
Patisiran in hATTR Amyloidosis: Six-Month Latency Period before Efficacy

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients and Outcome Measures
2.2. Statistical Analysis
- -
- Group 1 (all patients) from M-6 to M6.
- -
- Group 2 (14 patients) from M-12 to M12.
- -
- Group 3 (11 patients) from M-18 to M18.
- -
- Group 4 (4 patients with tafamidis as concomitant medication) from M-12 to M12.
- -
- Group 5 (8 patients in PND stage 1–2) from M-12 to M12.
- -
- Group 6 (6 patients in PND stage 3A and 3B) from M-12 to M12.
3. Results
3.1. Demographics
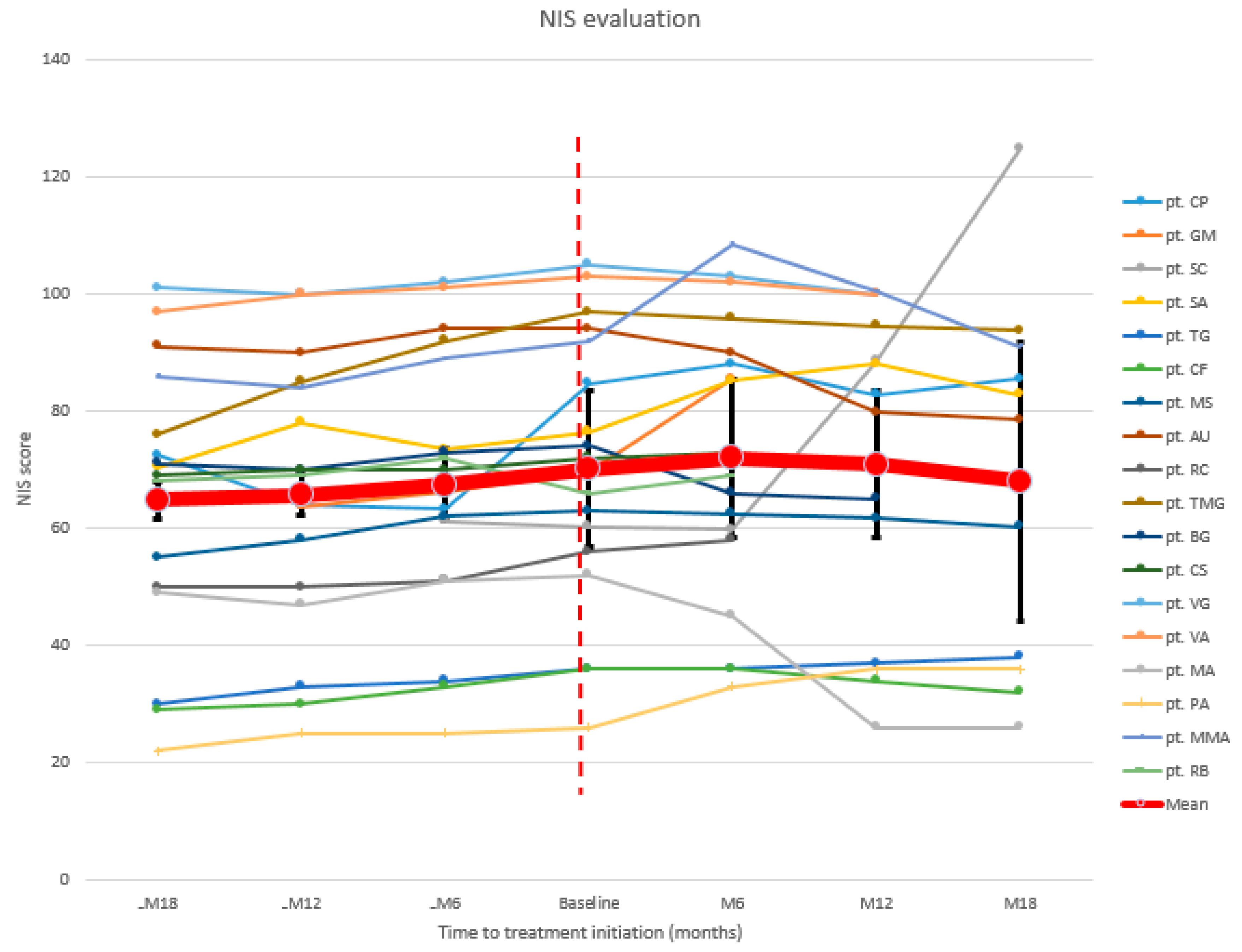
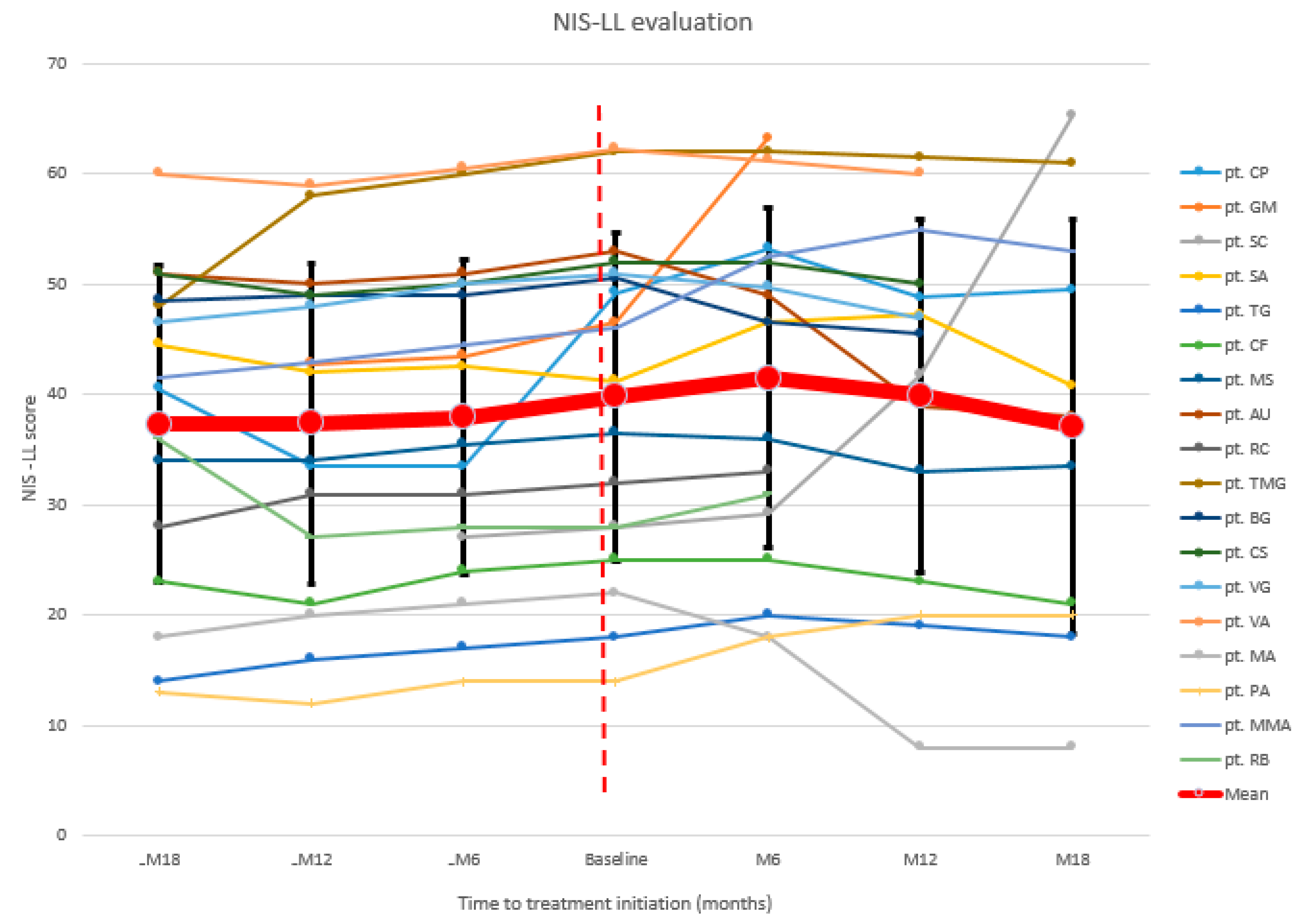
3.2. Neurologic Evaluation
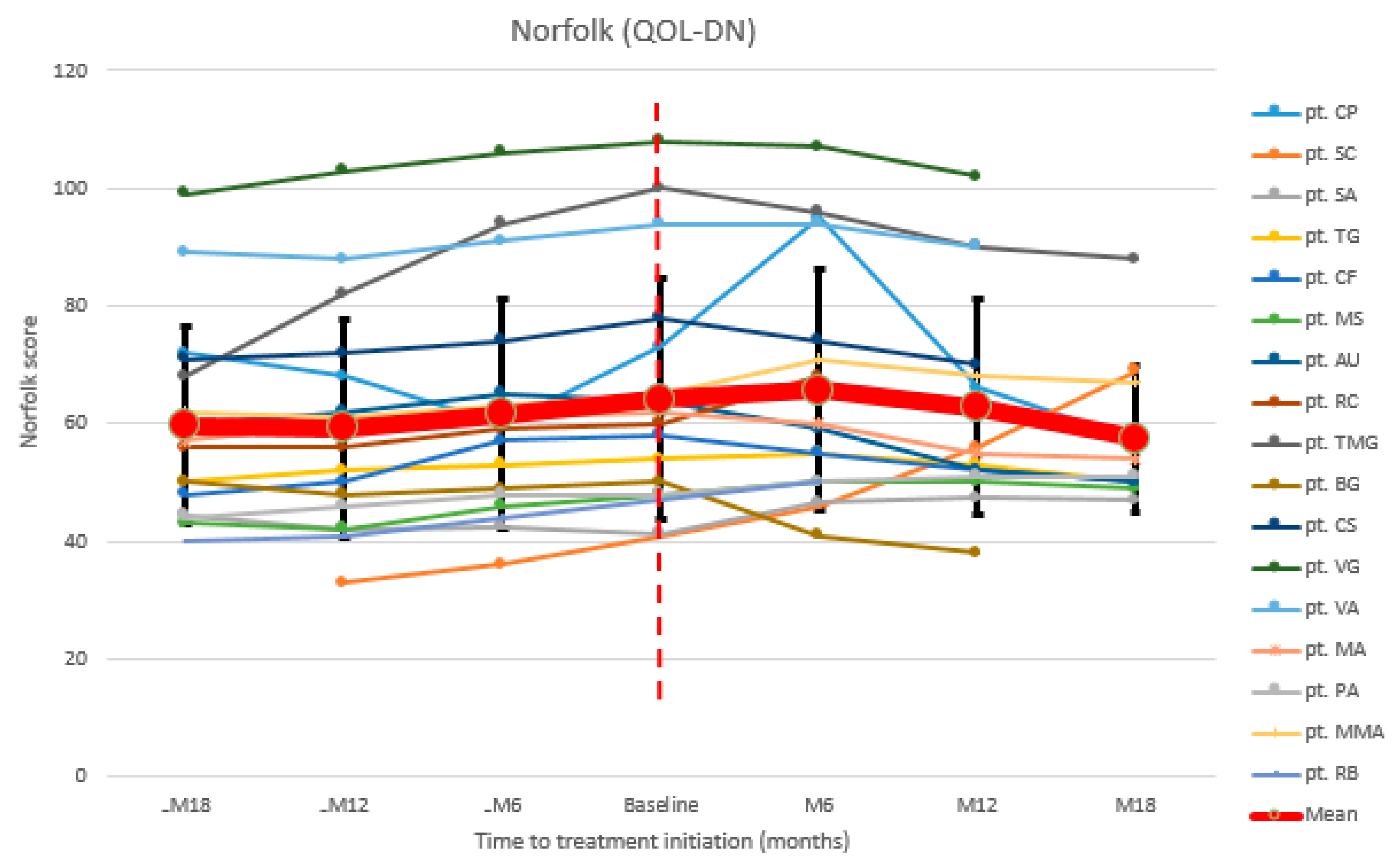
3.3. Quality of Life Assessment
3.4. Adverse Events
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Patient | PND at Baseline | PND at Last Follow-Up |
|---|---|---|
| pt. CP | 2 | 3a |
| pt. GM (d) 1 | 2 | 2 |
| pt. SC | 2 | 3b |
| pt. SA | 2 | 2 |
| pt. TG | 1 | 1 |
| pt. CF | 1 | 1 |
| pt. MS | 2 | 2 |
| pt. AU | 3b | 2 |
| pt. RC (d) 1 | 3b | 3b |
| pt. TMG | 3a | 2 |
| pt. BG | 3a | 2 |
| pt. CS | 3b | 3b |
| pt. VG | 3a | 3a |
| pt. VA | 2 | 2 |
| pt. MA | 2 | 2 |
| pt. PA | 1 | 1 |
| pt. MMA | 3a | 3a |
| pt. RB (d) 1 | 2 | 2 |
| Patient | -M18 | -M12 | -M6 | Baseline | M6 | M12 | M18 |
|---|---|---|---|---|---|---|---|
| pt. CP | 72.5 | 64 | 63.25 | 84.75 | 88 | 82.75 | 85.5 |
| pt. GM (d) 1 | 63.75 | 66.25 | 69.75 | 85.5 | |||
| pt. SC | 61.25 | 60.25 | 59.75 | 88.5 | 124.75 | ||
| pt. SA | 70.5 | 78 | 73.5 | 76.5 | 85.25 | 88 | 82.75 |
| pt. TG | 30 | 33 | 34 | 36 | 36 | 37 | 38 |
| pt. CF | 29 | 30 | 33 | 36 | 36 | 34 | 32 |
| pt. MS | 55 | 58 | 62 | 63 | 62.5 | 61.75 | 60.25 |
| pt. AU | 91 | 90 | 94 | 94 | 90 | 79.75 | 78.5 |
| pt. RC (d) 1 | 50 | 50 | 51 | 56 | 58 | ||
| pt. TMG | 76 | 85 | 92 | 97 | 95.75 | 94.5 | 93.75 |
| pt. BG | 71 | 70 | 73 | 74 | 66 | 65 | |
