Integrated Somatic and Germline Whole-Exome Sequencing Analysis in Women with Lung Cancer after a Previous Breast Cancer

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
2.1. Patients and Clinical Characteristics
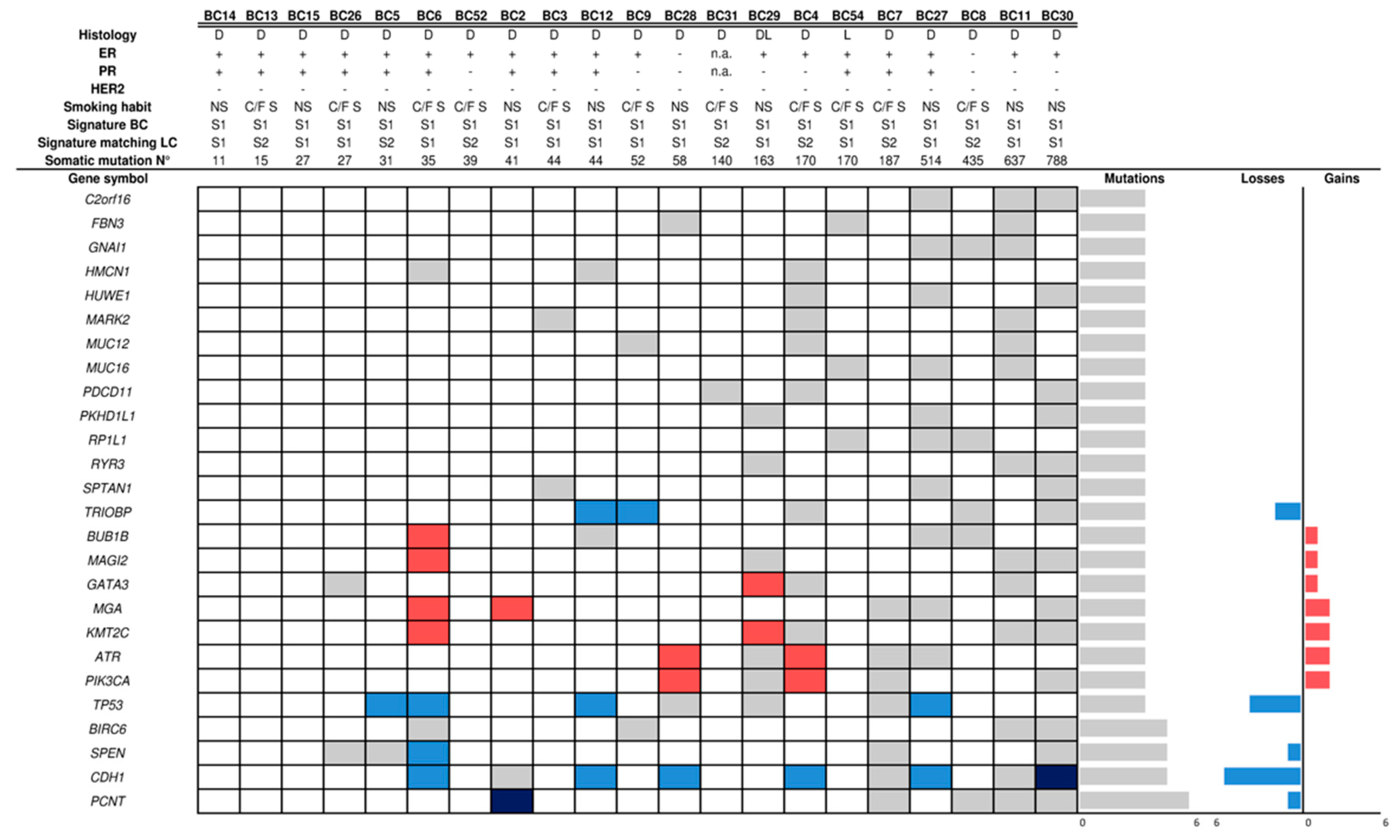
2.2. Mutational Profile and Copy Number Alteration (CNA) of SP Breast Cancers
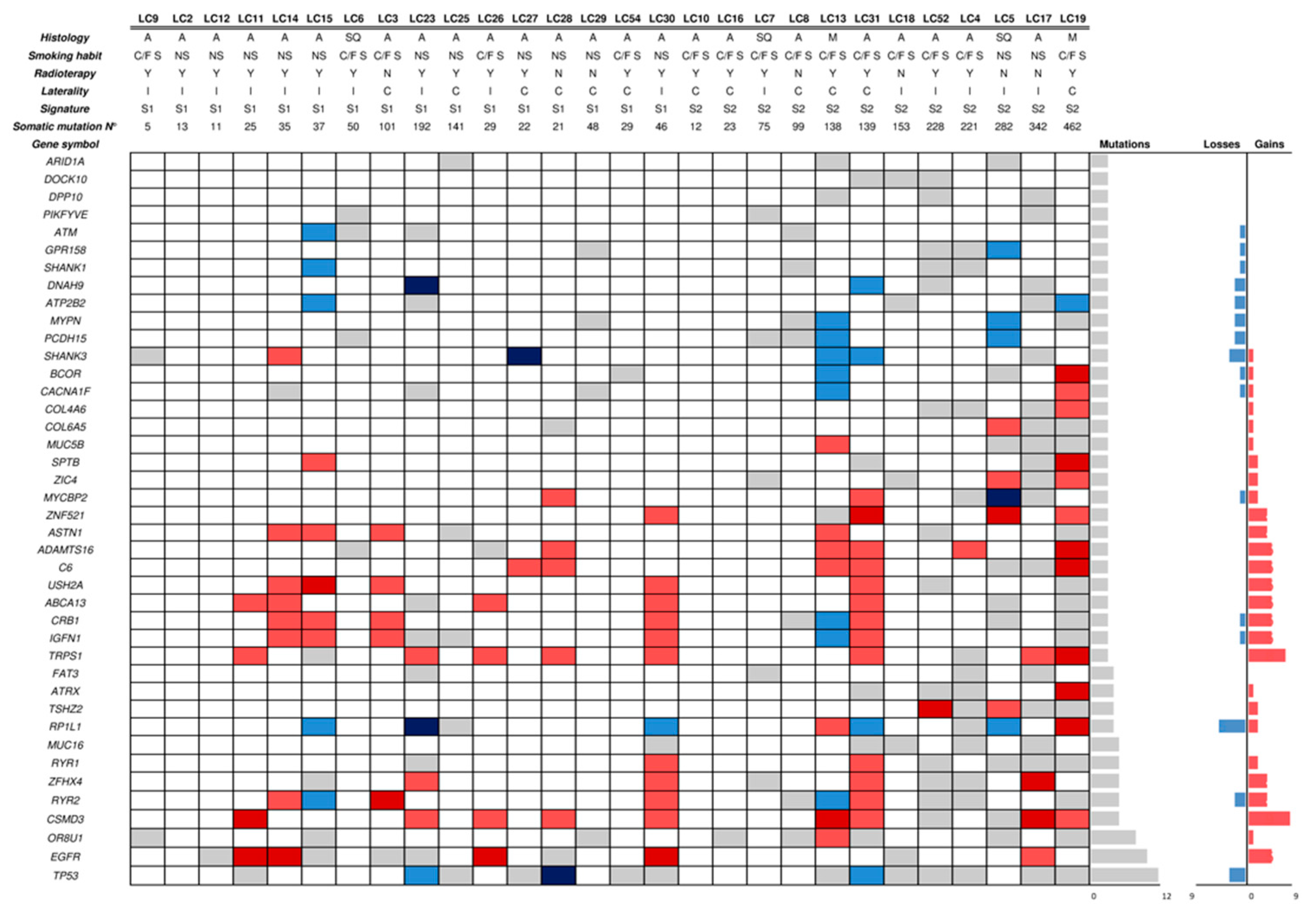
2.3. Mutational Profile and CNAs of SP Lung Cancers
2.4. Microsatellite Instability (MSI) Analysis
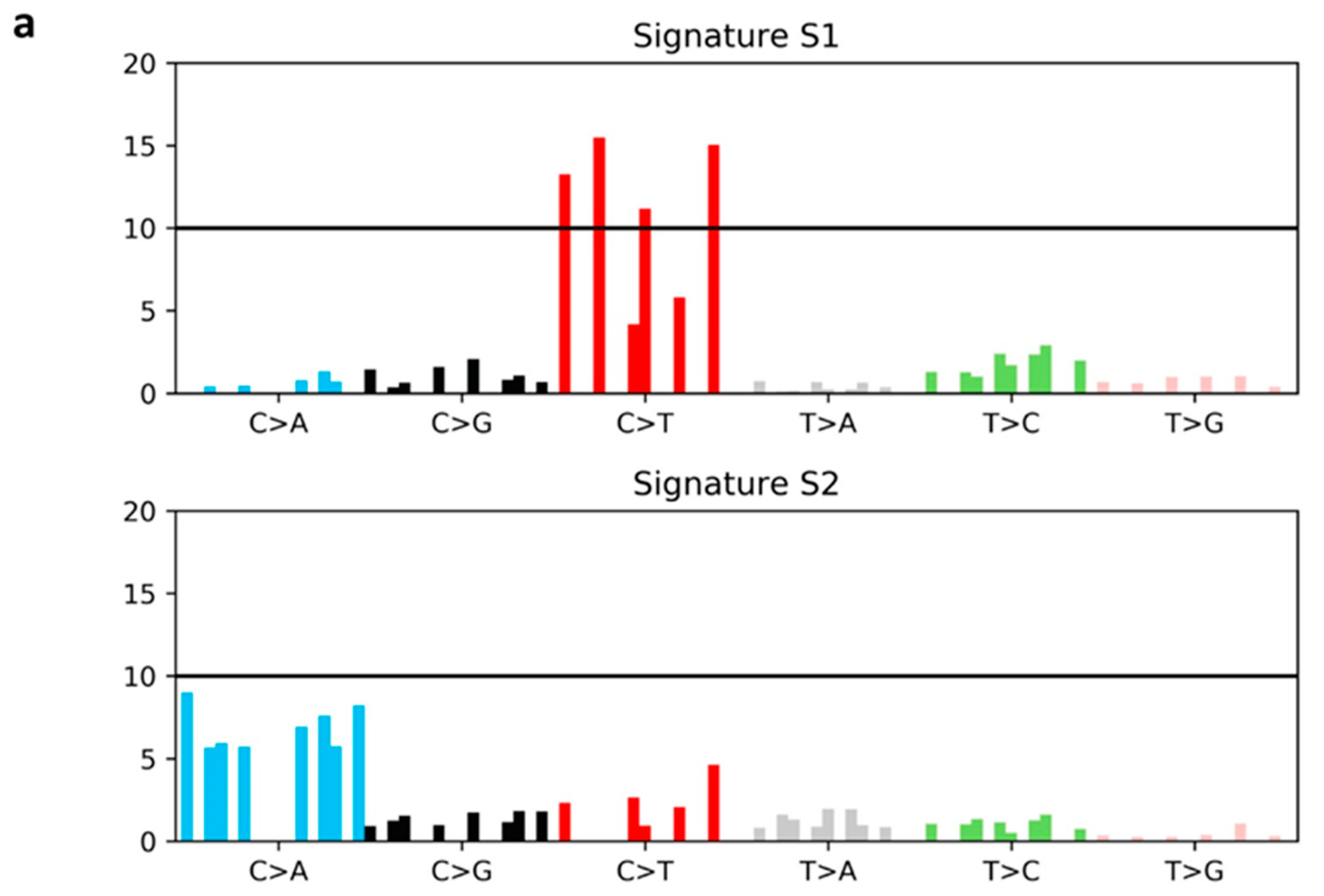
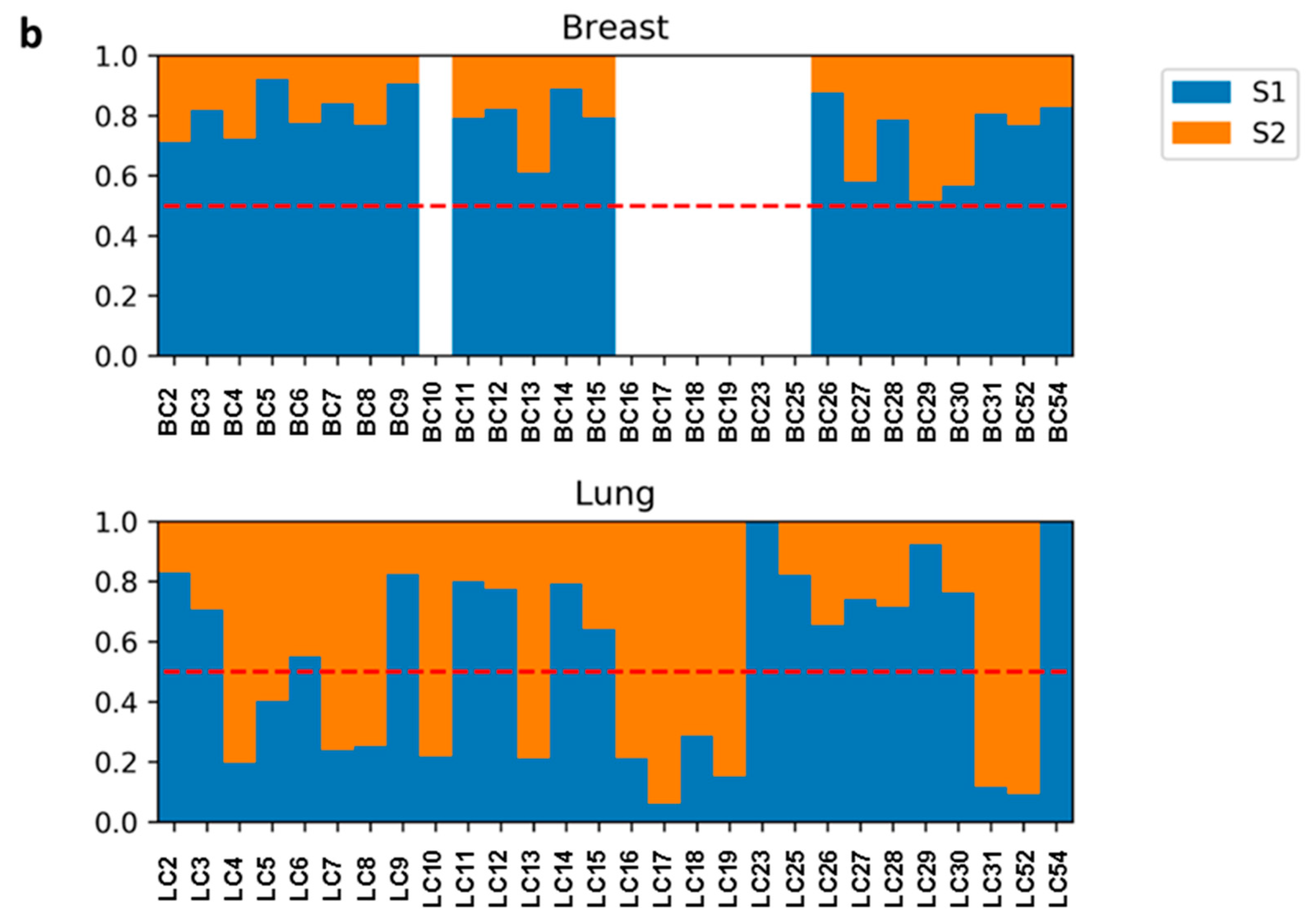
2.5. Mutational Signatures
2.6. WES Germline Analysis and Validation
3. Discussion
4. Materials and Methods
4.1. Patient Enrolment and Sample Collection
4.2. Genomic DNA Extraction
4.3. WES and Custom NGS Panel Library Preparation and Sequencing
4.4. Burden Test
4.5. MSI Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
WES Bioinformatics Analysis
Somatic Analysis
Copy Number Alteration Analysis
Germline Analysis
Custom Panel Bioinformatics Analysis
References
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Surveillance, Epidemiology, and End Results Program (SEER). SEER Stat Fact Sheets: Female Breast Cancer. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 4 October 2017).
- Raymond, J.S.; Hogue, C.J. Multiple primary tumours in women following breast cancer, 1973–2000. Br. J. Cancer 2006, 94, 1745–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, F.; Randimbison, L.; Te, V.C.; La Vecchia, C. Cancer risk after radiotherapy for breast cancer. Br. J. Cancer 2006, 95, 390–392. [Google Scholar] [CrossRef] [Green Version]
- Neugut, A.I.; Robinson, E.; Lee, W.C.; Murray, T.; Karwoski, K.; Kutcher, G.J. Lung cancer after radiation therapy for breast cancer. Cancer 1993, 71, 3054–3057. [Google Scholar] [CrossRef] [Green Version]
- Neugut, A.I.; Murray, T.; Santos, J.; Murray, T.; Karwoski, K.; Kutcher, G.J. Increased risk of lung cancer after breast cancer radiation therapy in cigarette smokers. Cancer 1994, 73, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Kirova, Y.M.; Gambotti, L.; De Rycke, Y.; Vilcoq, J.R.; Asselain, B.; Fourquet, A. Risk of second malignancies after adjuvant radiotherapy for breast cancer: A large-scale, single-institution review. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 359–363. [Google Scholar] [CrossRef]
