A Unique Pattern of Mesothelial-Mesenchymal Transition Induced in the Normal Peritoneal Mesothelium by High-Grade Serous Ovarian Cancer


Abstract
:
1. Introduction
2. Results
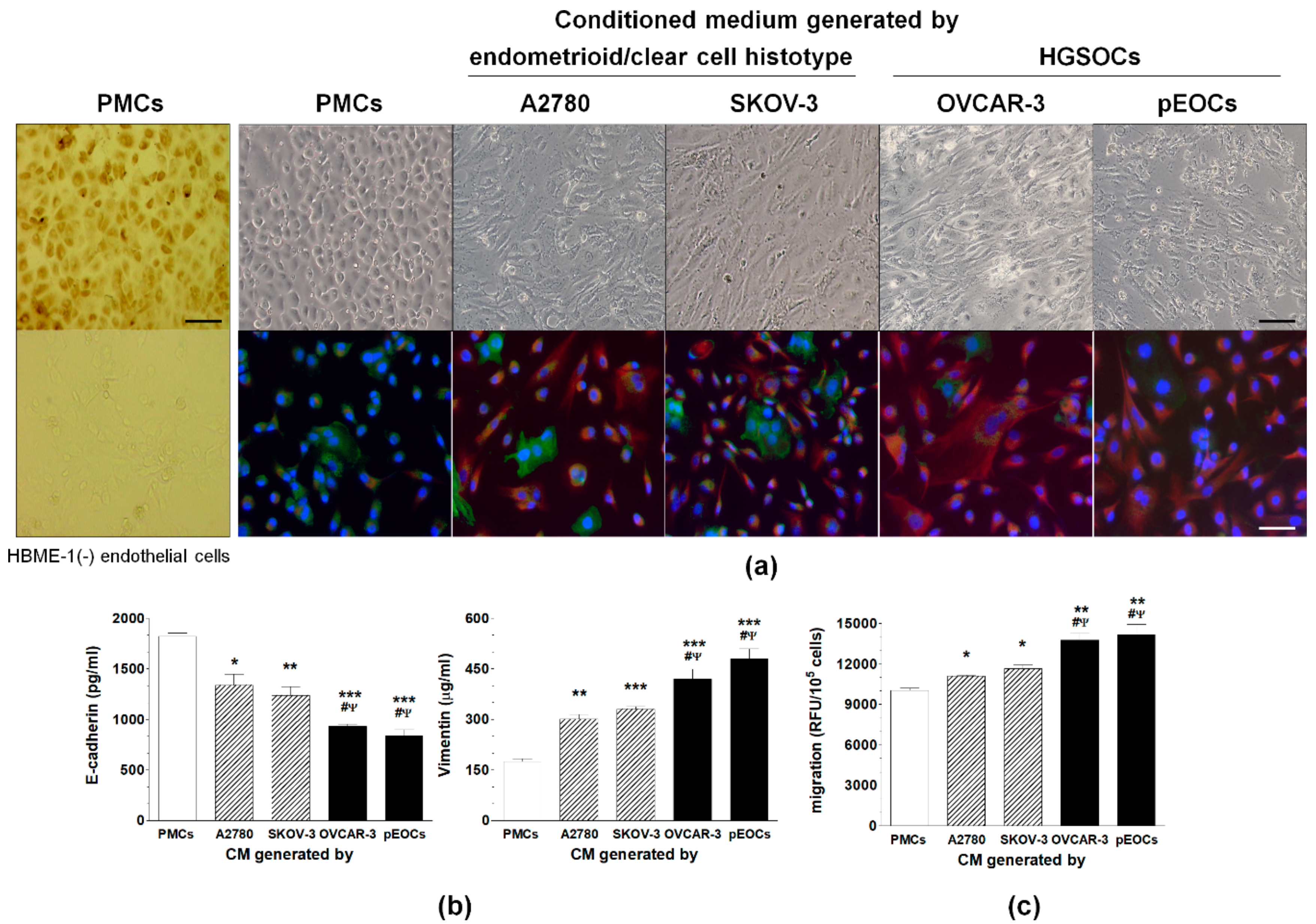
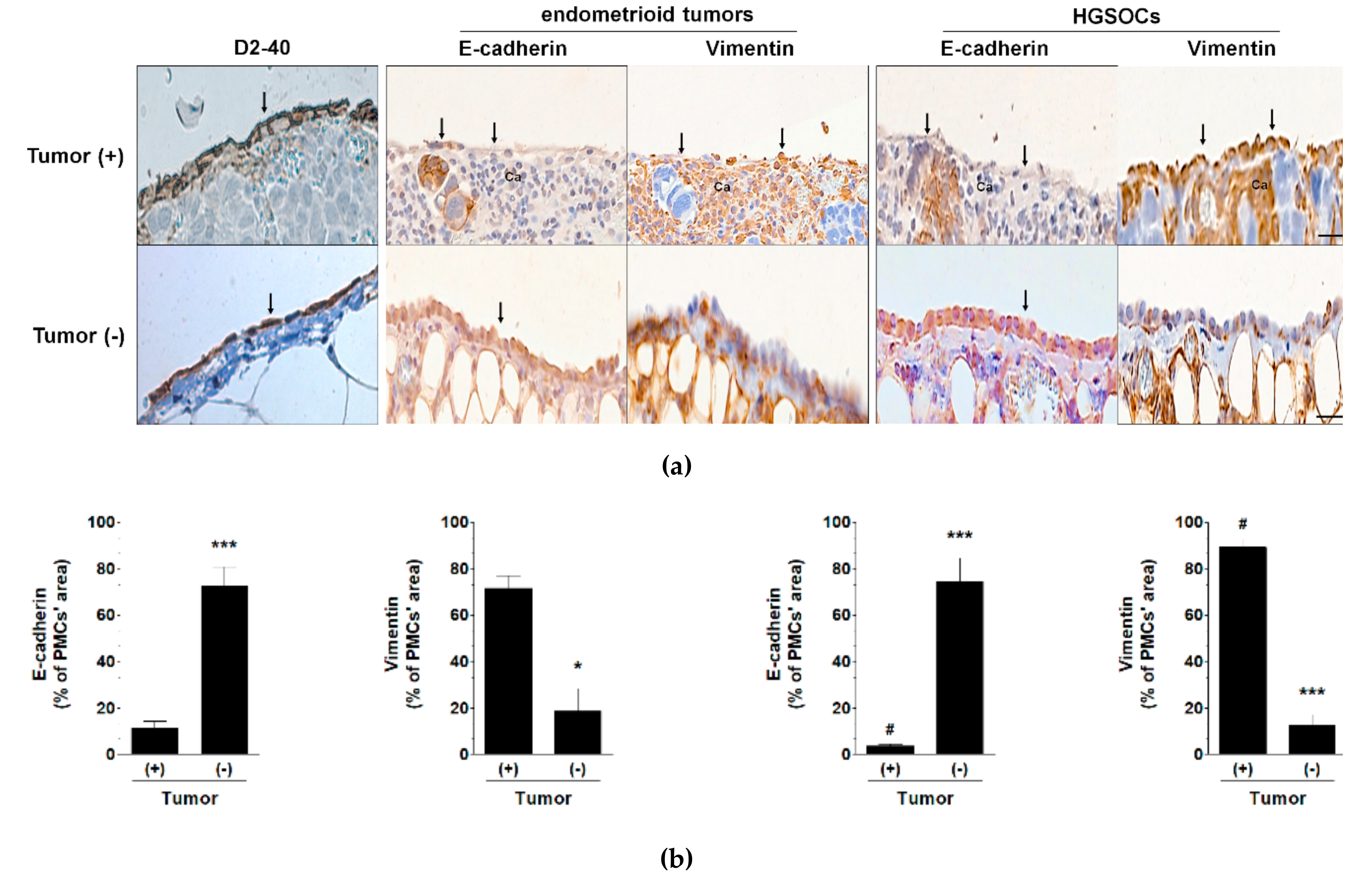
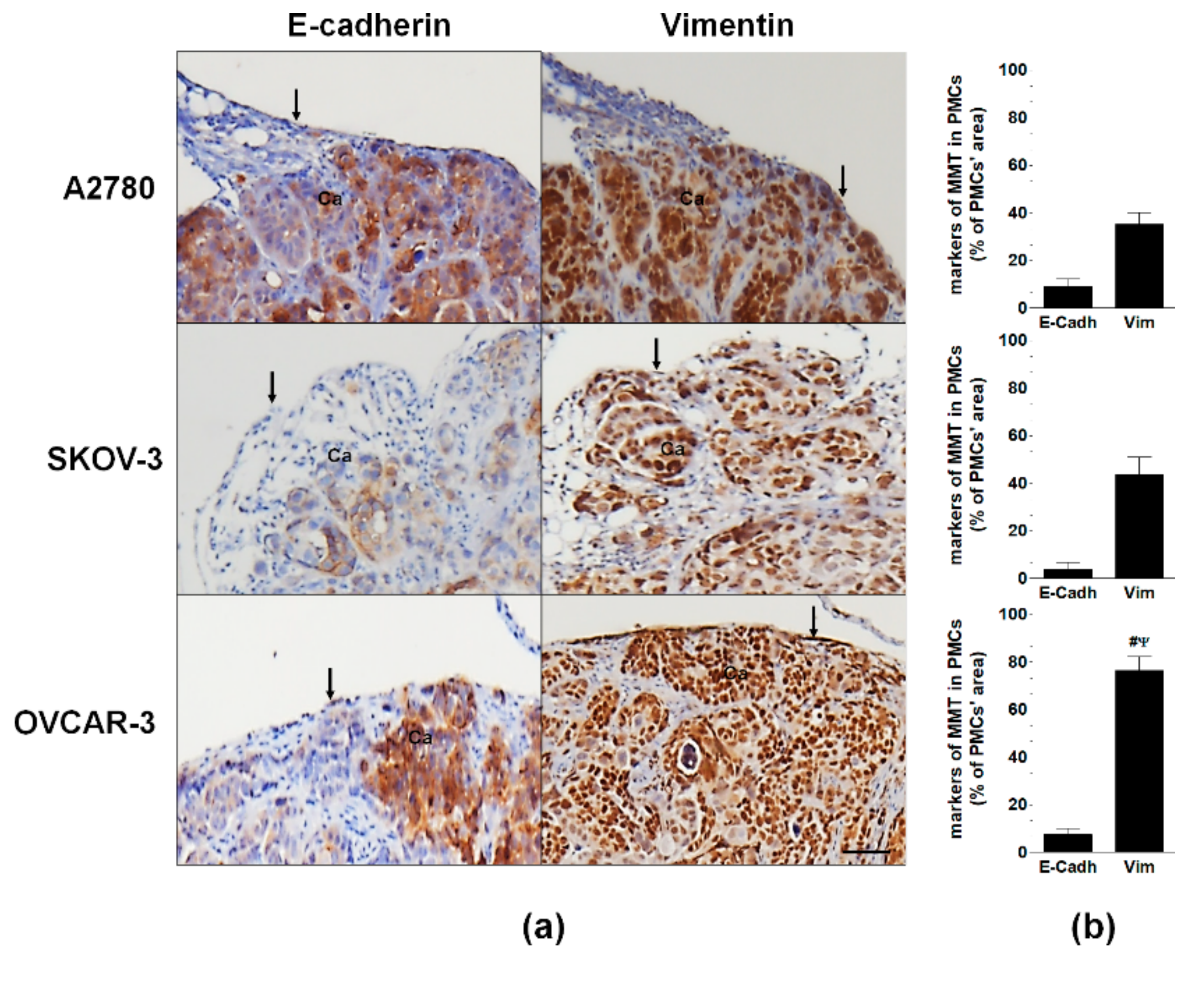
2.1. HGSOC Potentiates MMT in PMCs In Vitro and In Vivo
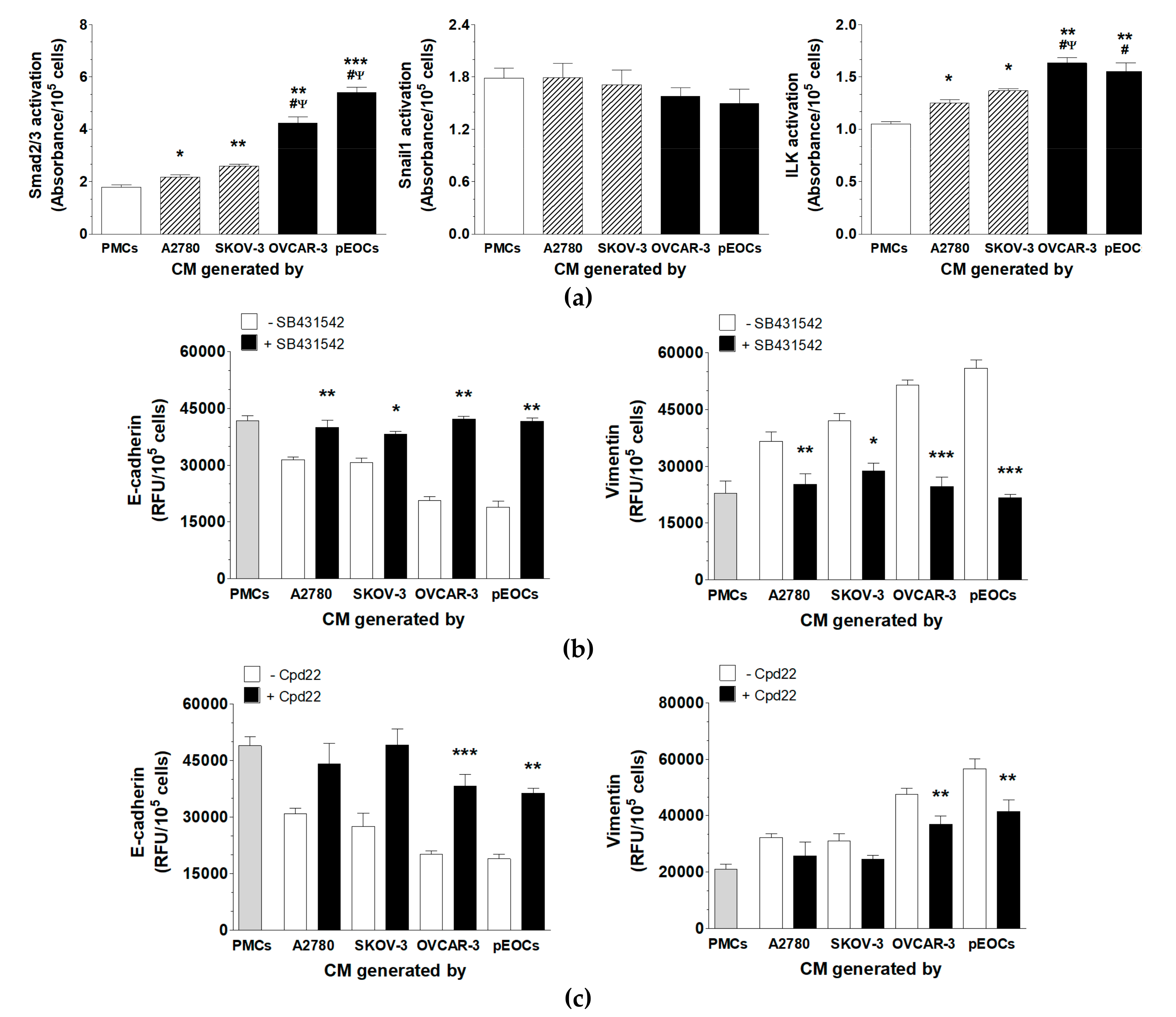
2.2. HGSOC-Driven MMT Involves Smad 2/3 and ILK
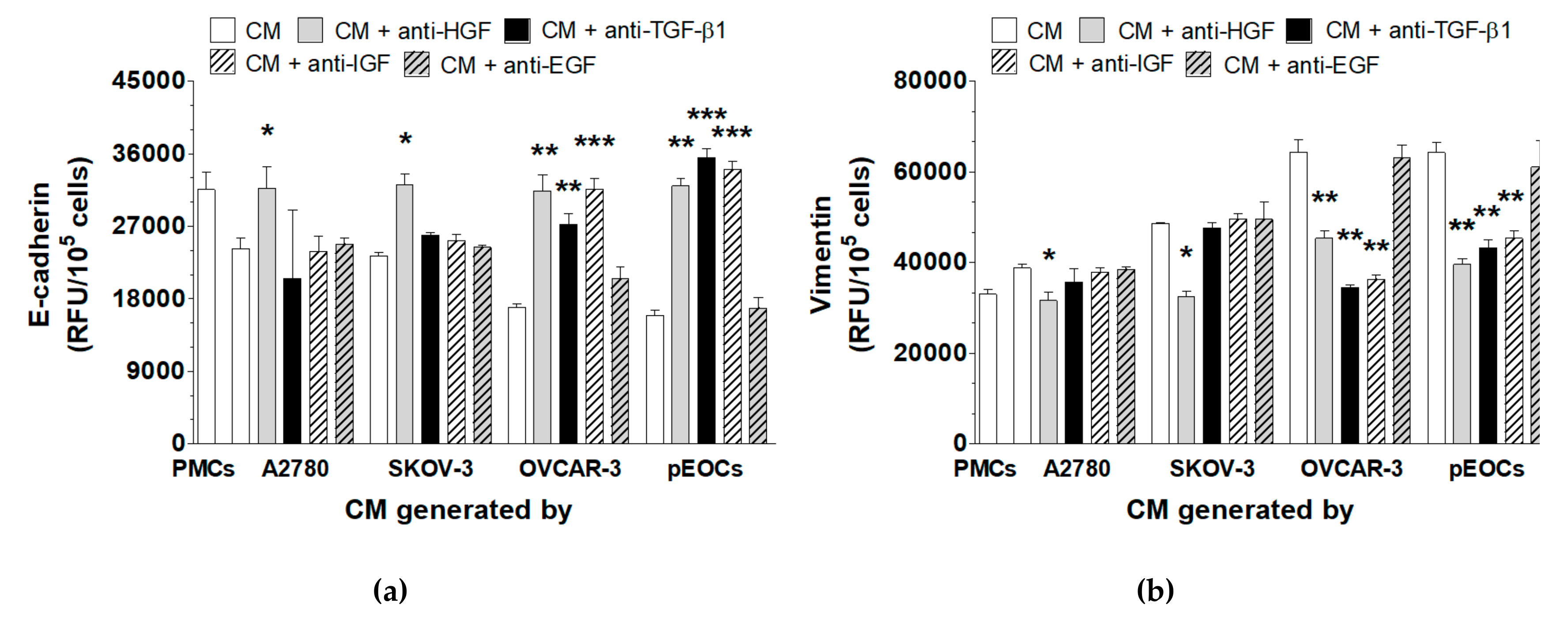
2.3. HGSOC-Driven MMT Is Triggered by IGF-1, HGF, and TGF-β1
2.4. PMCs Undergoing HGSOC-Induced MMT Are Strongly Tumor-Promoting
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Patients
4.3. Animal Studies
4.4. Cell Cultures
4.5. Conditioned Medium
4.6. Mesothelial-Mesenchymal Transition (MMT)
4.7. Signaling Pathways
4.8. Cell Proliferation, Migration, and Invasion
4.9. Cell Secretome
4.10. Intervention Studies
4.11. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Mikula-Pietrasik, J.; Uruski, P.; Tykarski, A.; Ksiazek, K. The peritoneal “soil” for a cancerous “seed”: A comprehensive review of the pathogenesis of intraperitoneal cancer metastases. Cell Mol. Life Sci. 2018, 75, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Veatch, A.L.; Carson, L.F.; Ramakrishnan, S. Differential expression of the cell-cell adhesion molecule E-cadherin in ascites and solid human ovarian tumor cells. Int. J. Cancer 1994, 58, 393–399. [Google Scholar] [CrossRef]
