
Modelling Parkinson’s Disease: iPSCs towards Better Understanding of Human Pathology


Physiology and CÚRAM SFI Centre for Research in Medical Devices, School of Medicine, National University of Ireland (NUI), H91 TK33 Galway, Ireland
*
Author to whom correspondence should be addressed.
Brain Sci. 2021, 11(3), 373; https://doi.org/10.3390/brainsci11030373
Submission received: 21 February 2021
/
Revised: 10 March 2021
/
Accepted: 10 March 2021
/
Published: 14 March 2021
(This article belongs to the Section Molecular and Cellular Neuroscience)
Abstract
:Parkinson’s Disease (PD) is a chronic neurodegenerative disorder characterized by motor and non-motor symptoms, among which are bradykinesia, rigidity, tremor as well as mental symptoms such as dementia. The underlying cause of Parkinson disease is degeneration of dopaminergic neurons. It has been challenging to develop an efficient animal model to accurately represent the complex phenotypes found with PD. However, it has become possible to recapitulate the myriad of phenotypes underlying the PD pathology by using human induced pluripotent stem cell (iPSC) technology. Patient-specific iPSC-derived dopaminergic neurons are available and present an opportunity to study many aspects of the PD phenotypes in a dish. In this review, we report the available data on iPSC-derived neurons derived from PD patients with identified gene mutations. Specifically, we will report on the key phenotypes of the generated iPSC-derived neurons from PD patients with different genetic background. Furthermore, we discuss the relationship these cellular phenotypes have to PD pathology and future challenges and prospects for iPSC modelling and understanding of the pathogenesis of PD.
1. Introduction
Parkinson’s disease (PD) is a complex, progressive neurological disorder characterized by degeneration of dopaminergic neurons (DA) in the substantia nigra pars compacta of the ventral mesencephalon [1]. The prevalence of PD is increasing with more than 6.1 million individuals reported globally to have PD in 2016 compared with 2.5 million in 1990 [2]. The increase in prevalence is due both to the ageing global population and associated changes in population behaviors such as smoking, decreased physical activity, and environmental factors such as air pollution [3,4]. PD occurrence is also sex-dependent with males displaying 1.5 times higher incidence compared to females [5]. The characteristic symptoms of PD are generally classified as motor (tremor, bradykinesia, and postural instability) and non-motor (dementia, depression, anxiety, fatigue, and pain) [6,7]. While the majority of PD