Externalized Keratin 8: A Target at the Interface of Microenvironment and Intracellular Signaling in Colorectal Cancer Cells

 , , , , , , add
Show full author list
, , , , , , add
Show full author list
Abstract
:1. Introduction
2. Results
2.1. Identification of eK8 from a Dynamic Proteomic Analysis
2.2. An Anti-K8 MAb Panel Exhibits Different eK8 Recognition Properties
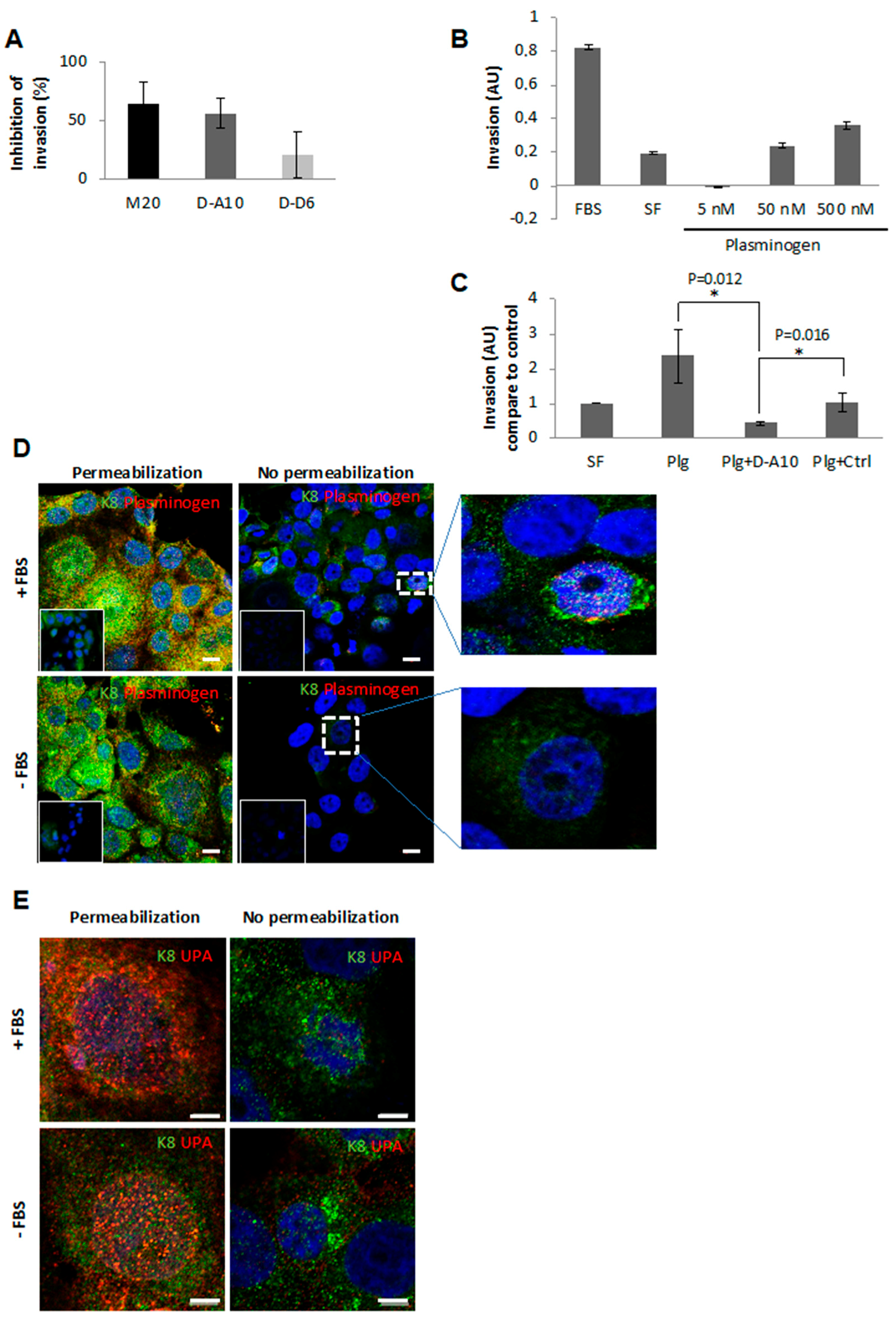
2.3. Anti-eK8 D-A10 MAb Limits the Plasminogen-Induced Invasion Process
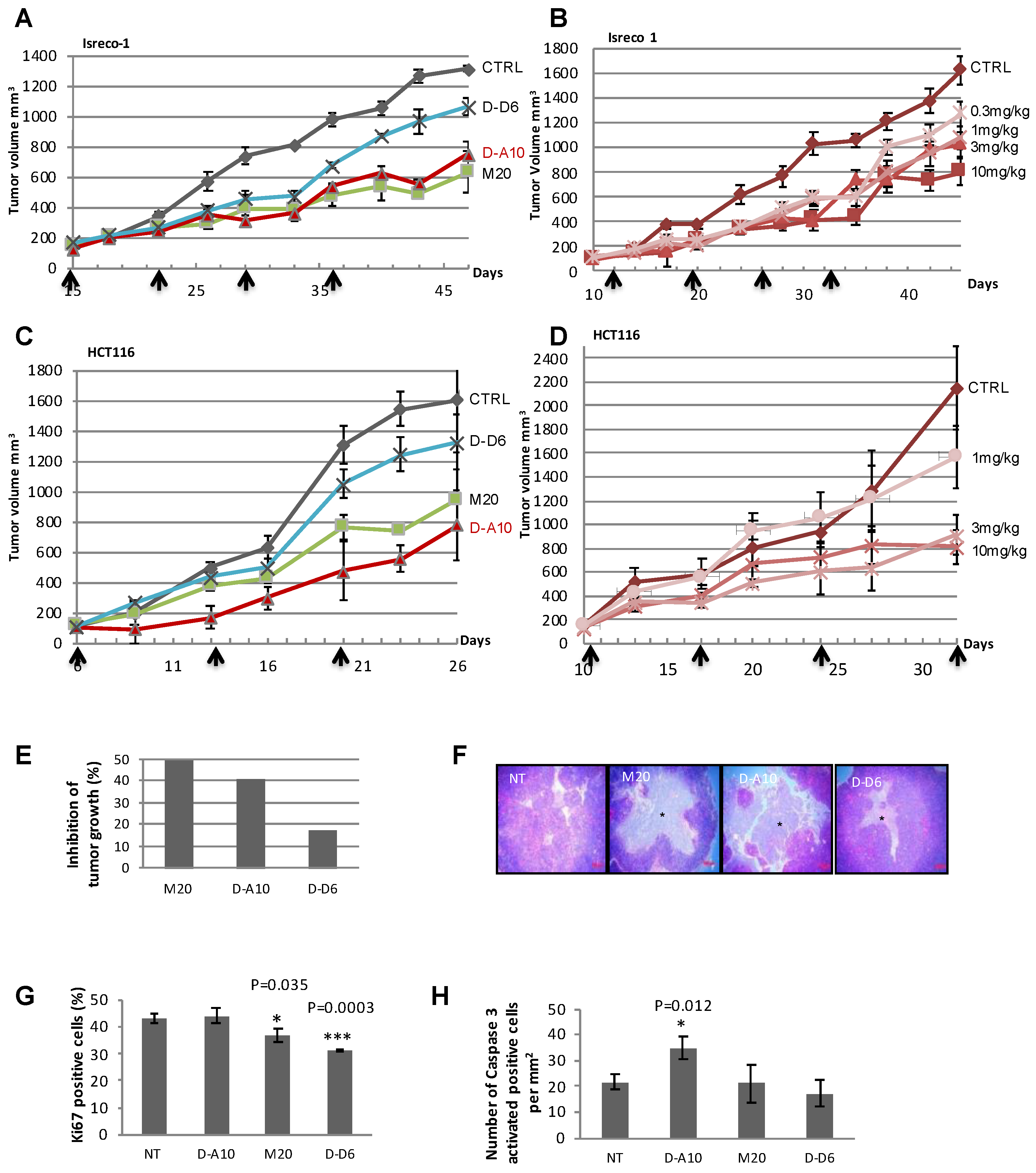
2.4. Anti-eK8 D-A10 MAb Induces Apoptosis of Colorectal Tumor Cells
2.5. The LSELEAAL Amino-Acid Motif of eK8 Is Key for Bidirectional Signaling
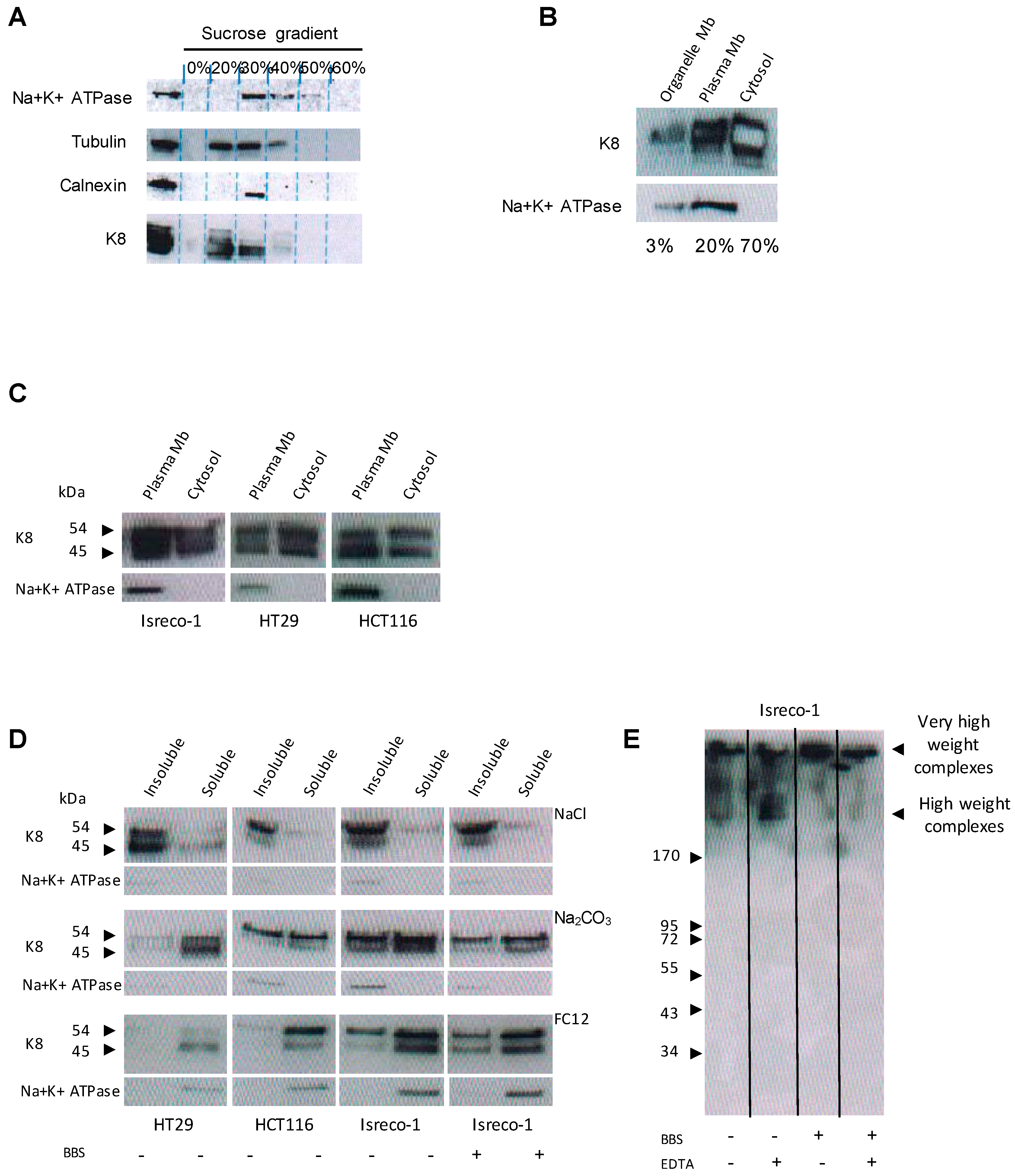
2.6. K8 Is Present at the Plasma Membrane
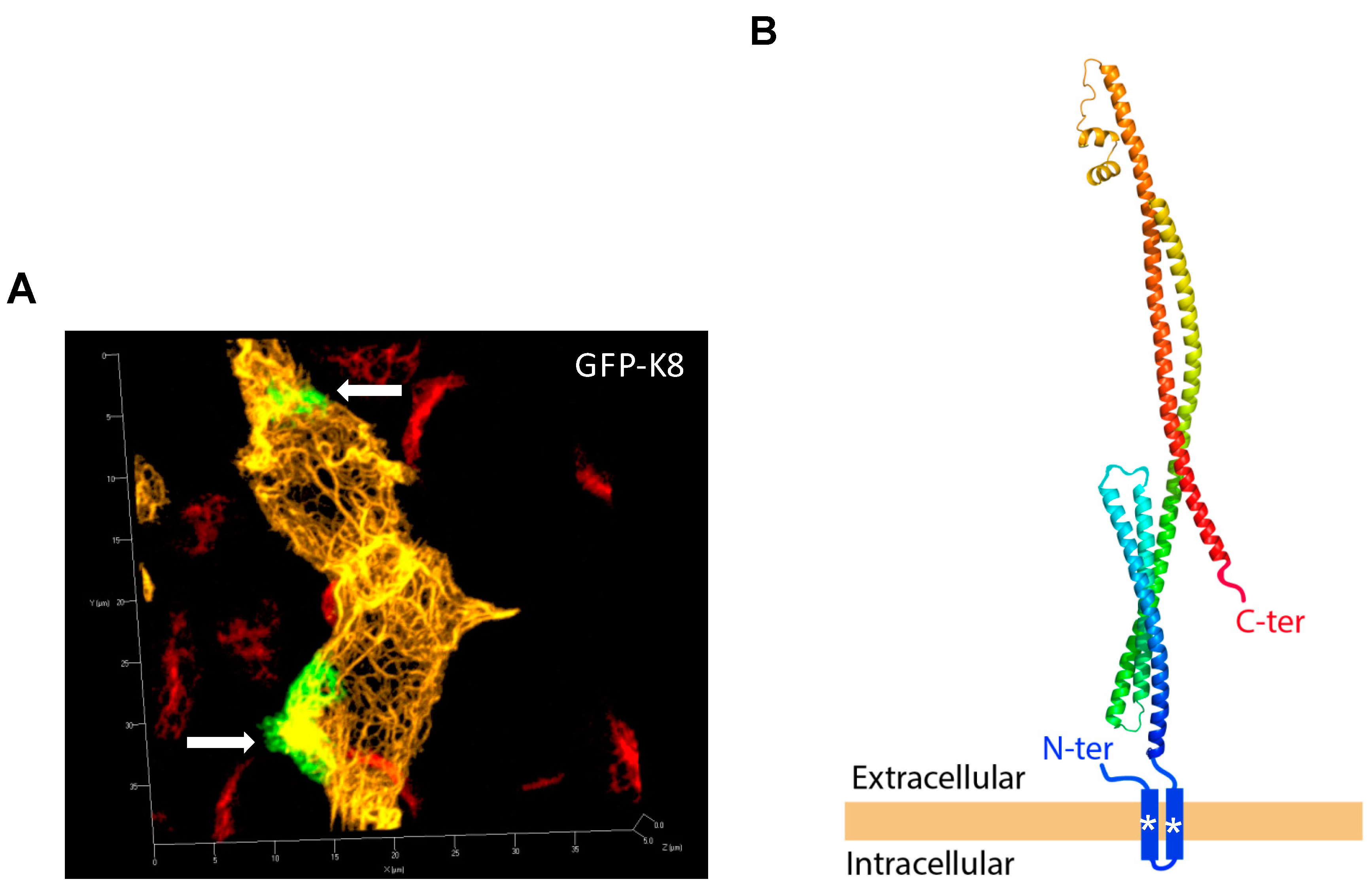
2.7. N- and C-Terminal Extremities of eK8 Are Exposed at the External Surface of the Plasma Membrane
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Antibodies
4.3. Cell Fractionation Procedures
4.4. Immunofluorescence Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plow, E.F.; Das, R. Enolase-1 as a plasminogen receptor. Blood 2009, 113, 5371–5372. [Google Scholar] [CrossRef] [PubMed]
- Farin, K.; Schokoroy, S.; Haklai, R.; Cohen-Or, I.; Elad-Sfadia, G.; Reyes-Reyes, M.E.; Bates, P.J.; Cox, A.D.; Kloog, Y.; Pinkas-Kramarski, R. Oncogenic synergism between ErbB1, nucleolin, and mutant Ras. Cancer Res. 2011, 71, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.T.; Kang, L.G.; Ding, L.; Vranic, S.; Gatalica, Z.; Wang, Z.Y. A positive feedback loop of ER-alpha36/EGFR promotes malignant growth of ER-negative breast cancer cells. Oncogene 2011, 30, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Deng, M.; Li, X.; Liu, W.; Chu, X.; Wang, J.; Chen, F.; Meng, S. Chaperone gp96 mediates ER-alpha36 cell membrane expression. Oncotarget 2015, 6, 31857–31867. