Intracellular Iron Chelation by a Novel Compound, C7, Reactivates Epstein–Barr Virus (EBV) Lytic Cycle via the ERK-Autophagy Axis in EBV-Positive Epithelial Cancers

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
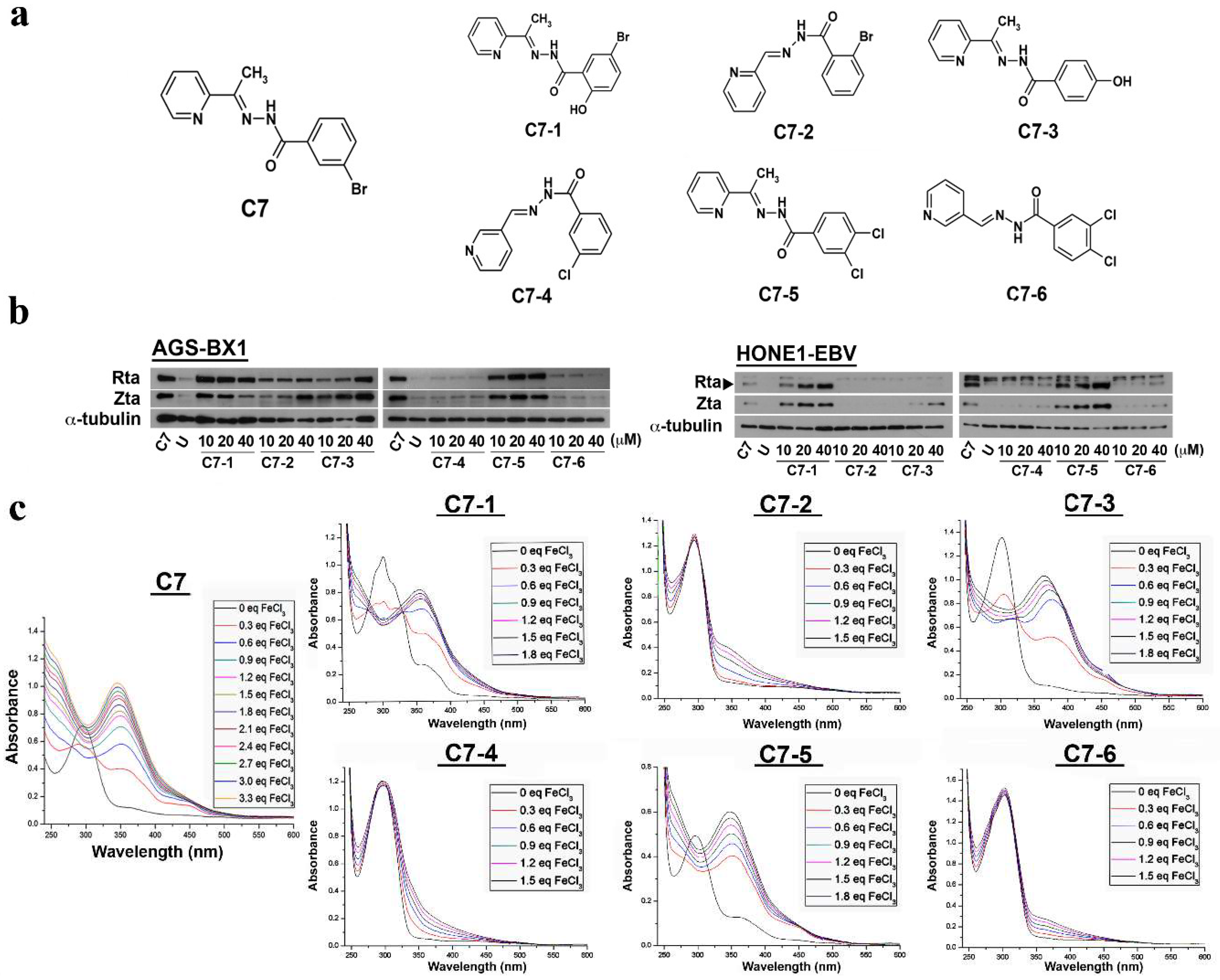
2.1. Effects of C7 and Its Analog Compounds on EBV Lytic Cycle Reactivation
2.2. Binding of C7 and Its Lytic-Inducing Analogs to Iron
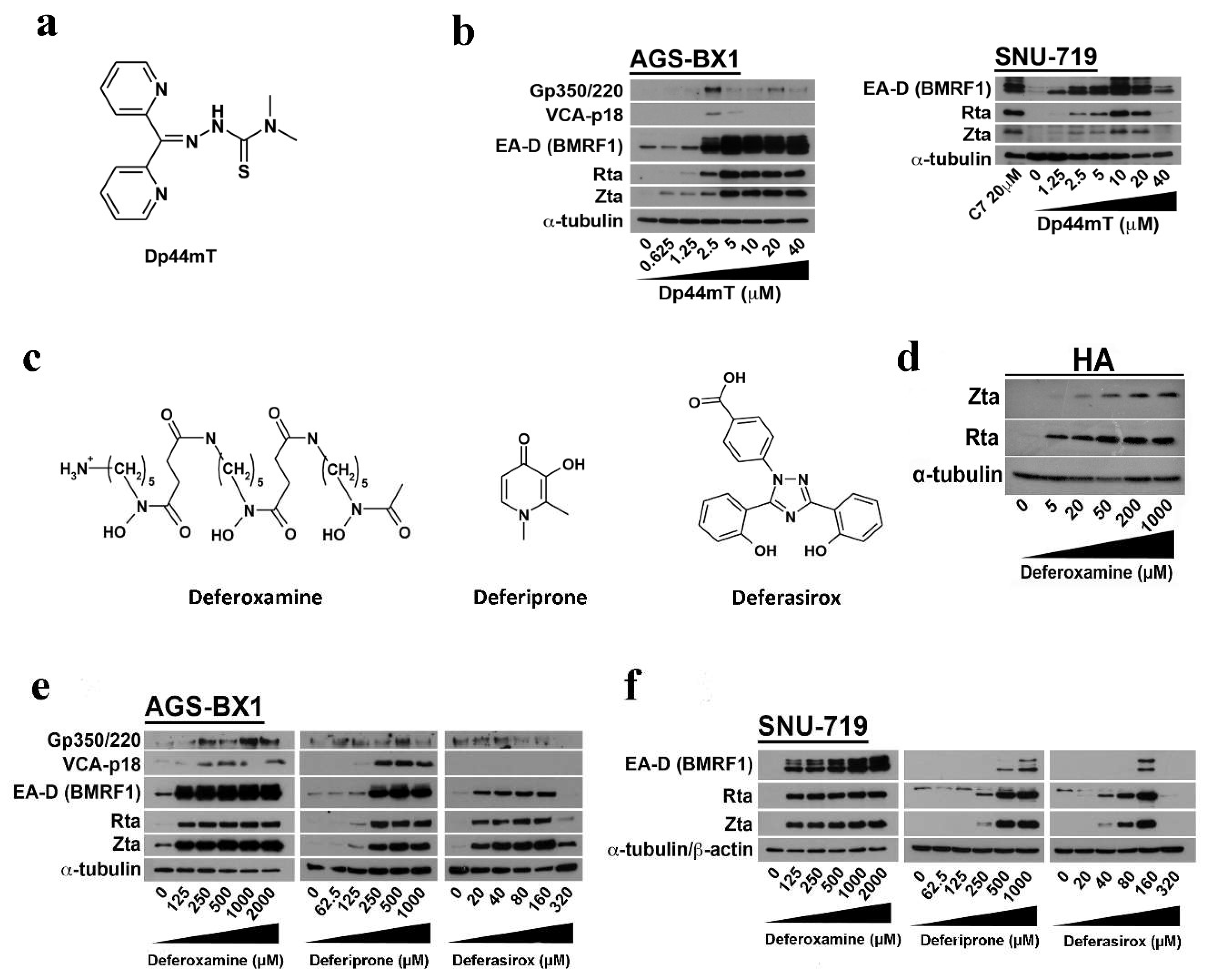
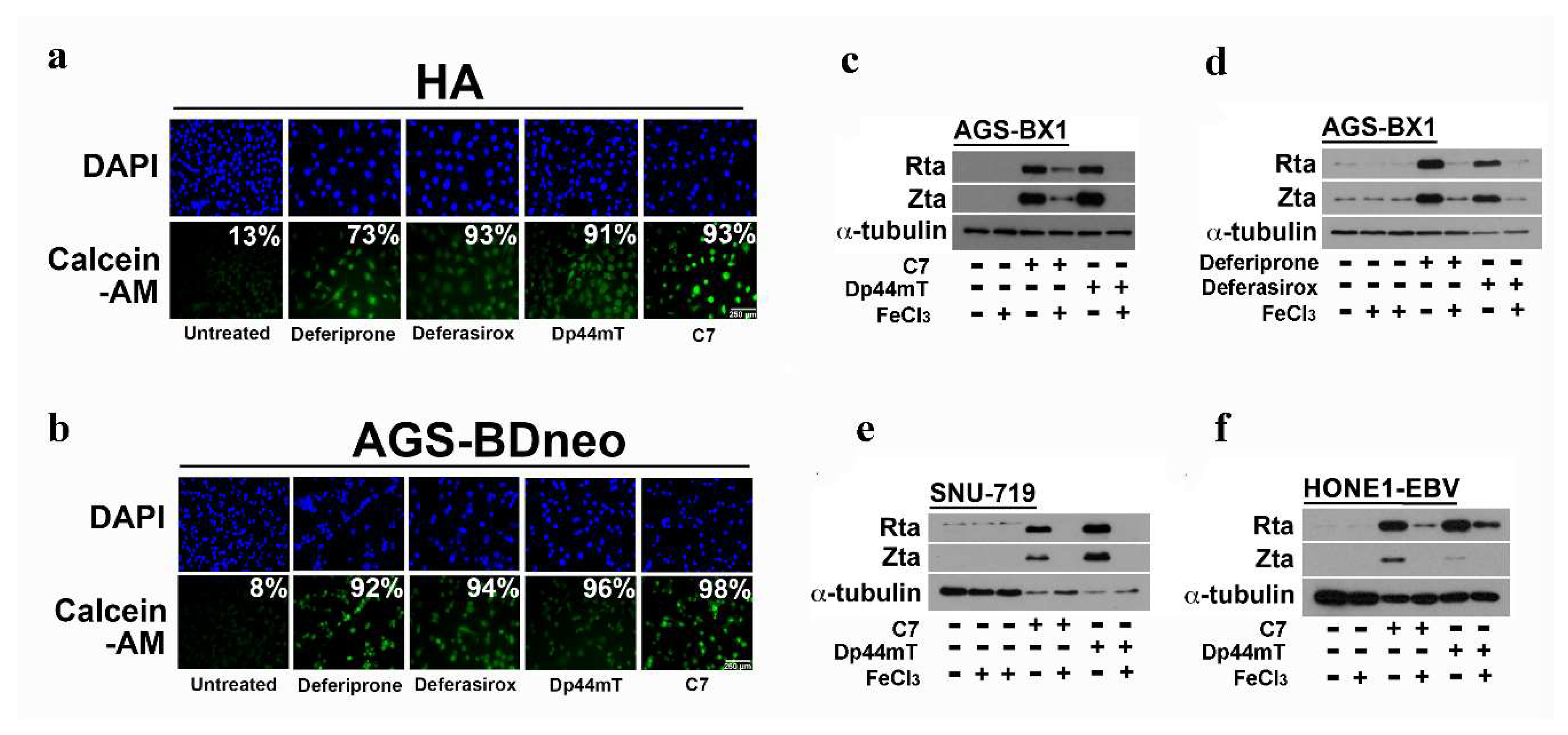
2.3. EBV Lytic Reactivation by Other Clinically Available Iron Chelators
2.4. Iron Chelation by C7 and Other Iron Chelators Is Required for EBV Lytic Reactivation
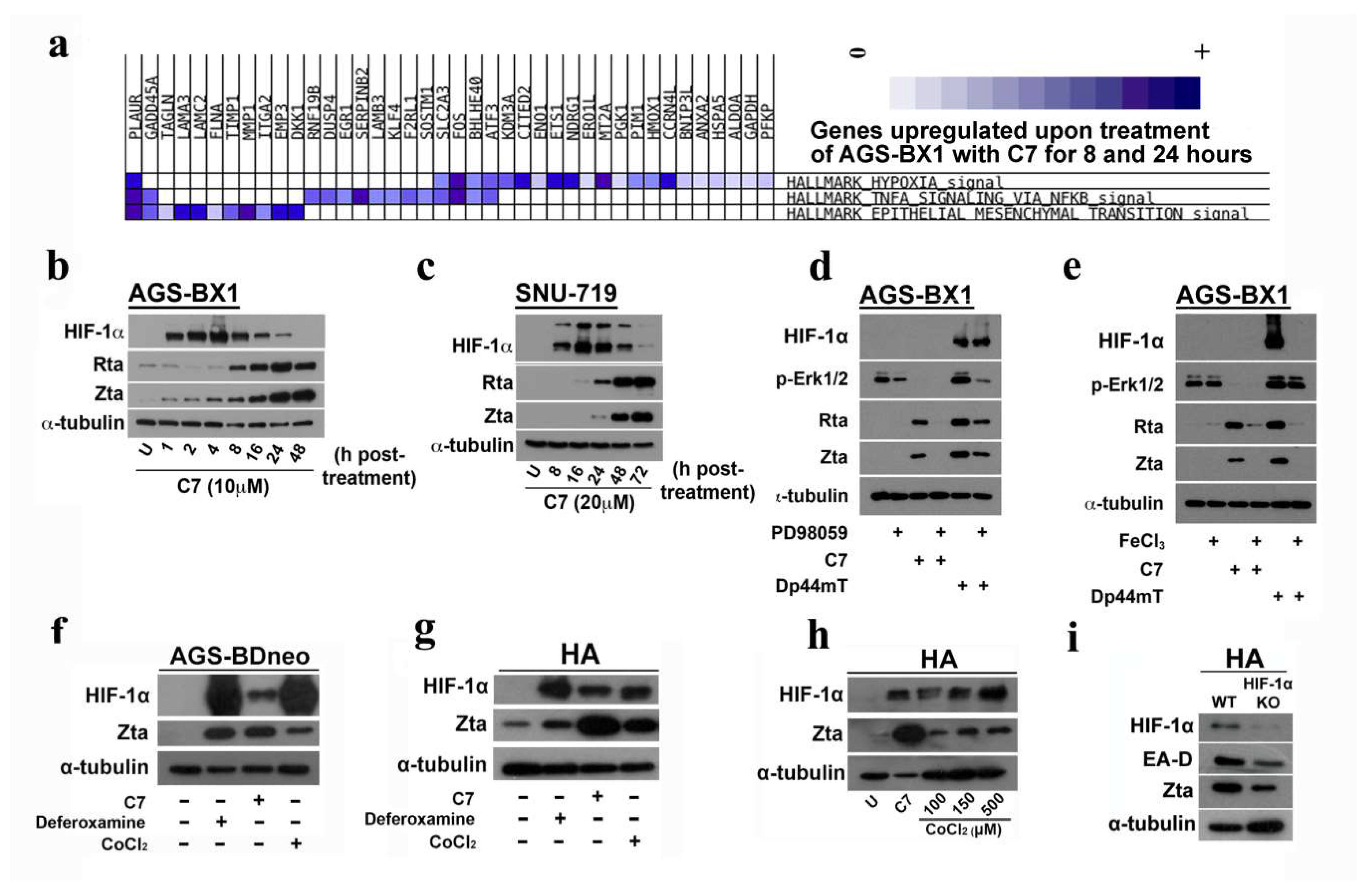
2.5. Enrichment of the Hallmark Gene Set in Hypoxia Signaling Pathway by C7
2.6. EBV Lytic Reactivation by C7 Requires the Activation of ERK in Addition to Hypoxia Signaling Pathway
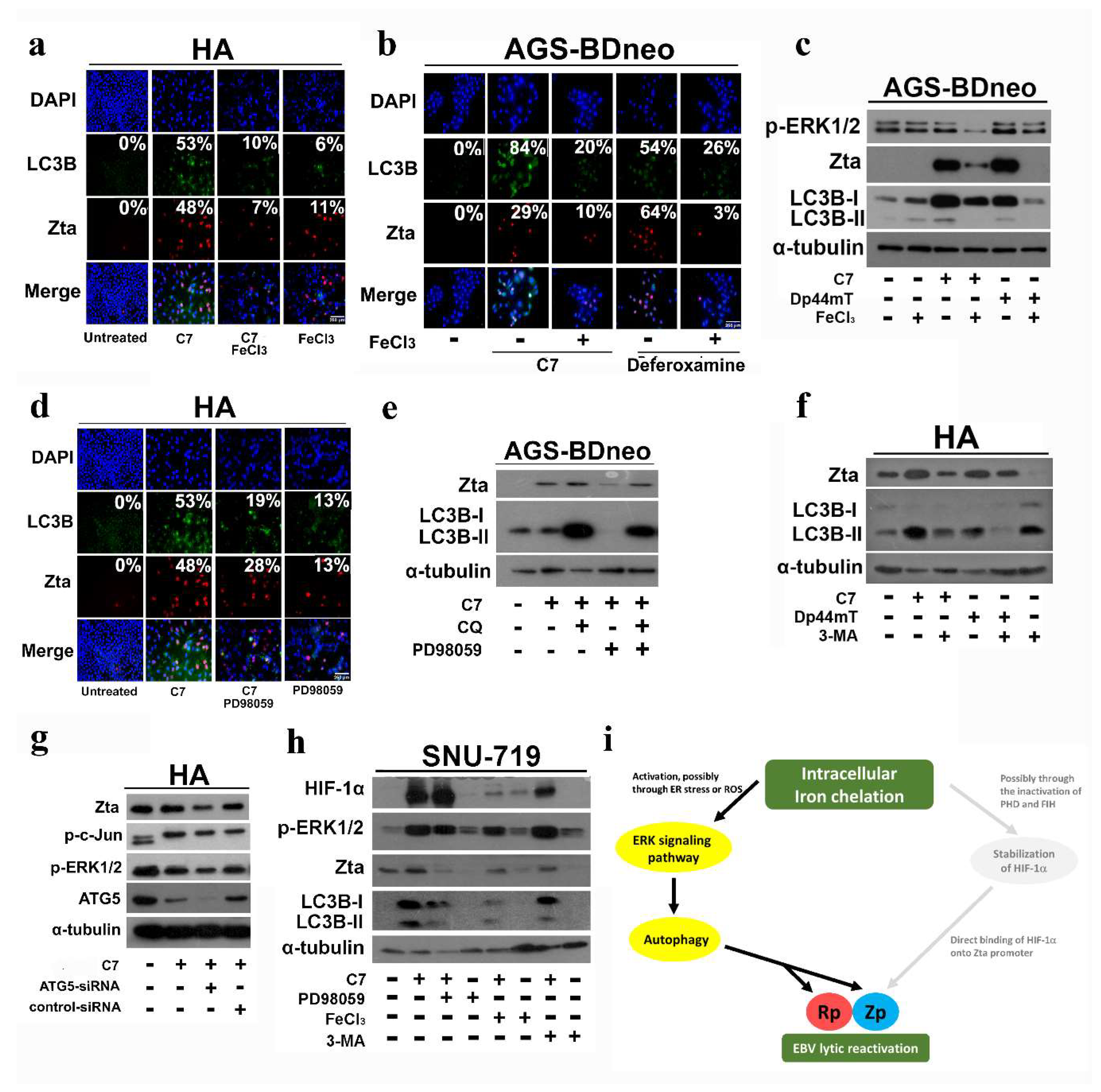
2.7. Intracellular Iron Chelation Leads to the Induction of the ERK-Autophagy Axis and Reactivation of EBV Lytic Cycle
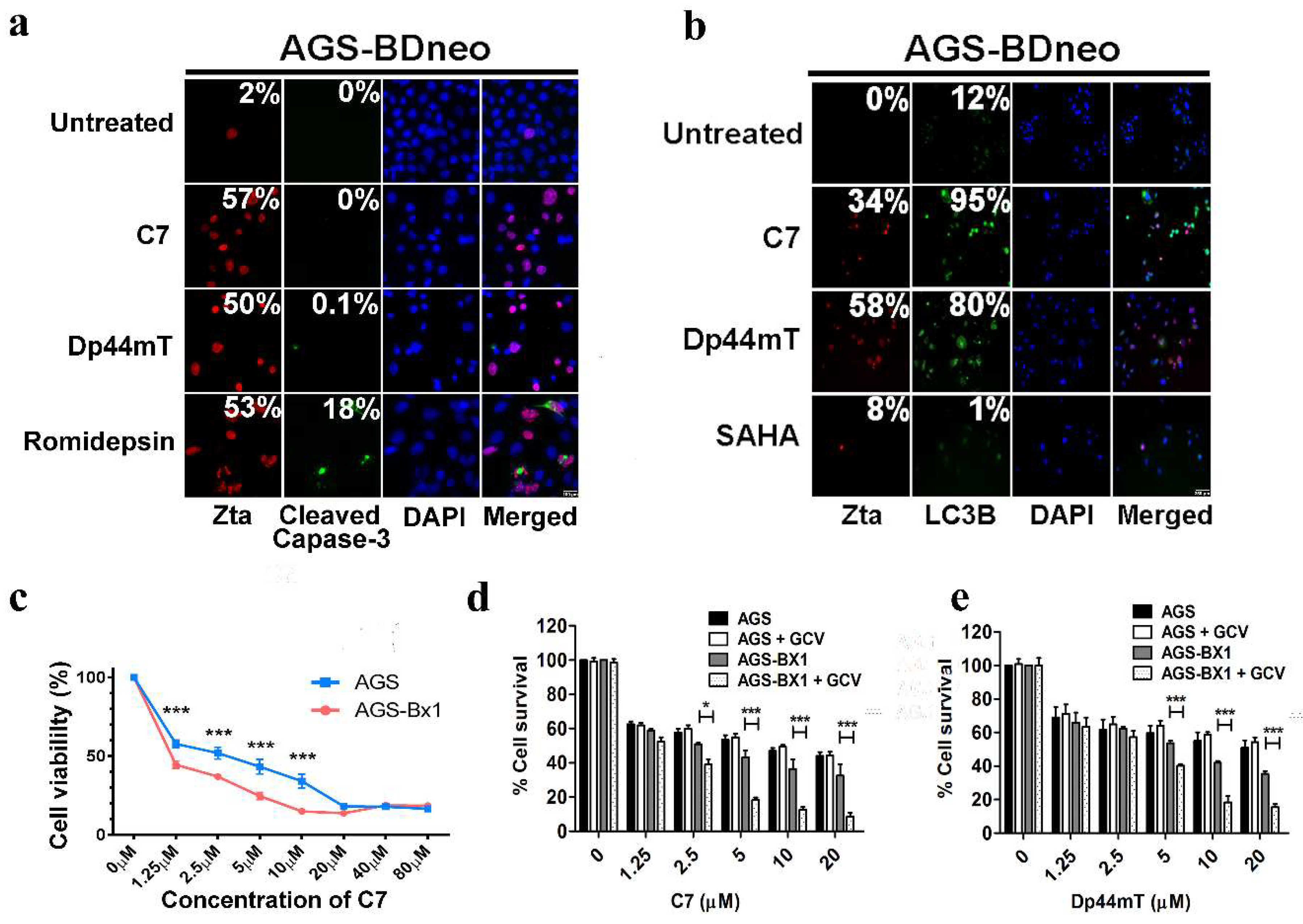
2.8. C7 Sensitizes EBV-Positive Gastric Carcinoma Cells to Killing by Ganciclovir
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culturing Conditions
4.2. Drug Treatment
4.3. Calcein-AM Measurement
4.4. Western Blot Analysis
4.5. RNA-Sequencing Analysis
4.6. Bioinformatics Analysis
4.7. Metal Binding Titration, Measurement, and Analysis
4.8. Immunofluorescence Staining
4.9. MTT Assay
4.10. CRISPR Gene Knockout
4.11. Transient Gene Knockdown
4.12. Quantitative PCR Assay