- Prochazka, M.; Granath, F.; Ekbom, A.; Shields, P.G.; Hall, P. Lung cancer risks in women with previous breast cancer. Eur. J. Cancer 2002, 38, 1520–1525. [Google Scholar] [CrossRef]
- Zablotska, L.B.; Neugut, A.I. Lung carcinoma after radiation therapy in women treated with lumpectomy or mastectomy for primary breast carcinoma. Cancer 2003, 97, 1404–1411. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.J.; Huang, T.W.; Lin, F.H.; Chung, C.H.; Tsao, C.H.; Chien, W.C. Radiation Therapy for Invasive Breast Cancer Increases the Risk of Second Primary Lung Cancer: A Nationwide Population-Based Cohort Analysis. J. Thorac. Oncol. 2017, 12, 782–790. [Google Scholar] [CrossRef]
- Kaufman, E.L.; Jacobson, J.S.; Hershman, D.L.; Desai, M.; Neugut, A.I. Effect of breast cancer radiotherapy and cigarette smoking on risk of second primary lung cancer. J. Clin. Oncol. 2008, 26, 392–398. [Google Scholar] [CrossRef]
- Cybulski, C.; Nazarali, S.; Narod, S.A. Multiple primary cancers as a guide to heritability. Int. J. Cancer 2014, 135, 1756–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, J.D.; Hung, R.J.; Gaborieau, V.; Boffetta, P.; Chabrier, A.; Byrnes, G.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J. Lung cancer susceptibility locus at 5p15.33. Nat. Genet. 2008, 40, 1404–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timofeeva, M.N.; Hung, R.J.; Rafnar, T.; Christiani, D.C.; Field, J.K.; Bickeböller, H.; Risch, A.; McKay, J.D.; Wang, Y.; Dai, J.; et al. Influence of common genetic variation on lung cancer risk: Meta-analysis of 14 900 cases and 29 485 controls. Hum. Mol. Genet. 2012, 21, 4980–4995. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Coco, S.; Truini, A.; Vanni, I.; Dal Bello, M.G.; Alama, A.; Rijavec, E.; Genova, C.; Barletta, G.; Sini, C.; Burrafato, G.; et al. Next generation sequencing in non-small cell lung cancer: New avenues toward the personalized medicine. Curr. Drug Targets 2015, 16, 47–59. [Google Scholar] [CrossRef]
- Mathur, P.; Medicherla, K.M.; Chaudhary, S.; Patel, M.; Bagali, P.; Suravajhala, P. Whole exome sequencing reveals rare variants linked to congenital pouch colon. Sci. Rep. 2018, 8, 6646. [Google Scholar] [CrossRef] [PubMed]