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, L.; Hou, J.; Gui, M.; Ying, J.; Zhao, H.; Lv, N.; Meng, S. Cell membrane gp96 facilitates HER2 dimerization and serves as a novel target in breast cancer. Int. J. Cancer 2015, 137, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Didiasova, M.; Wujak, L.; Wygrecka, M.; Zakrzewicz, D. From plasminogen to plasmin: Role of plasminogen receptors in human cancer. Int. J. Mol. Sci. 2014, 15, 21229–21252. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gronow, M.; Gomez, C.F.; de Ridder, G.G.; Ray, R.; Pizzo, S.V. Binding of tissue-type plasminogen activator to the glucose-regulated protein 78 (GRP78) modulates plasminogen activation and promotes human neuroblastoma cell proliferation in vitro. J. Biol. Chem. 2014, 289, 25166–25176. [Google Scholar] [CrossRef] [PubMed]
- Godfroid, E.; Geuskens, M.; Dupressoir, T.; Parent, I.; Szpirer, C. Cytokeratins are exposed on the outer surface of established human mammary carcinoma cells. J. Cell Sci. 1991, 99, 595–607. [Google Scholar] [PubMed]
- Weidle, U.H.; Maisel, D.; Klostermann, S.; Schiller, C.; Weiss, E.H. Intracellular proteins displayed on the surface of tumor cells as targets for therapeutic intervention with antibody-related agents. Cancer Genom. Proteom. 2011, 8, 49–63. [Google Scholar]
- Kralovich, K.R.; Li, L.; Hembrough, T.A.; Webb, D.J.; Karns, L.R.; Gonias, S.L. Characterization of the binding sites for plasminogen and tissue-type plasminogen activator in cytokeratin 8 and cytokeratin 18. J. Protein Chem. 1998, 17, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Saurin, J.C.; Fallavier, M.; Sordat, B.; Gevrey, J.C.; Chayvialle, J.A.; Abello, J. Bombesin stimulates invasion and migration of Isreco1 colon carcinoma cells in a Rho-dependent manner. Cancer Res. 2002, 62, 4829–4835. [Google Scholar] [PubMed]
- Hembrough, T.A.; Li, L.; Gonias, S.L. Cell-surface cytokeratin 8 is the major plasminogen receptor on breast cancer cells and is required for the accelerated activation of cell-associated plasminogen by tissue-type plasminogen activator. J. Biol. Chem. 1996, 271, 25684–25691. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Cell surface remodeling by plasmin: A new function for an old enzyme. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Loranger, A.; Gilbert, S.; Brouard, J.S.; Magin, T.M.; Marceau, N. Keratin 8 modulation of desmoplakin deposition at desmosomes in hepatocytes. Exp. Cell Res. 2006, 312, 4108–4119. [Google Scholar] [CrossRef] [PubMed]
- Hembrough, T.A.; Vasudevan, J.; Allietta, M.M.; Glass, W.F.; Gonias, S.L. A cytokeratin 8-like protein with plasminogen-binding activity is present on the external surfaces of hepatocytes, HepG2 cells and breast carcinoma cell lines. J. Cell Sci. 1995, 108, 1071–1082. [Google Scholar] [PubMed]
- Ditzel, H.J.; Garrigues, U.; Andersen, C.B.; Larsen, M.K.; Garrigues, H.J.; Svejgaard, A.; Hellström, I.; Hellström, K.E.; Jensenius, J.C. Modified cytokeratins expressed on the surface of carcinoma cells undergo endocytosis upon binding of human monoclonal antibody and its recombinant Fab fragment. Proc. Natl. Acad. Sci. USA 1997, 94, 8110–8115. [Google Scholar] [CrossRef] [PubMed]