4.13. Luciferase Reporter Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Niedobitek, G.; Meru, N.; Delecluse, H.J. Epstein-barr virus infection and human malignancies. Int. J. Exp. Pathol. 2001, 82, 149–170. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Ho, D.N.; Tsang, C.M.; Middeldorp, J.M.; Tsao, G.S.; Chiang, A.K. Activation of lytic cycle of epstein-barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int. J. Cancer 2012, 131, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.H.; Hong, G.; Delecluse, H.J.; Kenney, S.C. Lytic induction therapy for epstein-barr virus-positive b-cell lymphomas. J. Virol. 2004, 78, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Chiang, A.K. Suberoylanilide hydroxamic acid induces viral lytic cycle in epstein-barr virus-positive epithelial malignancies and mediates enhanced cell death. Int. J. Cancer 2010, 126, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Cheung, A.K.; Choi, C.K.; Yeung, P.L.; Middeldorp, J.M.; Lung, M.L.; Tsao, S.W.; Chiang, A.K. Inhibition of class i histone deacetylases by romidepsin potently induces epstein-barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int. J. Cancer 2016, 138, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Nutter, L.M.; Grill, S.P.; Li, J.S.; Tan, R.S.; Cheng, Y.C. Induction of virus enzymes by phorbol esters and n-butyrate in epstein-barr virus genome-carrying raji cells. Cancer Res. 1987, 47, 4407–4412. [Google Scholar]
- Gradoville, L.; Kwa, D.; El-Guindy, A.; Miller, G. Protein kinase c-independent activation of the epstein-barr virus lytic cycle. J. Virol. 2002, 76, 5612–5626. [Google Scholar] [CrossRef] [PubMed]
