Chasing the FOXO3: Insights into Its New Mitochondrial Lair in Colorectal Cancer Landscape

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. The FOXO Family and FOXO3a
2.1. Phosphorylation of FOXO3a
2.2. Acetylation/Deacetylation of FOXO3a
2.3. Ubiquitination of FOXO3a
2.4. Methylation of FOXO3a
3. FOXO3a: A Cancer Therapy Opportunity
4. FOXO3a at the Interface Between Cell Death and Survival
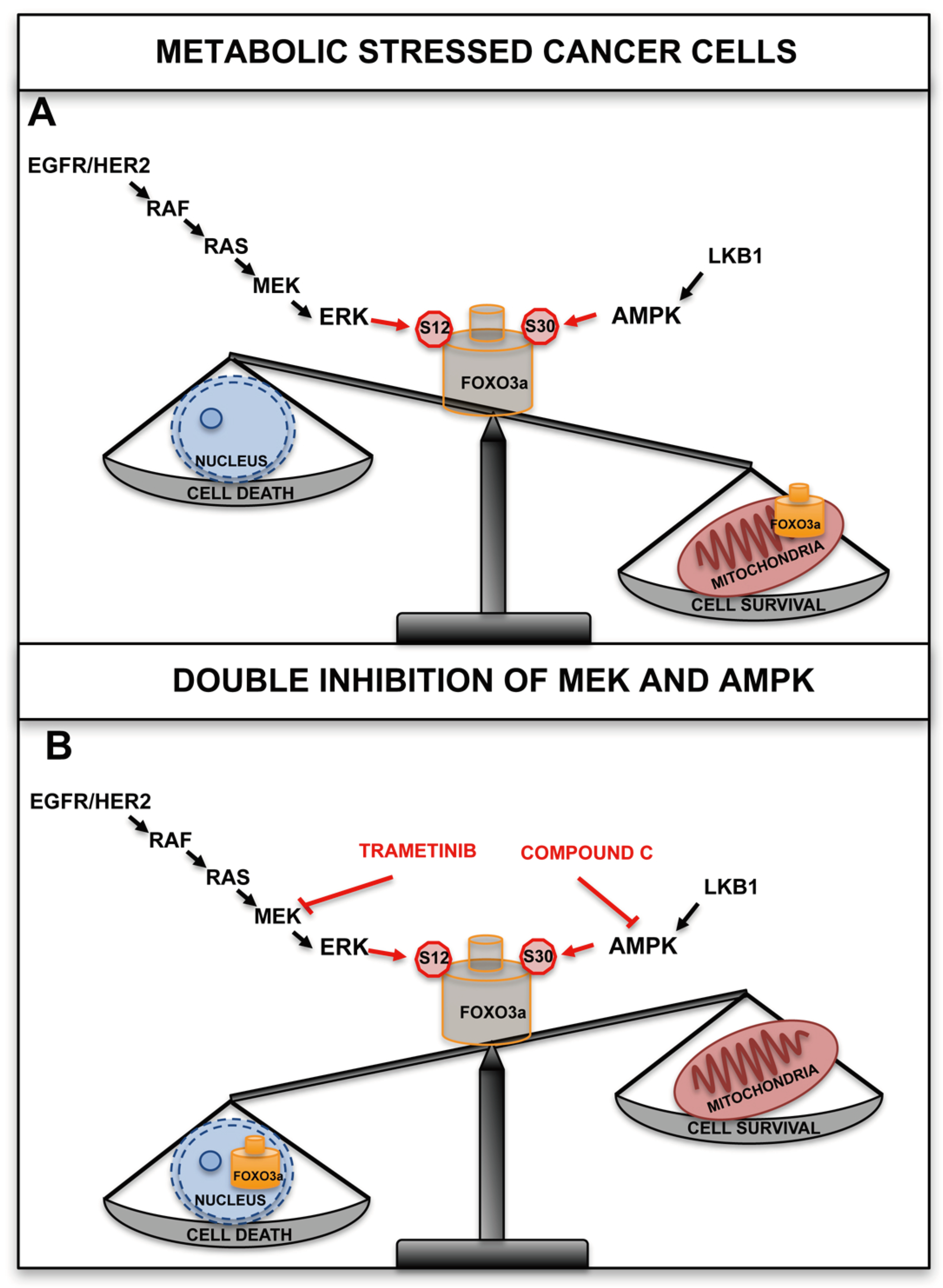
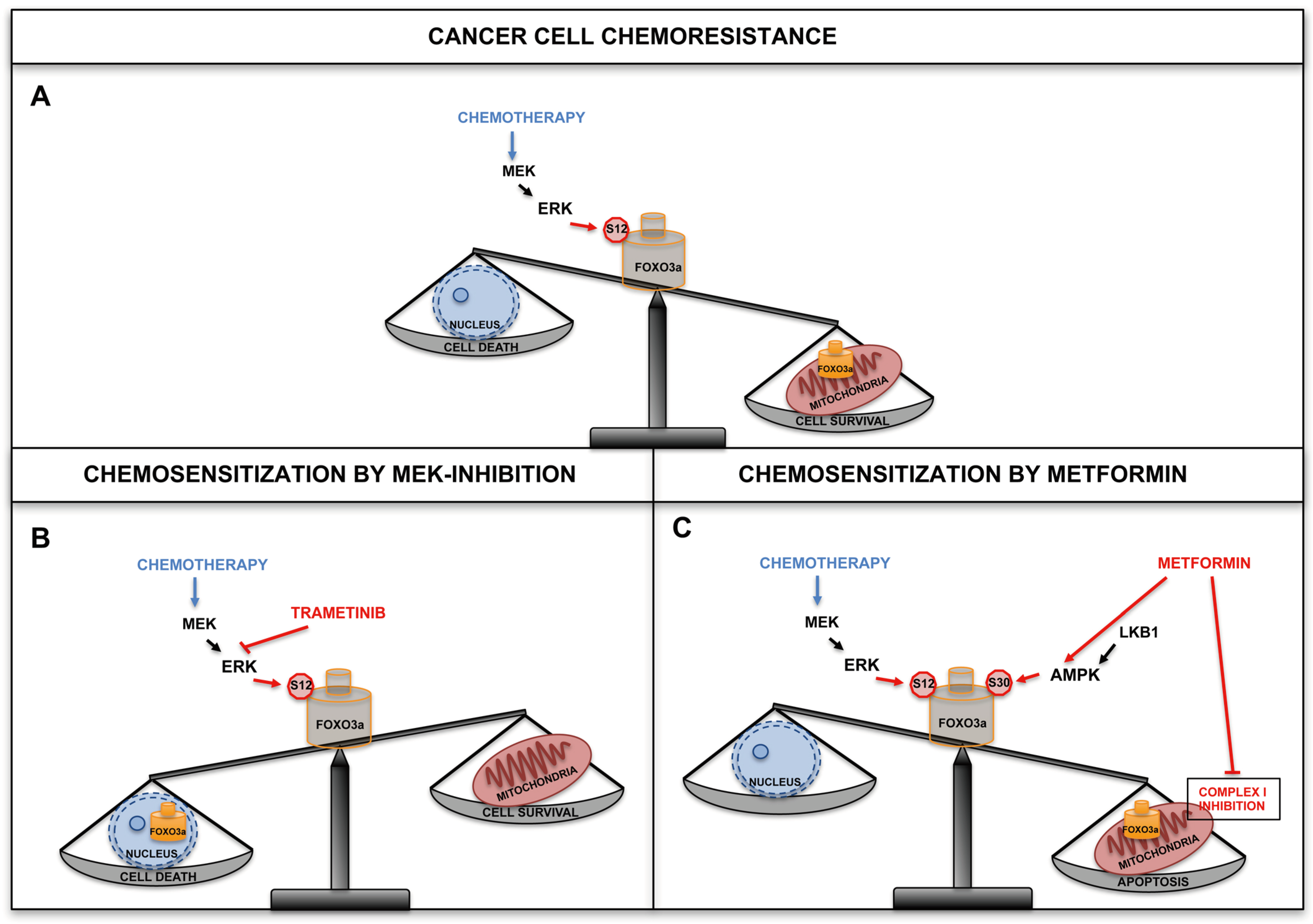
5. The Mitochondrial Arm of the AMPK-FOXO3a Axis
6. Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gunter, M.J.; Alhomound, S.; Arnold, M.; Brenner, H.; Burn, J.; Casey, G.; Chan, A.T.; Cross, A.J.; Giovannucci, E.; Hoover, R.; et al. Meeting Report from the joint IARC-NCI international cancer seminar series: A focus on colorectal cancer. Ann. Oncol. 2019. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2014, 499, 214–218. [Google Scholar] [CrossRef]
- Tamborero, D.; Lopez-Bigas, N.; Gonzalez-Perez, A. Oncodrive-CIS: A Method to Reveal Likely Driver Genes Based on the Impact of Their Copy Number Changes on Expression. PLoS ONE 2013, 8, e55489. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Rubio-Perez, C.; Tamborero, D.; Schroeder, M.P.; Antolín, A.A.; Deu-Pons, J.; Peremas, C.; Mestres, J.; Gonzalez-Perez, A.; Lopez-Biga, N. In Silico Prescription of Anticancer Drugs to Cohorts of 28 Tumor Types Reveals Targeting Opportunities. Cancer Cell 2015, 27, 382–396. [Google Scholar] [CrossRef] [Green Version]
- Letai, A. Functional precision cancer medicine-moving beyond pure genomics. Nat. Med. 2017, S23, 1028–1035. [Google Scholar] [CrossRef]
- Fojo, T. Precision oncology: A strategy we were not ready to deploy. Semin. Oncol. 2016, 43, 9–12. [Google Scholar] [CrossRef]
- Friedman, A.A.; Letai, A.; Fisher, D.E.; Flaherty, K.T. Precision medicine for cancer with next-generation functional diagnostics. Nat. Rev. Cancer 2015, 15, 747–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, V. Perspective: The precision-oncology illusion. Nature 2016, 537, S63. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Fojo, T.; Brada, M. Precision oncology: Origins, optimism, and potential. Lancet Oncol. 2016, 17, e81–e86. [Google Scholar] [CrossRef]
- Tannock, I.F.; Hickman, J.A. Limits to personalized cancer medicine. N. Engl. J. Med. 2016, 375, 1289–1294. [Google Scholar] [CrossRef]
- West, H.J. No solid evidence, only hollow argument for universal tumor sequencing: Show me the data. JAMA Oncol. 2016, 2, 717–718. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, P.D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Benefit From Genome-Driven Oncology. JAMA Oncol. 2018, 4, 1093–1098. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Grossi, V.; Cappellari, M.; Peserico, A.; Simonatto, M.; Germani, A.; Russo, S.; Moyer, M.P.; Resta, N.; Murzilli, S.; et al. Blocking p38/ERK crosstalk affects colorectal cancer growth by inducing apoptosis in vitro and in preclinical mouse models. Cancer Lett. 2012, 324, 98–108. [Google Scholar] [CrossRef]