| pt. CS | 69 | 70 | 70 | 72 | 73 | 70 | |
| pt. VG | 101 | 100 | 102 | 105 | 103 | 100 | |
| pt. VA | 97 | 100 | 101 | 103 | 102 | 100 | |
| pt. MA | 49 | 47 | 51 | 52 | 45 | 26 | 26 |
| pt. PA | 22 | 25 | 25 | 26 | 33 | 36 | 36 |
| pt. MMA | 86 | 84 | 89 | 92 | 108.5 | 100.5 | 91 |
| pt. RB (d) 1 | 68 | 69 | 72 | 66 | 69 | ||
| Mean | 64.8 | 65.7 | 67.4 | 70.2 | 72 | 70.9 | 68 |
| Patient | -M18 | -M12 | -M6 | Baseline | M6 | M12 | M18 |
|---|---|---|---|---|---|---|---|
| pt. CP | 40.5 | 33.5 | 33.5 | 49.25 | 53.25 | 48.75 | 49.5 |
| pt. GM (d) 1 | 42.75 | 43.5 | 46.5 | 63.25 | |||
| pt. SC | 27 | 28 | 29.25 | 41.7 | 65.25 | ||
| pt. SA | 44.5 | 42 | 42.5 | 41.25 | 46.5 | 47.25 | 40.75 |
| pt. TG | 14 | 16 | 17 | 18 | 20 | 19 | 18 |
| pt. CF | 23 | 21 | 24 | 25 | 25 | 23 | 21 |
| pt. MS | 34 | 34 | 35.5 | 36.5 | 36 | 33 | 33.5 |
| pt. AU | 51 | 50 | 51 | 53 | 49 | 39 | 38 |
| pt. RC (d) 1 | 28 | 31 | 31 | 32 | 33 | ||
| pt. TMG | 48 | 58 | 60 | 62 | 62 | 61.5 | 61 |
| pt. BG | 48.5 | 49 | 49 | 50.5 | 46.5 | 45.5 | |
| pt. CS | 51 | 49 | 50 | 52 | 52 | 50 | |
| pt. VG | 46.5 | 48 | 50 | 51 | 49.75 | 47 | |
| pt. VA | 60 | 59 | 60.5 | 62.25 | 61.25 | 60 | |
| pt. MA | 18 | 20 | 21 | 22 | 18 | 8 | 8 |
| pt. PA | 13 | 12 | 14 | 14 | 18 | 20 | 20 |
| pt. MMA | 41.5 | 43 | 44.5 | 46 | 52.5 | 55 | 53 |
| pt. RB (d) 1 | 36 | 27 | 28 | 28 | 31 | ||
| Mean | 37.3 | 37.4 | 37.9 | 39.8 | 41.4 | 39.9 | 37.1 |
| Patient | -M18 | -M12 | -M6 | Baseline | M6 | M12 | M18 |
|---|---|---|---|---|---|---|---|
| pt. CP | 72 | 68 | 60 | 73 | 95 | 66 | 57 |
| pt. SC | 33 | 36 | 41 | 46 | 56 | 69 | |
| pt. SA | 44.5 | 42 | 42.5 | 41.25 | 46.5 | 47.25 | 47 |
| pt. TG | 50 | 52 | 53 | 54 | 55 | 53 | 50 |
| pt. CF | 48 | 50 | 57 | 58 | 55 | 52 | 50 |
| pt. MS | 43 | 42 | 46 | 48 | 50 | 50 | 49 |
| pt. AU | 60 | 62 | 65 | 64 | 59 | 52 | 50 |
| pt. RC(d) 1 | 56 | 56 | 59 | 60 | 68 | ||