- Daigle, D.; Megyola, C.; El-Guindy, A.; Gradoville, L.; Tuck, D.; Miller, G.; Bhaduri-McIntosh, S. Upregulation of stat3 marks burkitt lymphoma cells refractory to epstein-barr virus lytic cycle induction by hdac inhibitors. J. Virol. 2010, 84, 993–1004. [Google Scholar] [CrossRef]
- Hui, K.F.; Leung, Y.Y.; Yeung, P.L.; Middeldorp, J.M.; Chiang, A.K. Combination of saha and bortezomib up-regulates cdkn2a and cdkn1a and induces apoptosis of epstein-barr virus-positive wp-restricted burkitt lymphoma and lymphoblastoid cell lines. Br. J. Haematol. 2014, 167, 639–650. [Google Scholar] [CrossRef]
- Stoker, S.D.; Novalic, Z.; Wildeman, M.A.; Huitema, A.D.; Verkuijlen, S.A.; Juwana, H.; Greijer, A.E.; Tan, I.B.; Middeldorp, J.M.; de Boer, J.P. Epstein-barr virus-targeted therapy in nasopharyngeal carcinoma. J. Cancer Res. Clin. Oncol. 2015, 141, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.K.; Ho, D.N.; Hui, K.F.; Kao, R.Y.; Chiang, A.K. Identification of novel small organic compounds with diverse structures for the induction of epstein-barr virus (ebv) lytic cycle in ebv-positive epithelial malignancies. PLoS ONE 2015, 10, e0145994. [Google Scholar] [CrossRef]
- Kwiatkowski, J.L. Management of transfusional iron overload—Differential properties and efficacy of iron chelating agents. J. Blood Med. 2011, 2, 135–149. [Google Scholar] [CrossRef]
- Le, N.T.; Richardson, D.R. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: A link between iron metabolism and proliferation. Blood 2004, 104, 2967–2975. [Google Scholar] [CrossRef]