- Waseem, A.; Karsten, U.; Leigh, I.M.; Purkis, P.; Waseem, N.H.; Lane, E.B. Conformational changes in the rod domain of human keratin 8 following heterotypic association with keratin 18 and its implication for filament stability. Biochemistry 2004, 43, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Ceruti, P.; Principe, M.; Capello, M.; Cappello, P.; Novelli, F. Three are better than one: Plasminogen receptors as cancer theranostic targets. Exp. Hematol. Oncol. 2013, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.W.; Torchia, D.A.; Steinert, P.M. Solid-state NMR studies of the dynamics and structure of mouse keratin intermediate filaments. Biochemistry 1988, 27, 5418–5426. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed]
- Blumenberg, M. Concerted gene duplications in the two keratin gene families. J. Mol. Evol. 1988, 27, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Wang, Y. Quantitative analysis of surface plasma membrane proteins of primary and metastatic melanoma cells. J. Proteome Res. 2008, 7, 1904–1915. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Rose-Sperling, D.; Hellmich, U.A. Diverse relations between ABC transporters and lipids: An overview. Biochim. Biophys. Acta Biomembr. 2017, 1859, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Davezac, N.; Tondelier, D.; Lipecka, J.; Fanen, P.; Demaugre, F.; Debski, J.; Dadlez, M.; Schrattenholz, A.; Cahill, M.A.; Edelman, A. Global proteomic approach unmasks involvement of keratins 8 and 18 in the delivery of cystic fibrosis transmembrane conductance regulator (CFTR)/deltaF508-CFTR to the plasma membrane. Proteomics 2004, 4, 3833–3844. [Google Scholar] [CrossRef] [PubMed]