| pt. TMG | 68 | 82 | 94 | 100 | 96 | 90 | 88 |
| pt. BG | 50 | 48 | 49 | 50 | 41 | 38 | |
| pt. CS | 71 | 72 | 74 | 78 | 74 | 70 | |
| pt. VG | 99 | 103 | 106 | 108 | 107 | 102 | |
| pt. VA | 89 | 88 | 91 | 94 | 94 | 90 | |
| pt. MA | 57 | 60 | 61 | 62 | 60 | 55 | 54 |
| pt. PA | 44 | 46 | 48 | 48 | 50 | 51 | 51 |
| pt. MMA | 62 | 61 | 63 | 65 | 71 | 68 | 67 |
| pt. RB(d) 1 | 40 | 41 | 44 | 47 | 50 | ||
| Mean | 59.6 | 59.2 | 61.7 | 64.2 | 65.7 | 62.7 | 57.4 |
References
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.-I.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Mazzeo, A.; Russo, M.; Di Bella, G.; Minutoli, F.; Stancanelli, C.; Gentile, L.; Baldari, S.; Carerj, S.; Toscano, A.; Vita, G. Transthyretin-Related Familial Amyloid Polyneuropathy (TTR-FAP): A Single-Center Experience in Sicily, an Italian Endemic Area. J. Neuromuscul. Dis. 2015, 2, S39–S48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertz, M.A.; Mauermann, M.L.; Grogan, M.; Coelho, T. Advances in the treatment of hereditary transthyretin amyloidosis: A review. Brain Behav. 2019, 9, e01371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Obici, L.; Bartolomei, I.; Cappelli, F.; Luigetti, M.; Fenu, S.; Cavallaro, T.; Chiappini, M.G.; Gemelli, C.; Pradotto, L.G.; et al. ATTRv amyloidosis Italian Registry: Clinical and epidemiological data. Amyloid 2020, 27, 259–265. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Russo, M.; Mazzeo, A.; Stancanelli, C.; Di Leo, R.; Gentile, L.; Di Bella, G.; Minutoli, F.; Baldari, S.; Vita, G. Transthyretin-related familial amyloidotic polyneuropathy: Description of a cohort of patients with Leu64 mutation and late onset. J. Peripher. Nerv. Syst. 2012, 17, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Romozzi, M.; Bisogni, G.; Cardellini, D.; Cavallaro, T.; Di Paolantonio, A.; Fabrizi, G.M.; Fenu, S.; Gentile, L.; Grandis, M.; et al. hATTR Pathology: Nerve Biopsy Results from Italian Referral Centers. Brain Sci. 2020, 10, 780. [Google Scholar] [CrossRef]