cases are idiopathic without any clear family history, numerous genetic mutations have been found in individuals, with more rare and familial forms of PD also reported [8]. These genetic factors include autosomal dominant and recessive genes such as leucine-rich-repeat kinase 2 (LRRK2), PARK2 (encoding Parkin), PTEN-induced putative kinase (PINK1), PARK7 (encoding DJ-1), SNCA (encoding α-synuclein), and glucosidase beta acid (GBA). Each of these gene can be seen to present with variable clinical phenotypes.
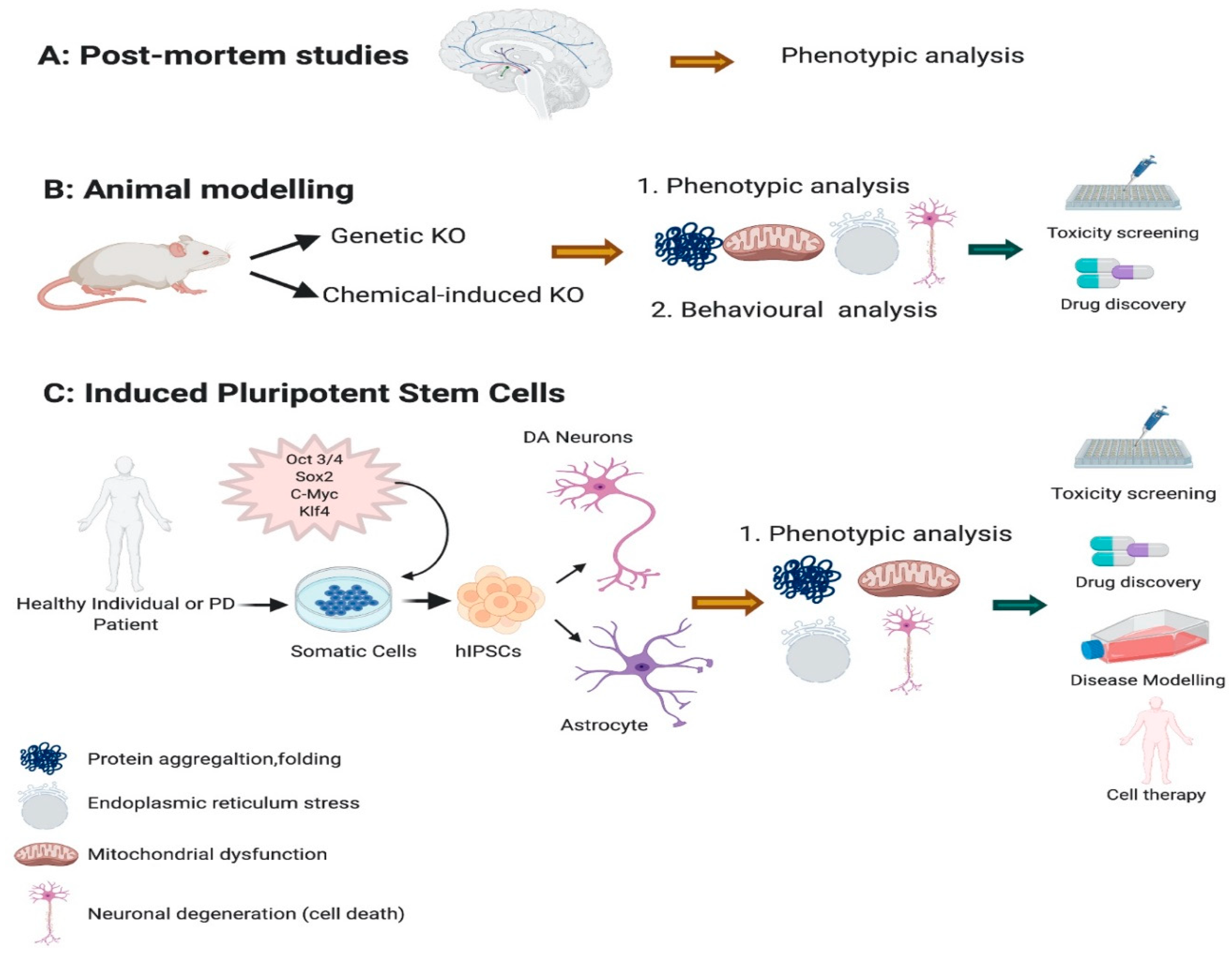
Previous studies have indicated that one of the main pathological features of PD is the formation of α-synuclein aggregation which leads to Lewy bodies development in both familial and sporadic PD [9,10]. Other key factors that are strongly associated with PD pathologies are oxidative stress, mitochondrial dysfunction, and neuroinflammation [11,12,13,14]. However, the precise pathogenesis of PD remains unknown. One of the major barriers in PD research is the lack of available brain tissue to study the problem in detail (Figure 1A). This has hampered investigations of the cellular and molecular mechanism underlying DA degeneration. As a substitute, DA ablation in animal models (Figure 1B) has been very useful, but as yet not fully recapitulating the complex phenotypes observed in PD [15,16]. Neurotoxins such as 6-hydroxydopamine (6-OHDA), and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) have been conventionally used in PD modelling [14]. These neurotoxins have been injected into animal brains, working by inhibition of complex I of the mitochondrial electron transport chain leading to oxidative stress and eventual neuronal death [17,18]. While these toxins cause neuronal damage but it does not yield aggregation of α-synuclein which is the major pathological marker of PD. Additionally, there are significant challenges in generating genetic animal models for selective degeneration of DA neurons [19].
A viable and exciting alternative approach to modelling PD seemed inevitable with the development of human pluripotent stem cells (iPSCs) [20,21] (Figure 1C). This was particularly exciting considering iPSC-derived mesencephalic DA neurons are indistinguishable from human fetal mesencephalic DA neurons in respect to their functionality, potency, maturity, and axonal outgrowth capacity [22]. While iPSCs represent a patient’s complete genomic background and as such provide a unique platform for modelling in particular, specific genes associated with disease, the potential of iPSCs to reveal important insights on the mechanism of PD pathogenesis is limited to date.