- Burleson, K.M.; Hansen, L.K.; Skubitz, A.P. Ovarian carcinoma spheroids disaggregate on type I collagen and invade live human mesothelial cell monolayers. Clin. Exp. Metastasis 2004, 21, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.D.; Saxena, A.; Bronner, M.E. Sip1 mediates an E-cadherin-to-N-cadherin switch during cranial neural crest EMT. J. Cell. Biol. 2013, 203, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Kim, O.; Stack, M.S. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers (Basel) 2017, 9, 104. [Google Scholar] [CrossRef]
- Sandoval, P.; Jimenez-Heffernan, J.A.; Rynne-Vidal, A.; Perez-Lozano, M.L.; Gilsanz, A.; Ruiz-Carpio, V.; Reyes, R.; Garcia-Bordas, J.; Stamatakis, K.; Dotor, J.; et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J. Pathol. 2013, 231, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ono, Y.J.; Kanemura, M.; Tanaka, T.; Hayashi, M.; Terai, Y.; Ohmichi, M. Hepatocyte growth factor secreted by ovarian cancer cells stimulates peritoneal implantation via the mesothelial-mesenchymal transition of the peritoneum. Gynecol. Oncol. 2015, 139, 345–354. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.A.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef]
- Mikula-Pietrasik, J.; Sosinska, P.; Kucinska, M.; Murias, M.; Maksin, K.; Malinska, A.; Ziolkowska, A.; Piotrowska, H.; Wozniak, A.; Ksiazek, K. Peritoneal mesothelium promotes the progression of ovarian cancer cells in vitro and in a mice xenograft model in vivo. Cancer Lett. 2014, 355, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Rynne-Vidal, A.; Au-Yeung, C.L.; Jimenez-Heffernan, J.A.; Perez-Lozano, M.L.; Cremades-Jimeno, L.; Barcena, C.; Cristobal-Garcia, I.; Fernandez-Chacon, C.; Yeung, T.L.; Mok, S.C.; et al. Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J. Pathol. 2017, 242, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.L.; Chen, L.Y.; Lai, H.C.; Chan, M.W. TGF-β: Friend or foe? The role of TGF-β/SMAD signaling in epigenetic silencing of ovarian cancer and its implication in epigenetic therapy. Expert. Opin. Ther. Targets 2010, 14, 1213–1223. [Google Scholar] [CrossRef]
- Tsoumas, D.; Nikou, S.; Giannopoulou, E.; Champeris Tsaniras, S.; Sirinian, C.; Maroulis, I.; Taraviras, S.; Zolota, V.; Kalofonos, H.P.; Bravou, V. ILK Expression in Colorectal Cancer Is Associated with EMT, Cancer Stem Cell Markers and Chemoresistance. Cancer Genomics Proteomics 2018, 15, 127–141. [Google Scholar] [CrossRef]