- Müller, M.L.; Butler, J.; Heidecker, B. Emerging therapies in transthyretin amyloidosis—A new wave of hope after years of stagnancy? Eur. J. Heart Fail. 2020, 22, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Gentile, L.; Di Bella, G.; Minutoli, F.; Cucinotta, F.; Obici, L.; Mussinelli, R.; Arimatea, I.; Russo, M.; Toscano, A.; Vita, G.; et al. Description of a large cohort of Caucasian patients with V122I ATTRv amyloidosis: Neurological and cardiological features. J. Peripher. Nerv. Syst. 2020, 25, 273–278. [Google Scholar] [CrossRef]
- Stancanelli, C.; Gentile, L.; Di Bella, G.; Minutoli, F.; Russo, M.; Vita, G.; Mazzeo, A. Phenotypic variability of TTR Val122Ile mutation: A Caucasian patient with axonal neuropathy and normal heart. Neurol. Sci. 2016, 38, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Louwsma, J.; Brunger, A.F.; Bijzet, J.; Kroesen, B.J.; Roeloffzen, W.W.H.; Bischof, A.; Kuhle, J.; Drost, G.; Lange, F.; Kuks, J.B.M.; et al. Neurofilament light chain, a biomarker for polyneuropathy in systemic amyloidosis. Amyloid 2021, 28, 50–55. [Google Scholar] [CrossRef]
- Ticau, S.; Sridharan, G.V.; Tsour, S.; Cantley, W.L.; Chan, A.; Gilbert, J.A.; Erbe, D.; Aldinc, E.; Reilly, M.M.; Adams, D.; et al. Neurofilament Light Chain as a Biomarker of Hereditary Transthyretin-Mediated Amyloidosis. Neurology 2021, 96, e412–e422. [Google Scholar] [CrossRef] [PubMed]
- Vita, G.L.; Aguennouz, M.; Polito, F.; Oteri, R.; Russo, M.; Gentile, L.; Barbagallo, C.; Ragusa, M.; Rodolico, C.; Di Giorgio, R.M.; et al. Circulating microRNAs Profile in Patients with Transthyretin Variant Amyloidosis. Front. Mol. Neurosci. 2020, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Salvalaggio, A.; Coraci, D.; Cacciavillani, M.; Obici, L.; Mazzeo, A.; Luigetti, M.; Pastorelli, F.; Grandis, M.; Cavallaro, T.; Bisogni, G.; et al. Nerve ultrasound in hereditary transthyretin amyloidosis: Red flags and possible progression biomarkers. J. Neurol. 2021, 268, 189–198. [Google Scholar] [CrossRef]