A number of recent reviews have focused on the advantage of using iPSCs in PD modelling which have contributed extensively to our knowledge [23,24]. However, a more comprehensive and detailed study of iPSC modelling and their related phenotypes to a specific genomic background and their ultimate relation to human pathology is lacking. This review presents the latest data on the modelling of iPSC-derived DA neurons with specific mutations in the key PD associated genes SNCA, LRRK2, PARK2, PINK1, GBA, and DJ-1. We will focus on the most common associated phenotypes that are strongly correlated and connected in human PD pathology: mitochondrial dysfunction, mitochondrial DNA damage, oxidative stress, and accumulation and aggregation of α-synuclein.
2. iPSC Generation and Differentiation of Dopaminergic Neurons
iPSC technology involves the conversion of human somatic cells into proliferative pluripotent stem cells, essentially equivalent to human embryonic stem cells [21]. The initial discovery was reported 15 years ago using mouse embryonic fibroblasts using the forced expression of so-called Yamanaka factors (Oct4, Sox2, Klf4, c-myc) [25]. The following year the same group further advanced this approach when they demonstrated that adult human dermal fibroblasts could also be reprogrammed into iPSCs [26]. These studies revolutionized stem cell research, making it possible to consider the use of patient-specific iPSC-derived cells to treat and/or investigate a myriad of human diseases.
In the context of PD, considering the loss of DA neurons as the primary pathological hallmark, the initial goal with iPSCs was to develop differentiation protocols to generate a homogenous population of tyrosine hydroxylase-positive (TH+) dopaminergic neurons. This has been achieved using numerous different approaches (reviewed in [27,28]), such as forced expression of the transcription factors ASCL1, NURR1 and LMX1A [29], dual SMAD inhibition-based floor plate (FP) protocols [30,31,32] or transient expression of transcription factors by means of AVV vectors [32]. To date more than 385 iPSC-derived neuronal lines from PD patients with different genetic mutations have been successfully generated across many independent laboratories all over the world (reviewed in [33]). In this review we synthesize the available data on phenotype characterization as the starting point in securing the iPSC platform for expanding a new paradigm for cellular therapy approaches and drug development and screening for both preclinical and clinical studies.
3. Modelling PD Using iPSCs
There has been remarkable advances in the discovery of genetic mutations associated with PD in recent years [34]. Familial PD collectively accounts for only 10% of all PD cases while the remainder of PD cases have unknown etiology [35,36]. Patient-specific iPSC-derived DA neurons with specific mutations allows the underlying mechanisms of a particular mutation to be investigated in depth. Here, we report on the available data based only on human iPSC-derived neuronal models, investigating specific mutation-associated phenotypes of PD.
3.1. iPSC Modelling of SNCA Mutation and Associated Phenotypes
The first reported mutation associated with autosomal-dominant PD was that of α-synuclein [37]. α-synuclein is involved in numerous important cellular functions such as modulation of intracellular vesicle trafficking [38], dopamine metabolism [39], microtubule nucleation and proliferation [40], as well as the regulation of synaptic vesicle recycling [41]. There are numerous specific mutations associated with SNCA-related PD pathology including A30P, G51D, E46K, A53T, and A53E [42,43,44,45]. The severity of symptoms for PD is proportional to the number of SNCA copies deleted. In human iPSC-derived neurons the most commonly studied alterations in SNCA are the A53T mutation and multiplication of SNCA as they are the most common mutations associated with PD. Alterations in α-synuclein physiology results in a myriad of cellular changes must commonly mediated through mitochondrial dysfunction and oxidative stress. These changes and abnormal aggregation of α-synuclein not only induce neuronal loss but are also seen to prevent neuronal regeneration.