- Haspel, M.V.; McCabe, R.P.; Pomato, N.; Janesch, N.J.; Knowlton, J.V.; Peters, L.C.; Hoover, H.C.; Hanna, M.G. Generation of tumor cell-reactive human monoclonal antibodies using peripheral blood lymphocytes from actively immunized colorectal carcinoma patients. Cancer Res. 1985, 45, 3951–3961. [Google Scholar] [PubMed]
- Ditzel, H.J.; Strik, M.C.; Larsen, M.K.; Willis, A.C.; Waseem, A.; Kejling, K.; Jensenius, J.C. Cancer-associated cleavage of cytokeratin 8/18 heterotypic complexes exposes a neoepitope in human adenocarcinomas. J. Biol. Chem. 2002, 277, 21712–21722. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Münz, M.; Schaffrik, M.; Kieu, C.; Rauch, J.; Ahlemann, M.; Eberle, D.; Mack, B.; Wollenberg, B.; Lang, S.; et al. Profile identification of disease-associated humoral antigens using AMIDA, a novel proteomics-based technology. Cell. Mol. Life Sci. 2004, 61, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Chen, Z.; Wang, J.; Shao, X.; Cui, Z.; Yang, C.; Zhu, Z.; Xiong, D. Overexpression of cell surface cytokeratin 8 in multidrug-resistant MCF-7/MX cells enhances cell adhesion to the extracellular matrix. Neoplasia 2008, 10, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Doljak, B.; Obermajer, N.; Jamnik, P.; Kos, J. Monoclonal antibody to cytokeratin VKIALEVEIATY sequence motif reduces plasminogen activation in breast tumour cells. Cancer Lett. 2008, 267, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Doljak, B.; Kos, J. Cytokeratin 8 ectoplasmic domain binds urokinase-type plasminogen activator to breast tumor cells and modulates their adhesion, growth and invasiveness. Mol. Cancer 2009, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.O.; Omary, M.B. A disease- and phosphorylation-related nonmechanical function for keratin 8. J. Cell Biol. 2006, 174, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Schutte, B.; Henfling, M.; Ramaekers, F.C. DEDD