- Artomov, M.; Stratigos, A.J.; Kim, I.; Kumar, R.; Lauss, M.; Reddy, B.Y.; Miao, B.; Daniela Robles-Espinoza, C.; Sankar, A.; Njauw, C.N.; et al. Rare Variant, Gene-Based Association Study of Hereditary Melanoma Using Whole-Exome Sequencing. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef] [PubMed]
- Vaca-Paniagua, F.; Alvarez-Gomez, R.M.; Maldonado-Martínez, H.A.; Pérez-Plasencia, C.; Fragoso-Ontiveros, V.; Lasa-Gonsebatt, F.; Herrera, L.A.; Cantú, D.; Bargallo-Rocha, E.; Mohar, A.; et al. Revealing the Molecular Portrait of Triple Negative Breast Tumors in an Understudied Population through Omics Analysis of Formalin-Fixed and Paraffin-Embedded Tissues. PLoS ONE 2015, 10, e0126762. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Coco, S.; Truini, A.; Vanni, I.; Genova, C.; Rosano, C.; Dal Bello, M.G.; Alama, A.; Venè, R.; Rijavec, E.; Barletta, G.; et al. Uncommon EGFR Exon 19 Mutations Confer Gefitinib Resistance in Advanced Lung Adenocarcinoma. J. Thorac. Oncol. 2015, 10, e50–e52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behjati, S.; Gundem, G.; Wedge, D.C.; Roberts, N.D.; Tarpey, P.S.; Cooke, S.L.; Van Loo, P.; Alexandrov, L.B.; Ramakrishna, M.; Davies, H.; et al. Mutational signatures of ionizing radiation in second malignancies. Nat. Commun. 2016, 7, 12605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calistri, D.; Presciuttini, S.; Buonsanti, G.; Radice, P.; Gazzoli, I.; Pensotti, V.; Sala, P.; Eboli, M.; Andreola, S.; Russo, A.; et al. Microsatellite instability in colorectal-cancer patients with suspected genetic predisposition. Int. J. Cancer 2000, 89, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef]
- Zhang, X.; Miao, X.; Sun, T.; Tan, W.; Qu, S.; Xiong, P.; Zhou, Y.; Lin, D. Functional polymorphisms in cell death pathway genes FAS and FASL contribute to risk of lung cancer. J. Med. Genet. 2005, 42, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Sun, T.; Xue, L.; Han, X.; Zhang, B.; Lu, N.; Shi, Y.; Tan, W.; Zhou, Y.; Zhao, D.; et al. Functional polymorphisms in FAS and FASL contribute to increased apoptosis of tumor infiltration lymphocytes and risk of breast cancer. Carcinogenesis 2007, 28, 1067–1073. [Google Scholar] [CrossRef]