- Nyholm, S.; Mann, G.J.; Johansson, A.G.; Bergeron, R.J.; Graslund, A.; Thelander, L. Role of ribonucleotide reductase in inhibition of mammalian cell growth by potent iron chelators. J. Biol. Chem. 1993, 268, 26200–26205. [Google Scholar]
- Moon, J.H.; Jeong, J.K.; Park, S.Y. Deferoxamine inhibits trail-mediated apoptosis via regulation of autophagy in human colon cancer cells. Oncol. Rep. 2015, 33, 1171–1176. [Google Scholar] [CrossRef]
- Pullarkat, V.; Meng, Z.; Donohue, C.; Yamamoto, V.N.; Tomassetti, S.; Bhatia, R.; Krishnan, A.; Forman, S.J.; Synold, T.W. Iron chelators induce autophagic cell death in multiple myeloma cells. Leuk. Res. 2014, 38, 988–996. [Google Scholar] [CrossRef]
- Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent roles of autophagy in virus infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Nowag, H.; Guhl, B.; Thriene, K.; Romao, S.; Ziegler, U.; Dengjel, J.; Munz, C. Macroautophagy proteins assist epstein barr virus production and get incorporated into the virus particles. EBioMedicine 2014, 1, 116–125. [Google Scholar] [CrossRef]
- Ma, Y.; Abbate, V.; Hider, R.C. Iron-sensitive fluorescent probes: Monitoring intracellular iron pools. Metallomics 2015, 7, 212–222. [Google Scholar] [CrossRef]
- Feng, W.H.; Israel, B.; Raab-Traub, N.; Busson, P.; Kenney, S.C. Chemotherapy induces lytic ebv replication and confers ganciclovir susceptibility to ebv-positive epithelial cell tumors. Cancer Res. 2002, 62, 1920–1926. [Google Scholar]
- Bryant, H.; Farrell, P.J. Signal transduction and transcription factor modification during reactivation of epstein-barr virus from latency. J. Virol. 2002, 76, 10290–10298. [Google Scholar] [CrossRef]