- Vita, G.L.; Stancanelli, C.; Gentile, L.; Barcellona, C.; Russo, M.; Di Bella, G.; Vita, G.; Mazzeo, A. 6MWT performance correlates with peripheral neuropathy but not with cardiac involvement in patients with hereditary transthyretin amyloidosis (hATTR). Neuromuscul. Disord. 2019, 29, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Misumi, Y.; Masuda, T.; Takashio, S.; Tasaki, M.; Matsushita, H.; Ueda, A.; Inoue, Y.; Nomura, T.; Nakajima, M.; et al. Plasma growth differentiation factor 15: A novel tool to detect early changes of hereditary transthyretin amyloidosis. ESC Heart Fail. 2020, 8, 1178–1185. [Google Scholar] [CrossRef]
- Minutoli, F.; Di Bella, G.; Mazzeo, A.; Laudicella, R.; Gentile, L.; Russo, M.; Vita, G.; Baldari, S. Serial scanning with 99mTc-3, 3-diphosphono-1, 2-propanodicarboxylic acid (99mTc-DPD) for early detection of cardiac amyloid deposition and prediction of clinical worsening in subjects carrying a transthyretin gene mutation. J. Nucl. Cardiol. 2019, 1–9. [Google Scholar] [CrossRef]
- Ericzon, B.-G.; Wilczek, H.E.; Larsson, M.; Wijayatunga, P.; Stangou, A.J.; Pena, J.R.; Furtado, E.; Barroso, E.; Daniel, J.M.; Samuel, D.; et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis. Transplantation 2015, 99, 1847–1854. [Google Scholar] [CrossRef]
- Vita, G.; Vita, G.L.; Stancanelli, C.; Gentile, L.; Russo, M.; Mazzeo, A. Genetic neuromuscular disorders: Living the era of a therapeutic revolution. Part 1: Peripheral neuropathies. Neurol. Sci. 2019, 40, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Planté-Bordeneuve, V.; Lin, H.; Gollob, J.; Agarwal, S.; Betts, M.; Fahrbach, K.; Chitnis, M.; Polydefkis, M. An indirect treatment comparison of the efficacy of patisiran and tafamidis for the treatment of hereditary transthyretin-mediated amyloidosis with polyneuropathy. Expert Opin. Pharmacother. 2018, 20, 473–481. [Google Scholar] [CrossRef]