- Celestini, V.; Tezil, T.; Russo, L.; Fasano, C.; Sanese, P.; Forte, G.; Peserico, A.; Lepore Signorile, M.; Longo, G.; De Rasmo, D.; et al. Uncoupling FoxO3A mitochondrial and nuclear functions in cancer cells undergoing metabolic stress and chemotherapy. Cell Death Dis. 2018, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, P.; Mahlapuu, M. Forkhead Transcription Factors: Key Players in Development and Metabolism. Dev. Biol. 2002, 250, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Arden, K.C. FoxO: Linking new signaling pathways. Mol. Cell 2004, 14, 416–418. [Google Scholar] [CrossRef]
- Arden, K.C. Multiple roles of FOXO transcription factors in mammalian cells point to multiple roles in cancer. Exp. Gerontol. 2006, 41, 709–717. [Google Scholar] [CrossRef]
- Van der Horst, A.; Burgering, B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell. Biol. 2007, 8, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Santo, E.E.; Paik, J. A splice junction-targeted CRISPR approach (spJCRISPR) reveals human FOXO3B to be a protein-coding gene. Gene 2018, 673, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, Z.; Zhang, M.Q. From worm to human: Bioinformatics approaches to identify FOXO target genes. Mech. Ageing Dev. 2005, 126, 209–215. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Nakamura, N.; Sansal, I.; Bergeron, L.; Sellers, W.R. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell 2002, 2, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Fernandez de Mattos, S.; Essafi, A.; Soeiro, I.; Pietersen, A.M.; Birkenkamp, K.U.; Edwards, C.S.; Martino, A.; Nelson, B.H.; Francis, J.M.; Jones, M.C.; et al. FoxO3a and BCR-ABL regulate cyclin D2 transcription through a STAT5/BCL6-dependent mechanism. Mol. Cell. Biol. 2004, 24, 10058–10071. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Le, H.V.; Shen, L.; Anderson, S.A.; Massague, J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef]
- Jensen, K.S.; Binderup, T.; Jensen, K.T.; Therkelsen, I.; Borup, R.; Nilsson, E.; Multhaupt, H.; Bouchard, C.; Quistorff, B.; Kjær, A.; et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011, 30, 4554–4570. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, C.; Marquardt, J.; Bras, A.; Medema, R.H.; Eilers, M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 2004, 23, 2830–2840. [Google Scholar] [CrossRef] [Green Version]
- Furukawa-Hibi, Y.; Yoshida-Araki, K.; Ohta, T.; Ikeda, K.; Motoyama, N. FOXO Forkhead Transcription Factors Induce G2-M Checkpoint in Response to Oxidative Stress. JBC 2002, 277, 26729–26732. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J.; Di Stefano, P.S., Jr.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Modur, V.; Nagarajan, R.; Evers, B.M.; Milbrandt, J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J. Biol. Chem. 2002, 277, 47928–47937. [Google Scholar] [CrossRef] [PubMed]
- Rokudai, S.; Fujita, N.; Kitahara, O.; Nakamura, Y.; Tsuruo, T. Involvement of FKHR-dependent TRADD expression in chemotherapeutic drug-induced apoptosis. Mol. Cell. Biol. 2002, 22, 8695–8708. [Google Scholar] [CrossRef]
- Dijkers, P.F.; Medema, R.H.; Lammers, J.W.; Koenderman, L.; Coffer, P.J. Expression of the proapoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 2000, 10, 1201–1204. [Google Scholar] [CrossRef]