- Yeh, I.T.; Lenci, R.E.; Qin, Y.; Buddavarapu, K.; Ligon, A.H.; Leteurtre, E.; Do Cao, C.; Cardot-Bauters, C.; Pigny, P.; Dahia, P.L. A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum. Genet. 2008, 124, 279–285. [Google Scholar] [CrossRef]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, S.H.; Zhou, J.; Wu, T.; Li, J. Contribution of PGR genetic polymorphisms to the pathogenesis of endometrial cancer: A meta-analysis. J. Cancer Res. Ther. 2015, 11, 810–817. [Google Scholar] [PubMed]
- Pritchard, A.L.; Johansson, P.A.; Nathan, V.; Howlie, M.; Symmons, J.; Palmer, J.M.; Hayward, N.K. Germline mutations in candidate predisposition genes in individuals with cutaneous melanoma and at least two independent additional primary cancers. PLoS ONE 2018, 13, e0194098. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef] [PubMed]
- King, R.J.; Yu, F.; Singh, P.K. Genomic alterations in mucins across cancers. Oncotarget 2017, 8, 67152–67168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes Fajardo, K.V.; Adams, D.; NISC Comparative Sequencing Program; Mason, C.E.; Sincan, M.; Tifft, C.; Toro, C.; Boerkoel, C.F.; Gahl, W.; Markello, T. Detecting false-positive signals in exome sequencing. Hum. Mutat. 2012, 33, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Lévy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 1, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2019, 534, 47–54, Erratum in 2019, 566, E1. [Google Scholar] [CrossRef] [PubMed]
- Li, G.H.; Arora, P.D.; Chen, Y.; McCulloch, C.A.; Liu, P. Multifunctional roles of gelsolin in health and diseases. Med. Res. Rev. 2012, 32, 999–1025. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Shirkoohi, R.; Nakagawa, K.; Qiao, H.; Fujita, H.; Okada, F.; Hamada, J.; Kuzumaki, S.; Takimoto, M.; Kuzumaki, N. siRNA gelsolin knockdown induces epithelial-mesenchymal transition with a cadherin switch in human mammary epithelial cells. Int. J. Cancer 2006, 118, 1680–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, R.M.; Mahjabeen, I.; Sabir, M.; Masood, N.; Ali, K.; Malik, F.A.; Kayani, M.A. Mutational spectrum of Gelsolin and its down regulation is associated with breast cancer. Dis. Markers 2013, 34, 71–80. [Google Scholar] [CrossRef]