- Cortese, A.; Vita, G.; Luigetti, M.; Russo, M.; Bisogni, G.; Sabatelli, M.; Manganelli, F.; Santoro, L.; Cavallaro, T.; Fabrizi, G.M.; et al. Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: A longitudinal multicenter study in a non-endemic area. J. Neurol. 2016, 263, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Russo, M.; Gentile, L.; Toscano, A.; Aguennouz, M.; Vita, G.; Mazzeo, A. Advances in Treatment of ATTRv Amyloidosis: State of the Art and Future Prospects. Brain Sci. 2020, 10, 952. [Google Scholar] [CrossRef] [PubMed]
- Milani, P.; Mussinelli, R.; Perlini, S.; Palladini, G.; Obici, L. An evaluation of patisiran: A viable treatment option for transthyretin-related hereditary amyloidosis. Expert Opin. Pharmacother. 2019, 20, 2223–2228. [Google Scholar] [CrossRef]
- González-Duarte, A.; Berk, J.L.; Quan, D.; Mauermann, M.L.; Schmidt, H.H.; Polydefkis, M.; Waddington-Cruz, M.; Ueda, M.; Conceição, I.M.; Kristen, A.V.; et al. Analysis of autonomic outcomes in APOLLO, a phase III trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. J. Neurol. 2019, 267, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Obici, L.; Berk, J.L.; González-Duarte, A.; Coelho, T.; Gillmore, J.; Schmidt, H.H.-J.; Schilling, M.; Yamashita, T.; Labeyrie, C.; Iii, T.H.B.; et al. Quality of life outcomes in APOLLO, the phase 3 trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. Amyloid 2020, 27, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Solomon, S.D.; Adams, D.; Kristen, A.; Grogan, M.; González-Duarte, A.; Maurer, M.S.; Merlini, G.; Damy, T.; Slama, M.S.; Brannagan, T.H.; et al. Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients With Hereditary Transthyretin-Mediated Amyloidosis. Circulation 2019, 139, 431–443. [Google Scholar] [CrossRef]