- Tsai, P.F.; Lin, S.J.; Weng, P.L.; Tsai, S.C.; Lin, J.H.; Chou, Y.C.; Tsai, C.H. Interplay between pkcdelta and sp1 on histone deacetylase inhibitor-mediated epstein-barr virus reactivation. J. Virol. 2011, 85, 2373–2385. [Google Scholar] [CrossRef]
- El-Guindy, A.S.; Heston, L.; Endo, Y.; Cho, M.S.; Miller, G. Disruption of epstein-barr virus latency in the absence of phosphorylation of zebra by protein kinase c. J. Virol. 2002, 76, 11199–11208. [Google Scholar] [CrossRef]
- Fahmi, H.; Cochet, C.; Hmama, Z.; Opolon, P.; Joab, I. Transforming growth factor beta 1 stimulates expression of the epstein-barr virus bzlf1 immediate-early gene product zebra by an indirect mechanism which requires the mapk kinase pathway. J. Virol. 2000, 74, 5810–5818. [Google Scholar] [CrossRef]
- Adamson, A.L.; Kenney, S.C. Rescue of the epstein-barr virus bzlf1 mutant, z(s186a), early gene activation defect by the brlf1 gene product. Virology 1998, 251, 187–197. [Google Scholar] [CrossRef]
- Hagemeier, S.R.; Barlow, E.A.; Meng, Q.; Kenney, S.C. The cellular ataxia telangiectasia-mutated kinase promotes epstein-barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J. Virol. 2012, 86, 13360–13370. [Google Scholar] [CrossRef]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans egl-9 and mammalian homologs define a family of dioxygenases that regulate hif by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Tong, Y.; Huang, H.; Pan, H. Inhibition of mek/erk activation attenuates autophagy and potentiates pemetrexed-induced activity against hepg2 hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2015, 456, 86–91. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, W.; Liu, S.; Wu, W.; Qin, M.; Huang, J. Activation of the sphk1/erk/p-erk pathway promotes autophagy in colon cancer cells. Oncol. Lett. 2018, 15, 9719–9724. [Google Scholar] [CrossRef]
- Tarhonskaya, H.; Hardy, A.P.; Howe, E.A.; Loik, N.D.; Kramer, H.B.; McCullagh, J.S.; Schofield, C.J.; Flashman, E. Kinetic investigations of the role of factor inhibiting hypoxia-inducible factor (fih) as an oxygen sensor. J. Biol. Chem. 2015, 290, 19726–19742. [Google Scholar] [CrossRef]
- Demidenko, Z.N.; Blagosklonny, M.V. The purpose of the hif-1/phd feedback loop: To limit mtor-induced hif-1alpha. Cell Cycle 2011, 10, 1557–1562. [Google Scholar] [CrossRef]
- Kraus, R.J.; Yu, X.; Cordes, B.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1alpha plays roles in epstein-barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early bzlf1 gene promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef]
- Lee, S.K.; Jang, H.J.; Lee, H.J.; Lee, J.; Jeon, B.H.; Jun, C.D.; Lee, S.K.; Kim, E.C. P38 and erk map kinase mediates iron chelator-induced apoptosis and -suppressed differentiation of immortalized and malignant human oral keratinocytes. Life Sci. 2006, 79, 1419–1427. [Google Scholar] [CrossRef]
- Choi, E.Y.; Kim, E.C.; Oh, H.M.; Kim, S.; Lee, H.J.; Cho, E.Y.; Yoon, K.H.; Kim, E.A.; Han, W.C.; Choi, S.C.; et al. Iron chelator triggers inflammatory signals in human intestinal epithelial cells: Involvement of p38 and extracellular signal-regulated kinase signaling pathways. J. Immunol. 2004, 172, 7069–7077. [Google Scholar] [CrossRef]