- Essafi, A.; Fernandez de Mattos, S.; Hassen, Y.A.; Soeiro, I.; Mufti, G.J.; Thomas NS Medema, R.H.; Lam, E.W. Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl expressing cells. Oncogene 2005, 24, 2317–2329. [Google Scholar] [CrossRef]
- Dansen, T.B.; Burgering, B.M. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol 2008, 18, 421–429. [Google Scholar] [CrossRef]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef] [Green Version]
- Flachsbart, F.; Caliebe, A.; Kleindorp, R.; Blanché, H.; von Eller-Eberstein, H.; Nikolaus, S.; Schreiber, S.; Nebel, A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl. Acad. Sci. USA 2009, 106, 2700–2705. [Google Scholar] [CrossRef] [Green Version]
- Anselmi, C.V.; Malovini, A.; Roncarati, R.; Novelli, V.; Villa, F.; Condorelli, G.; Bellazzi, R.; Puca, A.A. Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 2009, 12, 95–104. [Google Scholar] [CrossRef]
- Donlon, T.A.; Morris, B.J.; Chen, R.; Masaki, K.H.; Allsopp, R.C.; Willcox, D.C.; Elliott, A.; Willcox, B.J. FOXO3 longevity interactome on chromosome 6. Aging Cell 2017, 16, 1016–1025. [Google Scholar] [CrossRef] [Green Version]
- Flachsbart, F.; Dose, J.; Gentschew, L.; Geismann, C.; Caliebe, A.; Knecht, C.; Nygaard, M.; Badarinarayan, N.; ElSharawy, A.; May, S.; et al. Identification and characterization of two functional variants in the human longevity gene FOXO3. Nat. Commun. 2017, 8, 2063. [Google Scholar] [CrossRef] [Green Version]
- Forte, G.; Grossi, V.; Celestini, V.; Lucisano, G.; Scardapane, M.; Varvara, D.; Patruno, M.; Bagnulo, R.; Loconte, D.; Giunti, L.; et al. Characterization of the rs2802292 SNP identifies FOXO3A as a modifier locus predicting cancer risk in patients with PJS and PHTS hamartomatous polyposis syndromes. BMC Cancer 2014, 14, 661. [Google Scholar] [CrossRef] [PubMed]
- Grossi, V.; Forte, G.; Sanese, P.; Peserico, A.; Tezil, T.; Lepore Signorile, M.; Fasano, C.; Lovaglio, R.; Bagnulo, R.; Loconte, D.C.; et al. The longevity SNP rs2802292 uncovered: HSF1 activates stress-dependent expression of FOXO3 through an intronic enhancer. Nucleic Acids Res. 2018, 46, 5587–5600. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.J.; Tranah, G.J.; Chen, R.; Morris, B.J.; Masaki, K.H.; He, Q.; Willcox, D.C.; Allsopp, R.C.; Moisyadi, S.; Poon, L.W.; et al. The FoxO3 gene and cause-specific mortality. Aging Cell 2016, 15, 617–624. [Google Scholar] [CrossRef]
- Willcox, B.J.; Morris, B.J.; Tranah, G.J.; Chen, R.; Masaki, K.H.; He, Q.; Willcox, D.C.; Allsopp, R.C.; Moisyadi, S.; Gerschenson, M.; et al. Longevity-associated FOXO3 genotype and its impact on coronary artery disease mortality in japanese, whites, and blacks: A prospective study of three american populations. J. Gerontol. Biol. Sci. Med. Sci. 2017, 72, 724–728. [Google Scholar] [CrossRef]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef]
- Furukawa-Hibi, Y.; Kobayashi, Y.; Chen, C.; Motoyama, N. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid. Redox Signal. 2005, 7, 752–760. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell. Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Zhao, J.L.; Sinha, N.; Marinov, G.K.; Mann, M.; Kowalczyk, M.S.; Galimidi, R.P.; Du, X.; Erikci, E.; Regev, A.; et al. The microRNA-132 and microRNA-212 cluster regulates hematopoietic stem cell maintenance and survival with age by buffering FOXO3 expression. Immunity 2015, 42, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J.; Willcox, D.C.; Donlon, T.A.; Willcox, B.J. FOXO3: A major gene for human longevity—A mini-review. Gerontology 2015, 61, 515–525. [Google Scholar] [CrossRef]