- Adams, D.; Polydefkis, M.; González-Duarte, A.; Wixner, J.; Kristen, A.V.; Schmidt, H.H.; Berk, J.L.; López, I.A.L.; Dispenzieri, A.; Quan, D.; et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2021, 20, 49–59. [Google Scholar] [CrossRef]
- Luigetti, M.; Servidei, S. Patisiran in hereditary transthyretin-mediated amyloidosis. Lancet Neurol. 2021, 20, 21–23. [Google Scholar] [CrossRef]
- Russo, M.; Gentile, L.; Toscano, A.; Vita, G.; Mazzeo, A. From a misdiagnosis of anorexia nervosa to a dramatic patisiran-induced improvement in a patient with ATTRE89Q amyloidosis. Amyloid 2020, 27, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maia, L.F.; Da Silva, A.M.; Cruz, M.W.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceição, I.M.; Schmidt, H.H.-J.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef]



| Patient | Mutation | Age at Onset | Age at Diagnosis | Age at Baseline | Concomitant Treatments |
|---|---|---|---|---|---|
| pt. CP | Phe64Leu | 68 | 72 | 78 | \ |
| pt. GM (d) 1 | Phe64Leu | 63 | 68 | 68 | \ |
| pt. SC | Ala109Ser | 75 | 71 | 75 | \ |
| pt. SA | Val30Met | 59 | 62 | 68 | \ |
| pt. TG | Phe64Leu | 48 | 54 | 55 | Tafamidis |
| pt. CF | Glu89Gln | 48 | 54 | 55 | Tafamidis |
| pt. MS | Phe64Leu | 61 | 66 | 67 | Tafamidis |
| pt. AU | Phe64Leu | 75 | 77 | 77 | \ |
| pt. RC (d) 1 | Val122Ile | 65 | 69 | 70 | \ |
| pt. TMG | Glu89Gln | 56 | 57 | 58 | \ |
| pt. BG | Phe64Leu | 71 | 73 | 74 | Tafamidis |
| pt. CS | Phe64Leu | 66 | 68 | 69 | \ |
| pt. VG | Glu89Gln | 48 | 50 | 50 | \ |
| pt. VA | Val30Met | 71 | 75 | 75 | \ |
| pt. MA | Glu89Gln | 45 | 46 | 47 | \ |
| pt. PA | Thr49Ala | 55 | 56 | 57 | \ |
| pt. MMA | Thr49Ala | 46 | 47 | 50 | \ |
| pt. RB (d) 1 | Glu89Gln | 54 | 55 | 57 | \ |
| Group of Patients | Mean Monthly Change | p | |
|---|---|---|---|