- Wang, P.W.; Abedini, M.R.; Yang, L.X.; Ding, A.A.; Figeys, D.; Chang, J.Y.; Tsang, B.K.; Shieh, D.B. Gelsolin regulates cisplatin sensitivity in human head-and-neck cancer. Int. J. Cancer 2014, 135, 2760–2769. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.S.; Wang, W.; Li, J.P.; Liu, C.M.; He, L. Gelsolin Promotes Radioresistance in Non-Small Cell Lung Cancer Cells Through Activation of Phosphoinositide 3-Kinase/Akt Signaling. Technol Cancer Res. Treat. 2017, 16, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cui, F.; Cheng, Y.; Han, L.; Wang, J.; Sun, D.; Liu, Y.L.; Zhou, P.K.; Min, R. Gelsolin: Role of a functional protein in mitigating radiation injury. Cell Biochem. Biophys. 2015, 71, 389–396. [Google Scholar] [CrossRef]
- Yang, X.D.; Zhao, S.F.; Zhang, Q.; Li, W.; Wang, Y.X.; Hong, X.W.; Hu, Q.G. Gelsolin rs1078305 and rs10818524 polymorphisms were associated with risk of oral squamous cell carcinoma in a Chinese Han population. Biomarkers 2016, 21, 267–271. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Stitziel, N.O.; Kiezun, A.; Sunyaev, S. Computational and statistical approaches to analyzing variants identified by exome sequencing. Genome Biol. 2011, 12, 227. [Google Scholar] [CrossRef]
- Neale, B.M.; Rivas, M.A.; Voight, B.F.; Altshuler, D.; Devlin, B.; Orho-Melander, M.; Kathiresan, S.; Purcell, S.M.; Roeder, K.; Daly, M.J. Testing for an unusual distribution of rare variants. PLoS Genet. 2011, 7, e1001322. [Google Scholar] [CrossRef]
- Dang, N.; Meng, X.; Song, H. Nicotinic acetylcholine receptors and cancer. Biomed. Rep. 2016, 4, 515–518. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y. The Oncogenic Functions of Nicotinic Acetylcholine Receptors. J. Oncol. 2016, 2016, 9650481. [Google Scholar] [CrossRef]
- Reisz, J.A.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C.M. Effects of ionizing radiation on biological molecules mechanisms of damage and emerging methods of detection. Antioxid. Redox Signal. 2014, 21, 260–292. [Google Scholar] [CrossRef]
- Bonfiglio, S.; Vanni, I.; Rossella, V.; Truini, A.; Lazarevic, D.; Dal Bello, M.G.; Alama, A.; Mora, M.; Rijavec, E.; Genova, C.; et al. Performance comparison of two commercial human whole-exome capture systems on formalin-fixed paraffin-embedded lung adenocarcinoma samples. BMC Cancer 2016, 16, 692. [Google Scholar] [CrossRef]
- Madsen, B.E.; Browning, S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009, 5, e1000384. [Google Scholar] [CrossRef]