- Yang, W.B.; Chen, P.H.; Hsu, T.S.; Fu, T.F.; Su, W.C.; Liaw, H.; Chang, W.C.; Hung, J.J. Sp1-mediated microRNA-182 expression regulates lung cancer progression. Oncotargets 2014, 5, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Segura, M.F.; Hanniford, D.; Menendez, S.; Reavie, L.; Zou, X.; Alvarez-Diaz, S.; Zakrzewski, J.; Blochin, E.; Rose, A.; Bogunovic, D.; et al. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc. Natl. Acad. Sci. USA 2009, 106, 1814–1819. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Zhu, C.F.; Ma, M.Z.; Chen, G.; Song, M.; Zeng, Z.L.; Lu, W.H.; Yang, J.; Wen, S.; Chiao, P.J.; et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotargets 2015, 6, 21148–21158. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Du, Y.; Yang, C.; Zhang, D.; Zhang, N.; Liu, X.; Cho, W.C.; Yang, Y. An oncogenic role of miR-592 in tumorigenesis of human colorectal cancer by targeting Forkhead Box O3A (FoxO3A). Expert Opin. Ther. Targets 2016, 20, 771–782. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, Y.; Wang, Y.; Zhang, Y.; Luo, Q.; Man, X.; Wei, F.; Yu, X. Downregulation of Foxo3 and TRIM31 by miR-551b in side population promotes cell proliferation, invasion, and drug resistance of ovarian cancer. Med. Oncol. 2016, 33, 126. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Brownawell, A.M.; Kops, G.J.; Macara, I.G.; Burgering, B.M. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol. Cell. Biol. 2001, 21, 3534–3546. [Google Scholar] [CrossRef]
- Rena, G.; Guo, S.; Cichy, S.C.; Unterman, T.G.; Cohen, P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 1999, 274, 17179–17183. [Google Scholar] [CrossRef]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 2001, 21, 952–965. [Google Scholar] [CrossRef]
- Hu, M.C.; Lee, D.F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IκB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef]
- Tezil, T.; Bodur, C.; Kutuk, O.; Basaga, H. IKK-β mediates chemoresistance by sequestering FOXO3; A critical factor for cell survival and death. Cell Signal 2012, 24, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villen, J.; Becker, E.B.; DiBacco, S.; de la Iglesia, N.; Gygi, S.; Blackwell, T.K.; et al. A conserved MST-FOXO signalling pathway mediates oxidative-stress responses and extends life span. Cell 2006, 125, 987–1001. [Google Scholar] [CrossRef]
- Sunters, A.; Madureira, P.A.; Pomeranz, K.M.; Aubert, M.; Brosens, J.J.; Cook, S.J.; Burgering, B.M.; Coombes, R.C.; Lam, E.W. Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res. 2006, 66, 212–220. [Google Scholar] [CrossRef]
- Ho, K.K.; McGuire, V.A.; Koo, C.Y.; Muir, K.W.; de Olano, N.; Maifoshie, E.; Kelly, D.J.; McGovern, U.B.; Monteiro, L.J.; Gomes, A.R.; et al. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J. Biol. Chem. 2012, 287, 1545–1555. [Google Scholar] [CrossRef]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef]