- Zhou, W.; Peng, Z.; Zhang, C.; Liu, S.; Zhang, Y. ILK-induced epithelial-mesenchymal transition promotes the invasive phenotype in adenomyosis. Biochem. Biophys. Res. Commun. 2018, 497, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Kondo, S.; Urushihara, M.; Takamatsu, M.; Kanemoto, K.; Nagata, M.; Kagami, S. Role of integrin-linked kinase in epithelial-mesenchymal transition in crescent formation of experimental glomerulonephritis. Nephrol. Dial. Transplant 2006, 21, 2380–2390. [Google Scholar] [CrossRef]
- Barbera, M.J.; Puig, I.; Dominguez, D.; Julien-Grille, S.; Guaita-Esteruelas, S.; Peiro, S.; Baulida, J.; Franci, C.; Dedhar, S.; Larue, L.; et al. Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene 2004, 23, 7345–7354. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Zhao, X.M.; Shuai, Z.F.; Li, C.Y.; Bai, Q.Y.; Yu, X.W.; Wen, Q.T. Snail promotes epithelial-mesenchymal transition and invasiveness in human ovarian cancer cells. Int. J. Clin. Exp. Med. 2015, 8, 7388–7393. [Google Scholar] [PubMed]
- Pang, M.; Wang, H.; Rao, P.; Zhao, Y.; Xie, J.; Cao, Q.; Wang, Y.; Wang, Y.M.; Lee, V.W.; Alexander, S.I.; et al. Autophagy links β-catenin and Smad signaling to promote epithelial-mesenchymal transition via upregulation of integrin linked kinase. Int. J. Biochem. Cell. Biol. 2016, 76, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kong, J.; Chang, H.; Kim, H.; Kim, A. EGF induces epithelial-mesenchymal transition through phospho-Smad2/3-Snail signaling pathway in breast cancer cells. Oncotarget 2016, 7, 85021–85032. [Google Scholar] [CrossRef]
- Liu, F.; Song, S.; Yi, Z.; Zhang, M.; Li, J.; Yang, F.; Yin, H.; Yu, X.; Guan, C.; Liu, Y.; et al. HGF induces EMT in non-small-cell lung cancer through the hBVR pathway. Eur. J. Pharmacol. 2017, 811, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, Q.; Wang, B.; Sun, Q.; He, Z.; Hong, J.; Kuehn, F.; Liu, E.; Zhang, Z. IGF-1 induces the epithelial-mesenchymal transition via Stat5 in hepatocellular carcinoma. Oncotarget 2017, 8, 111922–111930. [Google Scholar] [CrossRef]
- Romagnoli, M.; Belguise, K.; Yu, Z.; Wang, X.; Landesman-Bollag, E.; Seldin, D.C.; Chalbos, D.; Barille-Nion, S.; Jezequel, P.; Seldin, M.L.; et al. Epithelial-to-mesenchymal transition induced by TGF-beta1 is mediated by Blimp-1-dependent repression of BMP-5. Cancer Res. 2012, 72, 6268–6278. [Google Scholar] [CrossRef]
- Yu, M.A.; Shin, K.S.; Kim, J.H.; Kim, Y.I.; Chung, S.S.; Park, S.H.; Kim, Y.L.; Kang, D.H. HGF and BMP-7 ameliorate high glucose-induced epithelial-to-mesenchymal transition of peritoneal mesothelium. J. Am. Soc. Nephrol. 2009, 20, 567–581. [Google Scholar] [CrossRef]