association with cytokeratin filaments correlates with sensitivity to apoptosis. Apoptosis 2006, 11, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Schickling, O.; Stegh, A.H.; Oshima, R.G.; Dinsdale, D.; Cohen, G.M.; Peter, M.E. DEDD regulates degradation of intermediate filaments during apoptosis. J. Cell Biol. 2002, 158, 1051–1066. [Google Scholar] [CrossRef] [PubMed]
- Hembrough, T.A.; Kralovich, K.R.; Li, L.I.; Gonias, S.L. Cytokeratin 8 released by breast carcinoma cells in vitro binds plasminogen and tissue-type plasminogen activator and promotes plasminogen activation. Biochem. J. 1996, 317, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Scherl, A.; Couté, Y.; Déon, C.; Callé, A.; Kindbeiter, K.; Sanchez, J.C.; Greco, A.; Hochstrasser, D.; Diaz, J.J. Functional proteomic analysis of human nucleolus. Mol. Biol. Cell 2002, 13, 4100–4109. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide (N°) | Sequence K8 (aa) | Antibody Reactivity on Coated Free Peptides | |
|---|---|---|---|
| D-A10 | M20 | ||
| Peptide 1 | 338-AEQRGELAIKDANAKLSELEAALQRAKQD-C-366 | +++ | + |
| Peptide 5 | 338-AEQRGELAIKDANAKLSELEAALQRAKQD-366 | +++ | + |
| Peptide 6 | 358-AALQRAKQD-366 | - | - |
| Peptide 7 | 357-EAALQRAKQD-366 | - | - |
| Peptide 8 | 356-LEAALQRAKQD-366 | - | - |
| Peptide 9 | 355-ELEAALQRAKQD-366 | - | - |
| Peptide 10 | 354-SELEAALQRAKQD-366 | - | - |
| Peptide 11 | 353-LSELEAALQRAKQD-366 | ++ | - |
| Peptide 12 | 352-KLSELEAALQRAKQD-366 | ++ | - |
| Peptide 13 | 351-AKLSELEAALQRAKQD-366 | ++ | - |
| Peptide 14 | 350-NAKLSELEAALQRAKQD-366 | +++ | - |
| Peptide 15 | 349-ANAKLSELEAALQRAKQD-366 | +++ | - |
| Peptide 16 | 348-DANAKLSELEAALQRAKQD-366 | +++ | - |