- Colombo, S.L.; Moncada, S. AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. Biochem. J. 2009, 421, 163–169. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peserico, A.; Chiacchiera, F.; Grossi, V.; Matrone, A.; Latorre, D.; Simonatto, M.; Fusella, A.; Ryall, J.G.; Finley, L.W.; Haigis, M.C.; et al. A novel AMPK-dependent FoxO3A-SIRT3 intramitochondrial complex sensing glucose levels. Cell. Mol. Life Sci. 2013, 70, 2015–2029. [Google Scholar] [CrossRef] [PubMed]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004, 116, 551–563. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Furukawa-Hibi, Y.; Chen, C.; Horio, Y.; Isobe, K.; Ikeda, K.; Motoyama, N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int. J. Mol. Med. 2005, 16, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Daitoku, H.; Sakamaki, J.; Fukamizu, A. Regulation of FoxO transcription factors by acetylation and protein–protein interactions. Biochim. Biophys. Acta 2011, 1813, 1954–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria- localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef]
- Jacobs, K.M.; Pennington, J.D.; Bisht, K.S.; Aykin-Burns, N.; Kim, H.S.; Mishra, M.; Sun, L.; Nguyen, P.; Ahn, B.H.; Leclerc, J.; et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int. J. Biol. Sci. 2008, 4, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.H.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku-70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [PubMed]
- Scher, M.B.; Vaquero, A.; Reinberg, D. SIRT3 is a nuclear NAD+- dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [Green Version]
- Calnan, D.R.; Webb, A.E.; White, J.L.; Stowe, T.R.; Goswami, T.; Shi, X.; Espejo, A.; Bedford, M.T.; Gozani, O.; Gygi, S.P.; et al. Methylation by Set9 modulates FoxO3 stability and transcriptional activity. Aging 2012, 4, 462–479. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistic, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Fei, M.; Zhao, Y.; Wang, Y.; Lu, M.; Cheng, C.; Huang, X.; Zhang, D.; Lu, J.; He, S.; Shen, A. Low expression of Foxo3a is associated with poor prognosis in ovarian cancer patients. Cancer Investig. 2009, 27, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zou, L.; Lu, W.Q.; Zhang, Y.; Shen, A.G. Foxo3a expression is a prognostic marker in breast cancer. PLoS ONE 2013, 8, e70746. [Google Scholar] [CrossRef]
- Habashy, H.O.; Rakha, E.A.; Aleskandarany, M.; Ahmed, M.A.; Green, A.R.; Ellis, I.O.; Powe, D.G. FOXO3A nuclear localisation is associated with good prognosis in luminal- like breast cancer. Breast Cancer Res. Treat. 2011, 129, 11–21. [Google Scholar] [CrossRef]
- Yang, X.B.; Zhao, J.J.; Huang, C.Y.; Wang, Q.J.; Pan, K.; Wang, D.D.; Pan, Q.Z.; Jiang, S.-S.; Lv, L.; Gao, X.; et al. Decreased expression of the FOXO3A gene is associated with poor prognosis in primary gastric adenocarcinoma patients. PLoS ONE 2013, 8, e78158. [Google Scholar] [CrossRef]
- Yu, S.; Yu, Y.; Zhang, W.; Yuan, W.; Zhao, N.; Li, Q.; Cui, Y. FOXO3a promotes gastric cancer cell migration and invasion through the induction of cathepsin L. Oncotarget 2016, 7, 34773–34784. [Google Scholar] [CrossRef]
- Liu, H.B.; Gao, X.X.; Zhang, Q.; Liu, J.; Cui, Y.; Zhu, Y.; Liu, Y.F. (2015). Expression and prognostic implications of FOXO3A and Ki67 in lung adenocarcinomas. Asian Pac. J. Cancer Prev. 2015, 16, 1443–1448. [Google Scholar] [CrossRef]