- Mikula-Pietrasik, J.; Uruski, P.; Sosinska, P.; Maksin, K.; Piotrowska-Kempisty, H.; Kucinska, M.; Murias, M.; Szubert, S.; Wozniak, A.; Szpurek, D.; et al. Senescent peritoneal mesothelium creates a niche for ovarian cancer metastases. Cell Death. Dis. 2016, 7, e2565. [Google Scholar] [CrossRef] [PubMed]
- Que, J.; Wilm, B.; Hasegawa, H.; Wang, F.; Bader, D.; Hogan, B.L. Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc. Natl. Acad. Sci. USA 2008, 105, 16626–16630. [Google Scholar] [CrossRef]
- Mikula-Pietrasik, J.; Sosinska, P.; Maksin, K.; Kucinska, M.G.; Piotrowska, H.; Murias, M.; Wozniak, A.; Szpurek, D.; Ksiazek, K. Colorectal cancer-promoting activity of the senescent peritoneal mesothelium. Oncotarget 2015, 6, 29178–29195. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, K.; Mikula-Pietrasik, J.; Korybalska, K.; Dworacki, G.; Jorres, A.; Witowski, J. Senescent peritoneal mesothelial cells promote ovarian cancer cell adhesion: The role of oxidative stress-induced fibronectin. Am. J. Pathol. 2009, 174, 1230–1240. [Google Scholar] [CrossRef]
- Mikula-Pietrasik, J.; Uruski, P.; Szubert, S.; Szpurek, D.; Sajdak, S.; Tykarski, A.; Ksiazek, K. Malignant ascites determine the transmesothelial invasion of ovarian cancer cells. Int. J. Biochem. Cell. Biol. 2017, 92, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Pakula, M.; Mikula-Pietrasik, J.; Witucka, A.; Kostka-Jeziorny, K.; Uruski, P.; Moszynski, R.; Naumowicz, E.; Sajdak, S.; Tykarski, A.; Ksiazek, K. The Epithelial-Mesenchymal Transition Initiated by Malignant Ascites Underlies the Transmesothelial Invasion of Ovarian Cancer Cells. Int. J. Mol. Sci. 2019, 20, 137. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soluble Agent (pg/mL) | PMCs | A2780 | SKOV-3 | OVCAR-3 | pEOCs |
|---|---|---|---|---|---|
| EGF | 40 ± 2 | 48 ± 1 * | 39 ± 1 | 53 ± 1 *Ψ | 46 ± 2 *Ψ |
| HGF | 1195 ± 20 * | 1342 ± 23 * | 1552 ± 21 ** | 2704 ± 42 ***#Ψ | 3055 ± 56 ***#Ψ |
| IGF-1 | 134 ± 3 | 145 ± 2 * | 151 ± 8 * | 296 ± 32 **#Ψ | 491 ± 16 ***#Ψ |
| TGF-β1 | 114 ± 11 | 167 ± 5 * | 151 ± 8 * | 311 ± 7 ***#Ψ | 434 ± 12 ***#Ψ |
| Soluble Agent (pg/105 Cells) | PMCs | A2780 | SKOV-3 | OVCAR-3 | pEOCs |
|---|---|---|---|---|---|
| CCL2 | 246 ± 4 | 288 ± 2 * | 288 ± 1 * | 386 ± 4 *#Ψ | 420 ± 42 **#Ψ |
| CXCL8 | 291 ± 12 | 245 ± 34 | 306 ± 8 * | 278 ± 12 | 422 ± 6 ***#Ψ |
| CXCL12 | 171 ± 8 | 205 ± 9 ** | 223 ± 10* | 385 ± 21 **#Ψ | 419 ± 18 ***#Ψ |
| HGF | 33 ± 4 | 170 ± 11 *** | 63 ± 7 * | 223 ± 7 ***#Ψ | 202 ± 10 ***#Ψ |
| ICAM-1 | 98 ± 2 | 82 ± 4 | 148 ± 1 * | 271 ± 16 ***#Ψ | 334 ± 3 ***#Ψ |
| IL-6 | 75 ± 2 | 73 ± 4 | 69 ± 6 | 81 ± 4 | 221 ± 2 ***#Ψ |