- Zauber, P.; Marotta, S.; Sabbath-Solitare, M. Copy number of the Adenomatous Polyposis Coli gene is not always neutral in sporadic colorectal cancers with loss of heterozygosity for the gene. BMC Cancer 2016, 16, 213. [Google Scholar] [CrossRef]
- Vanni, I.; Coco, S.; Bonfiglio, S.; Cittaro, D.; Genova, C.; Biello, F.; Mora, M.; Rossella, V.; Dal Bello, M.G.; Truini, A.; et al. Whole exome sequencing of independent lung adenocarcinoma, lung squamous cell carcinoma, and malignant peritoneal mesothelioma: A case report. Medicine (Baltim.) 2016, 95, e5447. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 1, 589–595. [Google Scholar] [CrossRef]
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.T.; Quinlan, A.R.; Hall, I.M. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat. Methods 2015, 12, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 4, D777–D783. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 18, 285–291. [Google Scholar] [CrossRef]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Perez, C.; Tamborero, D.; Schroeder, M.P.; Antolín, A.A.; Deu-Pons, J.; Perez-Llamas, C.; Mestres, J.; Gonzalez-Perez, A.; Lopez-Bigas, N. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals novel targeting opportunities. Cancer Cell 2015, 9, 382–396. [Google Scholar] [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Cheng, W.C.; Chung, I.F.; Chen, C.Y.; Sun, H.J.; Fen, J.J.; Tang, W.C.; Chang, T.Y.; Wong, T.T.; Wang, H.W. DriverDB: An exome sequencing database for cancer driver gene identification. Nucleic Acids Res. 2014, 42, D1048–D1054. [Google Scholar] [CrossRef]
- Kautto, E.A.; Bonneville, R.; Miya, J.; Yu, L.; Krook, M.A.; Reeser, J.W.; Roychowdhury, S. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 2017, 31, 7452–7463. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Population | Control Population | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Breast Cancer n. 60 | Lung Cancer n. 60 | Breast Cancer n. 42 | Lung Cancer n. 11 | ||||||||
| Age at diagnosis | median | range | Age at diagnosis | median | range | Age at diagnosis | median | range | Age at diagnosis | median | range |
| Breast Cancer | 60 | 37–76 | Lung Cancer | 68 | 48–88 | Breast Cancer | 60 | 41–81 | Lung Cancer | 59 | 37–81 |