- Potente, M.; Urbich, C.; Sasaki, K.; Hofmann, W.K.; Heeschen, C.; Aicher, A.; Kollipara, R.; DePinho, R.A.; Zeiher, A.M.; Dimmeler, S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Investig. 2005, 115, 2382–2392. [Google Scholar] [CrossRef] [Green Version]
- Hui, R.C.; Francis, R.E.; Guest, S.K.; Costa, J.R.; Gomes, A.R.; Myatt, S.S.; Brosens, J.J.; Lam, E.W. Doxorubicin activates FOXO3a to induce the expression of multidrug resistance gene ABCB1 (MDR1) in K562 leukemic cells. Mol. Cancer Ther. 2008, 7, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Karadedou, C.T.; Gomes, C.T.; Chen, J.; Petkovic, M.; Ho, K.K.; Zwolinska, A.K.; Feltes, A.; Wong, S.Y.; Chan, K.Y.; Cheung, Y.N.; et al. FOXO3a represses VEGF expression through FOXM1-dependent and -independent mechanisms in breast cancer. Oncogene 2012, 31, 1845–1858. [Google Scholar] [CrossRef] [PubMed]
- McGovern, U.B.; Francis, R.E.; Peck, B.; Guest, S.K.; Wang, J.; Myatt, S.S.; Krol, J.; Kwok, J.M.; Polychronis, A.; Coombes, R.C.; et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol. Cancer Ther. 2009, 8, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Bernard, R.; Getachew, R.; Kamato, D.; Thach, L.; Osman, N.; Chan, V.; Zheng, W.; Little, P.J. Evaluation of the potential synergism of imatinib-related poly kinase inhibitors using growth factor stimulated proteoglycan synthesis as a model response. J. Pharm. Pharmacol. 2016, 68, 368–378. [Google Scholar] [CrossRef]
- Khongkow p Gomes, A.R.; Gong, C.; Man, E.P.; Tsang, J.W.; Zhao, F.; Monteiro, L.J.; Coombes, R.C.; Medema, R.H.; Khoo, U.S.; Lam, E.W. Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene 2016, 35, 990–1002. [Google Scholar] [CrossRef]
- Fernández de Mattos, S.; Villalonga, P.; Clardy, J.; Lam, E.W.F. FOXO3a mediates the cytotoxic effects of Cisplatin in colon cancer cells. Mol. Cancer Ther. 2008, 7, 3237–3246. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Yang, X.; Jin, Y.; Pei, S.; Zhang, D.; Zhang, H.; Zhou, B.; Zhang, Y.; Lin, D. PUMA mediates the combinational therapy of 5- FU and NVP-BEZ235 in colon cancer. Oncotarget 2015, 6, 14385–14398. [Google Scholar] [CrossRef]
- Germani, A.; Matrone, A.; Grossi, V.; Peserico, A.; Sanese, P.; Liuzzi, M.; Palermo, R.; Murzilli, S.; Campese, A.F.; Ingravallo, G.; et al. Targeted therapy against chemoresistant colorectal cancers: Inhibition of p38α modulates the effect of Cisplatin in vitro and in vivo through the tumor suppressor FoxO3A. Cancer Lett. 2014, 344, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.C.; Kashida, Y.; Kulp, S.K.; Wang, D.; Brueggemeier, R.W.; Shapiro, C.L.; Chen, C.S. Sensitizing estrogen receptor-negative breast cancer cells totamoxifen with OSU-03012, a novel celecoxib-derived phosphoinositide-dependent protein kinase-1/Akt signaling inhibitor. Mol. Cancer Ther. 2008, 7, 800–808. [Google Scholar] [CrossRef]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Real, P.J.; Benito, A.; Cuevas, J.; Berciano, M.T.; de Juan, A.; Coffer, P.; Gomez-Roman, J.; Lafarga, M.; Lopez-Vega, J.M.; Fernandez-Luna, J.L. Blockade of epidermal growth factor receptors chemosensitizes breast cancer cells through up-regulation of Bnip3L. Cancer Res. 2005, 65, 8151–8157. [Google Scholar] [CrossRef]