| Pre-Treatment | Post-Tratment | ||
| G.1-NIS | 0.46 | 0.31 | 0.004 |
| G.2-NIS | 0.43 | −0.15 | 0.000 |
| G.3-NIS | 0.42 | −0.19 | 0.002 |
| G.4-NIS | 0.38 | −0.23 | 0.125 |
| G.5-NIS | 0.88 | −0.24 | 0.015 |
| G.6-NIS | 0.97 | −0.67 | 0.031 |
| G. 1-NIS-LL | 0.32 | 0.27 | 0.001 |
| G. 2-NIS-LL | 0.27 | −0.11 | 0.000 |
| G. 3-NIS-LL | 0.22 | −0.13 | 0.002 |
| G. 4-NIS-LL | 0.21 | −0.20 | 0.125 |
| G. 5-NIS-LL | 0.64 | −0.19 | 0.015 |
| G. 6-NIS-LL | 0.49 | −0.46 | 0.031 |
| Placebo group NIS-LL [33] | 0.32 | ||
| Tafamidis Group NIS-LL [33] | 0.16 | ||
| Group of Patients | Mean Monthly Change | p | |
|---|---|---|---|
| Pre-Treatment | Post-Tratment | ||
| G. 1-Norfolk Qol | 0.42 | 0.26 | 0.002 |
| G. 2-Norfolk Qol | 0.37 | −0.27 | 0.001 |
| G. 3-Norfolk Qol | 0.36 | −0.28 | 0.001 |
| G. 4-Norfolk Qol | 0.38 | −0.35 | 0.125 |
| G. 5-Norfolk Qol | 0.63 | −0.29 | 0.023 |
| G. 6-Norfolk Qol | 1.03 | −1.25 | 0.031 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, L.; Russo, M.; Luigetti, M.; Bisogni, G.; Di Paolantonio, A.; Romano, A.; Guglielmino, V.; Arimatea, I.; Sabatelli, M.; Toscano, A.; et al. Patisiran in hATTR Amyloidosis: Six-Month Latency Period before Efficacy. Brain Sci. 2021, 11, 515. https://doi.org/10.3390/brainsci11040515
Gentile L, Russo M, Luigetti M, Bisogni G, Di Paolantonio A, Romano A, Guglielmino V, Arimatea I, Sabatelli M, Toscano A, et al. Patisiran in hATTR Amyloidosis: Six-Month Latency Period before Efficacy. Brain Sciences. 2021; 11(4):515. https://doi.org/10.3390/brainsci11040515
Chicago/Turabian StyleGentile, Luca, Massimo Russo, Marco Luigetti, Giulia Bisogni, Andrea Di Paolantonio, Angela Romano, Valeria Guglielmino, Ilenia Arimatea, Mario Sabatelli, Antonio Toscano, and et al. 2021. "Patisiran in hATTR Amyloidosis: Six-Month Latency Period before Efficacy" Brain Sciences 11, no. 4: 515. https://doi.org/10.3390/brainsci11040515