| Smoking habit | N. | % | Smoking habit | N. | % | Smoking habit | N. | % | Smoking habit | N. | % |
| Smoker | 28 | 46.7 | Smoker | 25 | 41.7 | Smoker | 9 | 21.4 | Smoker | 3 | 27.3 |
| Former smoker | 8 | 13.3 | Former smoker | 11 | 18.3 | Former smoker | 2 | 4.8 | Former smoker | 1 | 9.1 |
| Never smoker | 23 | 38.3 | Never smoker | 23 | 38.3 | Never smoker | 27 | 64.3 | Never smoker | 6 | 54.5 |
| n.a. | 1 | 1.7 | n.a. | 1 | 1.7 | n.a. | 4 | 9.5 | n.a. | 1 | 9.1 |
| BC histology | N. | % | LC histology | N.* | % * | BC histology | N. | % | LC histology | N. | % |
| Ductal Infiltrating | 50 | 83.3 | Adenocarcinoma | 51 | 83.6 | Ductal Infiltrating | 33 | 78.6 | Adenocarcinoma | 11 | 100.0 |
| Lobular | 5 | 8.3 | Squamous cell carcinoma | 5 | 8.2 | Lobular | 7 | 16.7 | Squamous cell carcinoma | 0 | - |
| Ductal/Lobular | 1 | 1.7 | Large cell | 2 | 3.3 | Ductal/Lobular | 2 | 4.8 | Large cell | 0 | - |
| In situ ductal | 2 | 3.3 | Small cell lung cancer | 3 | 4.9 | In situ ductal | 0 | - | Small cell lung cancer | 0 | - |
| n.a. | 2 | 3.3 | n.a. | 0 | - | n.a. | 0 | - | n.a. | 0 | - |
| Surgery type | N. | % | Surgery type | N. | % | Surgery type | N. | % | Surgery Type | N. | % |
| Quadrantectomy | 35 | 58.3 | Lobectomy | 48 | 80.0 | Quadrantectomy | 12 | 28.6 | Lobectomy | 10 | 90.9 |
| Lumpectomy | 8 | 13.3 | Segmentectomy | 3 | 5.0 | Lumpectomy | 19 | 45.2 | Segmentectomy | 1 | 9.1 |
| Mastectomy | 17 | 28.3 | Resection | 2 | 3.3 | Mastectomy | 11 | 26.2 | Resection | 0 | - |
| No surgery 1 | 0 | - | No surgery 1 | 6 | 10.0 | No surgery 1 | 0 | - | No surgery 1 | 0 | - |
| n.a. | 0 | - | n.a. | 1 | 1.7 | n.a. | 0 | - | n.a. | 0 | - |
| Disease stage | N. | % | Disease stage | N. | % | Disease stage | N. | % | Disease stage | N. | % |
| Non-metastatic | 53 | 88.3 | Non-metastatic | 50 | 83.3 | Non-metastatic | 42 | 100.0 | Non-metastatic | 11 | 100.0 |
| Metastatic | 0 | - | Metastatic | 7 | 11.7 | Metastatic | 0 | - | Metastatic | 0 | - |
| n.a. | 7 | 11.7 | n.a./n.a. 2 | 3 | 5.0 | n.a. | 0 | - | n.a./n.a.2 | 0 | - |
| Study Population | Control Population | ||||
|---|---|---|---|---|---|
| Breast Cancer n. 60 | Breast Cancer n. 42 | ||||
| Adjuvant radiotherapy | N. | % | Adjuvant radiotherapy | N. | % |
| Yes | 42 | 70.0 | Yes | 34 | 81.0 |