| Peptide 17 | 345-C-AIKDANAKLSELEAALQRAKQD-366 | +++ | - |
| Peptide 18 | 345-AIKDANAKLSELEAALQRAKQ-365 | +++ | - |
| Peptide 19 | 345-AIKDANAKLSELEAALQRAK-364 | +++ | - |
| Peptide 20 | 345-AIKDANAKLSELEAALQRA-363 | +++ | - |
| Peptide 21 | 345-AIKDANAKLSELEAALQR-362 | +++ | - |
| Peptide 22 | 345-AIKDANAKLSELEAALQ-361 | +++ | - |
| Peptide 23 | 345-AIKDANAKLSELEAAL-360 | +++ | - |
| Peptide 24 | 345-AIKDANAKLSELEAA-359 | - | - |
| Peptide 25 | 345-AIKDANAKLSELEA-358 | - | - |
| Peptide 26 | 345-AIKDANAKLSELE-357 | - | - |
| Peptide 27 | 345-AIKDANAKLSEL-356 | - | - |
| Peptide 28 | 345-AIKDANAKLSE-355 | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albaret, M.A.; Vermot-Desroches, C.; Paré, A.; Roca-Martinez, J.-X.; Malet, L.; Esseily, J.; Gerossier, L.; Brière, J.; Pion, N.; Marcel, V.; et al. Externalized Keratin 8: A Target at the Interface of Microenvironment and Intracellular Signaling in Colorectal Cancer Cells. Cancers 2018, 10, 452. https://doi.org/10.3390/cancers10110452
Albaret MA, Vermot-Desroches C, Paré A, Roca-Martinez J-X, Malet L, Esseily J, Gerossier L, Brière J, Pion N, Marcel V, et al. Externalized Keratin 8: A Target at the Interface of Microenvironment and Intracellular Signaling in Colorectal Cancer Cells. Cancers. 2018; 10(11):452. https://doi.org/10.3390/cancers10110452
Chicago/Turabian StyleAlbaret, Marie Alexandra, Claudine Vermot-Desroches, Arnaud Paré, Jean-Xavier Roca-Martinez, Lucie Malet, Jad Esseily, Laetitia Gerossier, Johan Brière, Nathalie Pion, Virginie Marcel, and et al. 2018. "Externalized Keratin 8: A Target at the Interface of Microenvironment and Intracellular Signaling in Colorectal Cancer Cells" Cancers 10, no. 11: 452. https://doi.org/10.3390/cancers10110452