- Yang, J.Y.; Xia, W.; Hu, M.C. Ionizing radiation activates expression of FOXO3a, Fas ligand, and Bim, and induces cell apoptosis. Int. J. Oncol. 2006, 29, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.R.; Brosens, J.J.; Lam, E.W.F. Resist or die: FOXO transcription factors determine the cellular response to chemotherapy. Cell Cycle 2008, 7, 3133–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gan, B.; Liu, D.; Paik, J.H. FoxO family members in cancer. Cancer Biol. Ther. 2011, 12, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornsveld, M.; Smits, L.M.M.; Meerlo, M.; van Amersfoort, M.; Groot Koerkamp, M.J.A.; van Leenen, D.; Kloet, D.E.A.; Holstege, F.C.P.; Derksen, P.W.B.; Burgering, B.M.T.; et al. FOXO Transcription Factors Both Suppress and Support Breast Cancer Progression. Cancer Res. 2018, 78, 2356–2369. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Döppler, H.; Copland, J.A.; Simpson, K.J.; Toker, A. FOXO3A promotes tumor cell invasion through the induction of matrix metalloproteinases. Mol. Cell. Biol. 2009, 29, 4906–4917. [Google Scholar] [CrossRef] [PubMed]
- Sisci, D.; Maris, P.; Cesario, M.G.; Anselmo, W.; Coroniti, R.; Trombino, G.E.; Romeo, F.; Ferraro, A.; Lanzino, M.; Aquila, S.; et al. The estrogen receptor α is the key regulator of the bifunctional role of FoxO3a transcription factor in breast cancer motility and invasiveness. Cell Cycle 2013, 12, 3405–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Chen, W.; Zhi, X.; Ma, T.; Xia, X.; Liu, H.; Zhang, Q. Serotonin promotes the proliferation of serum-deprived hepatocellular carcinoma cells via upregulation of FOXO3A. Mol. Cancer 2013, 12, 14. [Google Scholar] [CrossRef]
- Ahn, H.; Kim, H.; Abdul, R.; Kim, Y.; Sim, J.; Choi, D.; Paik, S.S.; Shin, S.J.; Kim, D.H.; Jang, K. Overexpression of Forkhead Box O3a and Its Association with Aggressive Phenotypes and Poor Prognosis in Human Hepatocellular Carcinoma. Am. J. Clin. Pathol. 2018, 149, 117–127. [Google Scholar] [CrossRef]
- Qian, Z.; Ren, L.; Wu, D.; Yang, X.; Zhou, Z.; Nie, Q.; Jiang, G.; Xue, S.; Weng, W.; Qiu, Y.; et al. Overexpression of FoxO3a is associated with glioblastoma progression and predicts poor patient prognosis. Int. J. Cancer 2017, 140, 2792–2804. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Simone, C. Inhibition of p38alpha unveils an AMPK- FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy 2009, 5, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Comes, F.; Matrone, A.; Lastella, P.; Nico, B.; Susca, F.C.; Bagnulo, R.; Ingravallo, G.; Modica, S.; Lo Sasso, G.; Moschetta, A.; et al. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 2007, 14, 693–702. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Matrone, A.; Ferrari, E.; Ingravallo, G.; Lo Sasso, G.; Murzilli, S.; Petruzzelli, M.; Salvatore, L.; Moschetta, A.; Simone, C. p38α blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1α- to FoxO-dependent transcription. Cell Death Differ. 2009, 16, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- Grossi, V.; Liuzzi, M.; Murzilli, S.; Martelli, N.; Napoli, A.; Ingravallo, G.; Del Rio, A.; Simone, C. Sorafenib inhibits p38alpha activity in colorectal cancer cells and synergizes with the DFG-in inhibitor SB202190 to increase apoptotic response. Cancer Biol. Ther. 2012, 13, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.P.; East, P.; Schulze, A. FoxO3A regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [Google Scholar] [CrossRef]
