Pleural Mesothelioma. Clin. Cancer Res. 2017, 23, 1060–1067. [Google Scholar] [CrossRef]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef]
- Li, L.; Wei, Y.; To, C.; Zhu, C.-Q.; Tong, J.; Pham, N.-A.; Taylor, P.; Ignatchenko, V.; Ignatchenko, A.; Zhang, W.; et al. Integrated omic analysis of lung cancer reveals metabolism proteome signatures with prognostic impact. Nat. Commun. 2014, 5, 5469. [Google Scholar] [CrossRef]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Futreal, P.A.; Andrew Futreal, P.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- McGrail, D.J.; Federico, L.; Li, Y.; Dai, H.; Lu, Y.; Mills, G.B.; Yi, S.; Lin, S.-Y.; Sahni, N. Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat. Commun. 2018, 9, 1317. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Belvedere, O.; Berri, S.; Chalkley, R.; Conway, C.; Barbone, F.; Pisa, F.; MacLennan, K.; Daly, C.; Alsop, M.; Morgan, J.; et al. A computational index derived from whole-genome copy number analysis is a novel tool for prognosis in early stage lung squamous cell carcinoma. Genomics 2012, 99, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Miller, A.; Asmann, Y.; Cattaneo, L.; Braggio, E.; Keats, J.; Auclair, D.; Lonial, S.; MMRF CoMMpass Network; Russell, S.J.; Stewart, A.K. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 2017, 7, e612. [Google Scholar] [CrossRef]
- Campesato, L.F.; Barroso-Sousa, R.; Jimenez, L.; Correa, B.R.; Sabbaga, J.; Hoff, P.M.; Reis, L.F.L.; Galante, P.A.F.; Camargo, A.A. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget 2015, 6, 34221–34227. [Google Scholar] [CrossRef]
- Kinkead, H.L.; Hopkins, A.; Lutz, E.; Wu, A.A.; Yarchoan, M.; Cruz, K.; Woolman, S.; Vithayathil, T.; Glickman, L.H.; Ndubaku, C.O.; et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018, 3, 122857. [Google Scholar] [CrossRef]
- Miller, A.; Cattaneo, L.; Asmann, Y.W.; Braggio, E.; Keats, J.J.; Auclair, D.; Lonial, S.; Russell, S.J.; Keith Stewart, A. Correlation Between Somatic Mutation Burden, Neoantigen Load and Progression Free Survival in Multiple Myeloma: Analysis of MMRF CoMMpass Study. Blood 2016, 128, 193. [Google Scholar]
- John, T.; Kohler, D.; Pintilie, M.; Yanagawa, N.; Pham, N.-A.; Li, M.; Panchal, D.; Hui, F.; Meng, F.; Shepherd, F.A.; et al. The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non-small cell lung cancer. Clin. Cancer Res. 2011, 17, 134–141. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Jacobsen, A. Cgdsr: R-Based API for Accessing the MSKCC Cancer Genomics Data Server (CGDS), version 1.3.0; CRAN: Taipei City, Taiwan, 2017.