- De Leo, A.; Colavita, F.; Ciccosanti, F.; Fimia, G.M.; Lieberman, P.M.; Mattia, E. Inhibition of autophagy in ebv-positive burkitt’s lymphoma cells enhances ebv lytic genes expression and replication. Cell Death Dis. 2015, 6, e1876. [Google Scholar] [CrossRef]
- Hung, C.H.; Chen, L.W.; Wang, W.H.; Chang, P.J.; Chiu, Y.F.; Hung, C.C.; Lin, Y.J.; Liou, J.Y.; Tsai, W.J.; Hung, C.L.; et al. Regulation of autophagic activation by rta of epstein-barr virus via the extracellular signal-regulated kinase pathway. J. Virol. 2014, 88, 12133–12145. [Google Scholar] [CrossRef]
- Chaturvedi, U.C.; Shrivastava, R. Interaction of viral proteins with metal ions: Role in maintaining the structure and functions of viruses. FEMS Immunol. Med. Microbiol. 2005, 43, 105–114. [Google Scholar] [CrossRef]
- De Guzman, R.N.; Wu, Z.R.; Stalling, C.C.; Pappalardo, L.; Borer, P.N.; Summers, M.F. Structure of the hiv-1 nucleocapsid protein bound to the sl3 psi-rna recognition element. Science 1998, 279, 384–388. [Google Scholar] [CrossRef]
- Kalejta, R.F. Tegument proteins of human cytomegalovirus. Microbiol. Mol. Biol. Rev. 2008, 72, 249–265. [Google Scholar] [CrossRef]
- York, J.; Nunberg, J.H. A novel zinc-binding domain is essential for formation of the functional junin virus envelope glycoprotein complex. J. Virol. 2007, 81, 13385–13391. [Google Scholar] [CrossRef]
- Molesworth, S.J.; Lake, C.M.; Borza, C.M.; Turk, S.M.; Hutt-Fletcher, L.M. Epstein-barr virus gh is essential for penetration of b cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 2000, 74, 6324–6332. [Google Scholar] [CrossRef]