| No | 17 | 28.3 | No | 8 | 19.0 |
| n.a. | 1 | 1.7 | n.a. | 0 | - |
| Adjuvant chemotherapy | N. | % | Adjuvant chemotherapy | N. | % |
| Yes | 24 | 40.0 | Yes | 28 | 66.7 |
| No | 32 | 53.3 | No | 14 | 33.3 |
| n.a. | 4 | 6.7 | n.a. | 0 | - |
| Hormone therapy | N. | % | Hormone therapy | N. | % |
| Yes | 48 | 80.0 | Yes | 37 | 88.1 |
| No | 9 | 15.0 | No | 5 | 11.9 |
| n.a. | 3 | 5.0 | n.a. | 0 | - |
| Clinical Data of Lung Cancer | S1 | S2 | ||
|---|---|---|---|---|
| N. | % | N. | % | |
| Lung cancer | 16 | 57.1% | 12 | 42.9% |
| Age at diagnosis | 67.5 | 56–75 | 68 | 53–78 |
| LC occurrence after BC (years) | 3.7 | 0–11 | 7.7 | 2–21 |
| LC histotype | N. | % | N. | % |
| Adenocarcinoma | 15 | 93.8% | 8 | 66.7% |
| No adenocarcinoma | 1 | 6.3% | 4 | 33.3% |
| Smoking habit | N. | % | N. | % |
| Former/current smoker | 5 | 31.3% | 10 | 62.5% |
| Never smoker | 11 | 68.8% | 2 | 12.5% |
| Adjuvant radiotherapy for BC | N. | % | N. | % |
| Yes | 13 | 81.3% | 8 | 66.7% |
| No | 3 | 18.8% | 4 | 33.3% |
| Adjuvant chemotherapy for BC | N. | % | N. | % |
| Yes | 6 | 37.5% | 3 | 25.0% |
| No | 10 | 62.5% | 9 | 75.0% |
| Adjuvant hormone therapy for BC | N. | % | N. | % |
| Yes | 15 | 93.8% | 10 | 83.3% |
| No | 1 | 6.3% | 2 | 16.7% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coco, S.; Bonfiglio, S.; Cittaro, D.; Vanni, I.; Mora, M.; Genova, C.; Dal Bello, M.G.; Boccardo, S.; Alama, A.; Rijavec, E.; et al. Integrated Somatic and Germline Whole-Exome Sequencing Analysis in Women with Lung Cancer after a Previous Breast Cancer. Cancers 2019, 11, 441. https://doi.org/10.3390/cancers11040441
Coco S, Bonfiglio S, Cittaro D, Vanni I, Mora M, Genova C, Dal Bello MG, Boccardo S, Alama A, Rijavec E, et al. Integrated Somatic and Germline Whole-Exome Sequencing Analysis in Women with Lung Cancer after a Previous Breast Cancer. Cancers. 2019; 11(4):441. https://doi.org/10.3390/cancers11040441
Chicago/Turabian StyleCoco, Simona, Silvia Bonfiglio, Davide Cittaro, Irene Vanni, Marco Mora, Carlo Genova, Maria Giovanna Dal Bello, Simona Boccardo, Angela Alama, Erika Rijavec, and et al. 2019. "Integrated Somatic and Germline Whole-Exome Sequencing Analysis in Women with Lung Cancer after a Previous Breast Cancer" Cancers 11, no. 4: 441. https://doi.org/10.3390/cancers11040441