- Zhou, L.; Li, R.; Liu, C.; Sun, T.; Htet Aung, L.H.; Chen, C.; Gao, J.; Zhao, Y.; Wang, K. Foxo3a inhibits mitochondrial fission and protects against doxorubicin-induced cardiotoxicity by suppressing MIEF2. Free Radic. Biol. Med. 2017, 104, 360–370. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Xu, Z.X.; Ding, Z.; Lu, Y.; Yu, Q.; Werle, K.D.; Zhou, G.; Park, Y.Y.; Peng, G.; Gambello, M.J.; Mills, G.B. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat. Commun 2015, 6, 7926. [Google Scholar] [CrossRef] [PubMed]
- Caballero-Caballero, A.; Engel, T.; Martinez-Villarreal, J.; Sanz-Rodriguez, A.; Chang, P.; Dunleavy, M.; Mooney, C.M.; Jimenez-Mateos, E.M.; Schindler, C.K.; Henshall, D.C. Mitochondrial localization of the forkhead box class O transcription factor FOXO3a in brain. J. Neurochem. 2013, 124, 749–756. [Google Scholar] [CrossRef]
- Schwer, B.; North, B.J.; Frye, R.A.; Ott, M.; Verdin, E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide–dependent deacetylase. J. Cell Biol. 2002, 158, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Olsen, J.V.; Daub, H.; Mann, M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol. Cell. Proteom. 2009, 8, 2796–2808. [Google Scholar] [CrossRef] [PubMed]
- Klammer, M.; Kaminski, M.; Zedler, A.; Oppermann, F.; Blencke, S.; Marx, S.; Müller, S.; Tebbe, A.; Godl, K.; Schaab, C. Phosphosignature Predicts Dasatinib Response in Non-small Cell Lung Cancer. Mol. Cell. Proteom. 2012, 11, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Adjei, A. The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol. 2014, 11, 385–400. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing Metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef]
- Schulten, H.J. Pleiotropic effect of metformin on cancer. Int. J. Mol. Sci. 2018, 19, 2850. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, J.Y.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Laissue, P. The forkhead-box family of transcription factors: Key molecular players in colorectal cancer pathogenesis. Mol. Cancer 2019, 18, 5. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grossi, V.; Fasano, C.; Celestini, V.; Lepore Signorile, M.; Sanese, P.; Simone, C. Chasing the FOXO3: Insights into Its New Mitochondrial Lair in Colorectal Cancer Landscape. Cancers 2019, 11, 414. https://doi.org/10.3390/cancers11030414
Grossi V, Fasano C, Celestini V, Lepore Signorile M, Sanese P, Simone C. Chasing the FOXO3: Insights into Its New Mitochondrial Lair in Colorectal Cancer Landscape. Cancers. 2019; 11(3):414. https://doi.org/10.3390/cancers11030414
Chicago/Turabian StyleGrossi, Valentina, Candida Fasano, Valentina Celestini, Martina Lepore Signorile, Paola Sanese, and Cristiano Simone. 2019. "Chasing the FOXO3: Insights into Its New Mitochondrial Lair in Colorectal Cancer Landscape" Cancers 11, no. 3: 414. https://doi.org/10.3390/cancers11030414