- Hui, K.F.; Chiang, A.K. Combination of proteasome and class i hdac inhibitors induces apoptosis of npc cells through an hdac6-independent er stress-induced mechanism. Int. J. Cancer 2014, 135, 2950–2961. [Google Scholar] [CrossRef]
- Hui, K.F.; Lam, B.H.; Ho, D.N.; Tsao, S.W.; Chiang, A.K. Bortezomib and saha synergistically induce ros-driven caspase-dependent apoptosis of nasopharyngeal carcinoma and block replication of epstein-barr virus. Mol. Cancer Ther. 2013, 12, 747–758. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yiu, S.P.T.; Hui, K.F.; Choi, C.K.; Kao, R.Y.T.; Ma, C.W.; Yang, D.; Chiang, A.K.S. Intracellular Iron Chelation by a Novel Compound, C7, Reactivates Epstein–Barr Virus (EBV) Lytic Cycle via the ERK-Autophagy Axis in EBV-Positive Epithelial Cancers. Cancers 2018, 10, 505. https://doi.org/10.3390/cancers10120505
Yiu SPT, Hui KF, Choi CK, Kao RYT, Ma CW, Yang D, Chiang AKS. Intracellular Iron Chelation by a Novel Compound, C7, Reactivates Epstein–Barr Virus (EBV) Lytic Cycle via the ERK-Autophagy Axis in EBV-Positive Epithelial Cancers. Cancers. 2018; 10(12):505. https://doi.org/10.3390/cancers10120505
Chicago/Turabian StyleYiu, Stephanie Pei Tung, Kwai Fung Hui, Chung King Choi, Richard Yi Tsun Kao, Chi Wang Ma, Dan Yang, and Alan Kwok Shing Chiang. 2018. "Intracellular Iron Chelation by a Novel Compound, C7, Reactivates Epstein–Barr Virus (EBV) Lytic Cycle via the ERK-Autophagy Axis in EBV-Positive Epithelial Cancers" Cancers 10, no. 12: 505. https://doi.org/10.3390/cancers10120505