- Deng, M.; Brägelmann, J.; Kryukov, I.; Saraiva-Agostinho, N.; Perner, S. FirebrowseR: An R client to the Broad Institute’s Firehose Pipeline. Database 2017, 2017, 160. [Google Scholar] [CrossRef]
- Der, S.D.; Sykes, J.; Pintilie, M.; Zhu, C.-Q.; Strumpf, D.; Liu, N.; Jurisica, I.; Shepherd, F.A.; Tsao, M.-S. Validation of a histology-independent prognostic gene signature for early-stage, non-small-cell lung cancer including stage IA patients. J. Thorac. Oncol. 2014, 9, 59–64. [Google Scholar] [CrossRef]
- Nielsen, M.; Andreatta, M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016, 8, 33. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Szolek, A.; Schubert, B.; Mohr, C.; Sturm, M.; Feldhahn, M.; Kohlbacher, O. OptiType: Precision HLA typing from next-generation sequencing data. Bioinformatics 2014, 30, 3310–3316. [Google Scholar] [CrossRef]
- Sidney, J.; Peters, B.; Frahm, N.; Brander, C.; Sette, A. HLA class I supertypes: A revised and updated classification. BMC Immunol. 2008, 9, 1. [Google Scholar] [CrossRef]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, O.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2010, 39, D685–D690. [Google Scholar] [CrossRef]
- Radulovich, N.; Leung, L.; Ibrahimov, E.; Navab, R.; Sakashita, S.; Zhu, C.-Q.; Kaufman, E.; Lockwood, W.W.; Thu, K.L.; Fedyshyn, Y.; et al. Coiled-coil domain containing 68 (CCDC68) demonstrates a tumor-suppressive role in pancreatic ductal adenocarcinoma. Oncogene 2015, 34, 4238–4247. [Google Scholar] [CrossRef]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.-Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.-Q.; Tsao, M.-S. Prognostic markers in lung cancer: Is it ready for prime time? Transl. Lung Cancer Res. 2014, 3, 149–158. [Google Scholar]
- Hai, J.; Zhu, C.-Q.; Bandarchi, B.; Wang, Y.-H.; Navab, R.; Shepherd, F.A.; Jurisica, I.; Tsao, M.-S. L1 cell adhesion molecule promotes tumorigenicity and metastatic potential in non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1914–1924. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variation | HR | 95% CI | Wald Test p-Value |
|---|---|---|---|
| Age (>65 vs. ≤65) | 1.04 | 0.62–1.73 | 0.89 |
| Sex (F vs. M) | 0.85 | 0.51–1.43 | 0.54 |
| Tobacco (smoker vs. never) | 1.75 | 0.74–4.12 | 0.2 |
| Histology (adeno vs. squamous) | 0.96 | 0.52–1.79 | 0.91 |
| Overall NAG score (low vs. high burden) | 2.46 | 1.27–4.75 | 0.0077 * |
| Variation | HR | 95% CI | Wald Test p-Value |
|---|---|---|---|
| Age of diagnosis (≤65 y vs. >65 y) | 0.929 | 0.545–1.583 | 0.7865 |
| Gender (male vs. female) | 0.871 | 0.524–1.447 | 0.5929 |
| Smoking history (last 15 y; yes vs. no) | 0.751 | 0.434–1.302 | 0.3085 |
| Histology (adeno vs. squamous cell) | 0.677 | 0.392–1.169 | 0.1615 |
| Immunogenicity (≥1 neoantigen vs. 0 neoantigens) | 0.296 | 0.119–0.740 | 0.00919 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Pham, N.-A.; Freeman, T.M.; Raghavan, V.; Navab, R.; Chang, J.; Zhu, C.-Q.; Ly, D.; Tong, J.; Wouters, B.G.; et al. Somatic Alteration Burden Involving Non-Cancer Genes Predicts Prognosis in Early-Stage Non-Small Cell Lung Cancer. Cancers 2019, 11, 1009. https://doi.org/10.3390/cancers11071009
Wang D, Pham N-A, Freeman TM, Raghavan V, Navab R, Chang J, Zhu C-Q, Ly D, Tong J, Wouters BG, et al. Somatic Alteration Burden Involving Non-Cancer Genes Predicts Prognosis in Early-Stage Non-Small Cell Lung Cancer. Cancers. 2019; 11(7):1009. https://doi.org/10.3390/cancers11071009
Chicago/Turabian StyleWang, Dennis, Nhu-An Pham, Timothy M. Freeman, Vibha Raghavan, Roya Navab, Jonathan Chang, Chang-Qi Zhu, Dalam Ly, Jiefei Tong, Bradly G. Wouters, and et al. 2019. "Somatic Alteration Burden Involving Non-Cancer Genes Predicts Prognosis in Early-Stage Non-Small Cell Lung Cancer" Cancers 11, no. 7: 1009. https://doi.org/10.3390/cancers11071009