| PAI-1 | 1932 ± 32 | 2019 ± 63 | 1899 ± 28 | 2321 ± 1 *#Ψ | 4103 ± 92 ***#Ψ |
| TGF-β1 | 127 ± 1 | 148 ± 1 * | 163 ± 2 * | 311 ± 22 ***#Ψ | 380 ± 1 ***#Ψ |
| uPA | 6 ± 1 | 7 ± 1 | 8 ± 1 | 7 ± 1 | 18 ± 1 **#Ψ |
| VEGF | 227 ± 4 | 225 ± 11 | 262 ± 12 * | 287 ± 18 *# | 454 ± 4 ***#Ψ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pakuła, M.; Uruski, P.; Niklas, A.; Woźniak, A.; Szpurek, D.; Tykarski, A.; Mikuła-Pietrasik, J.; Książek, K. A Unique Pattern of Mesothelial-Mesenchymal Transition Induced in the Normal Peritoneal Mesothelium by High-Grade Serous Ovarian Cancer. Cancers 2019, 11, 662. https://doi.org/10.3390/cancers11050662
Pakuła M, Uruski P, Niklas A, Woźniak A, Szpurek D, Tykarski A, Mikuła-Pietrasik J, Książek K. A Unique Pattern of Mesothelial-Mesenchymal Transition Induced in the Normal Peritoneal Mesothelium by High-Grade Serous Ovarian Cancer. Cancers. 2019; 11(5):662. https://doi.org/10.3390/cancers11050662
Chicago/Turabian StylePakuła, Martyna, Paweł Uruski, Arkadiusz Niklas, Aldona Woźniak, Dariusz Szpurek, Andrzej Tykarski, Justyna Mikuła-Pietrasik, and Krzysztof Książek. 2019. "A Unique Pattern of Mesothelial-Mesenchymal Transition Induced in the Normal Peritoneal Mesothelium by High-Grade Serous Ovarian Cancer" Cancers 11, no. 5: 662. https://doi.org/10.3390/cancers11050662