3.1.1. SNCA Alteration Disrupts Normal Mitochondrial Function in iPSC-Derived Neurons
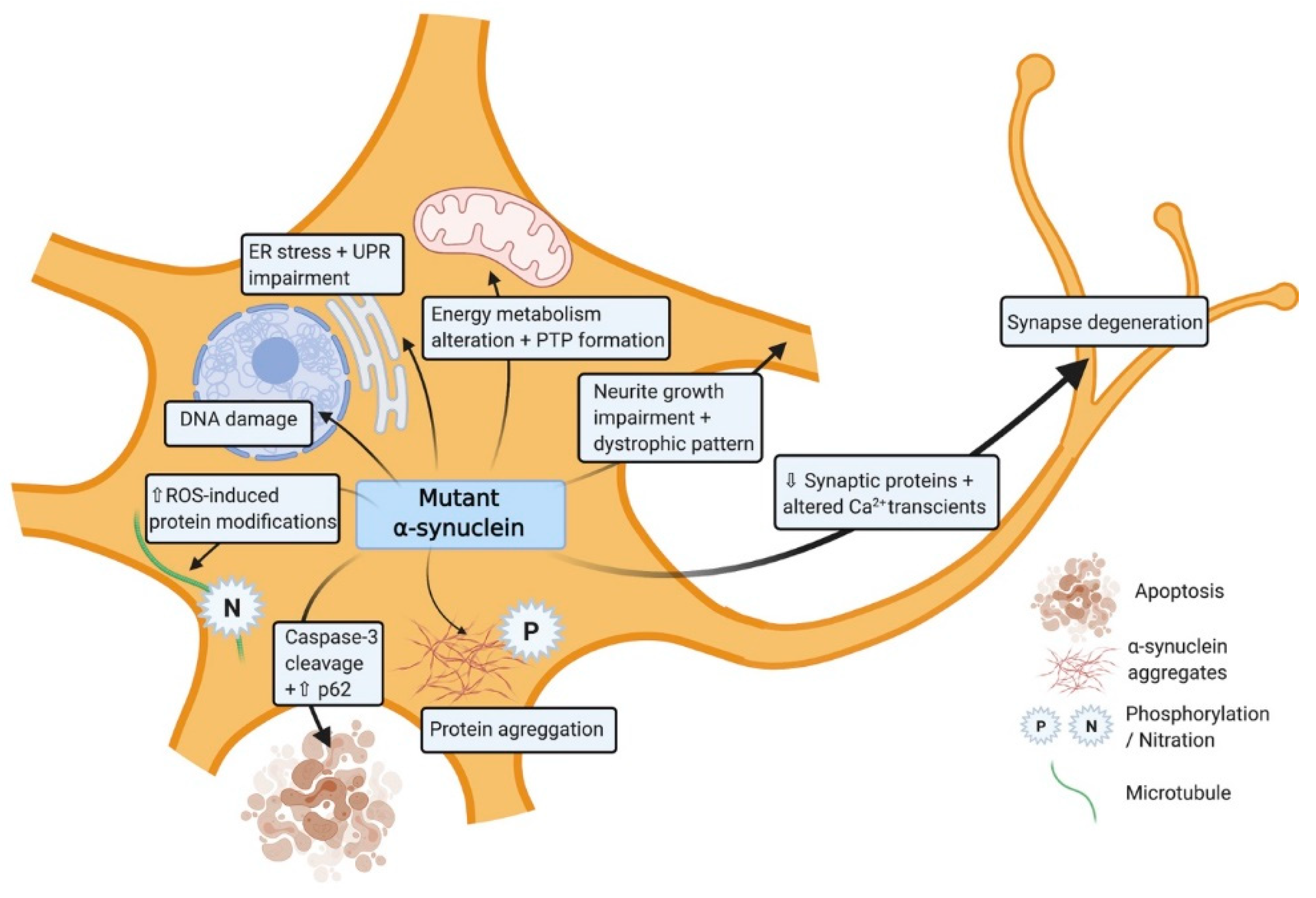
Mitochondrial impairment is very common in all SNCA altered iPSC-derived neurons [46,47] (Table 1). For SNCA-triplication iPSC-derived neuronal progenitors (NPCs), mitochondrial dysfunction presents as altered energy metabolism associated with impaired basal and/or maximal respiration capacity, or ATP production in [48] (Figure 2). This is also seen in SNCA-A53T iPSC-derived neuroepithelial stem cells and neurons [46,49,50]. Certain pathologies result in structures such as permeability transition pores (PTP) appearing in the inner membrane of damaged mitochondria. In SNCA-triplication iPSC-derived neurons exposed to low concentrations of ferutinin or laser-induced ROS, suggesting SNCA alterations results in higher susceptibility to PTP formation in comparison to controls [51]. In addition, mitochondrial axonal transport is decreased in SNCA-duplication and also in A53T iPSC-derived neurons leading to energy deficits and synapse degeneration [52,53]. Upregulation of Miro1, a key protein in mitochondrial transport has also been detected in SNCA-A53T iPSC-derived neurons, suggesting mitophagy delay [54]. Furthermore, mitochondrial morphology is also altered in mutated neurons, manifesting as more circular and unbranched in structure with significant reductions in mitochondrial membrane potential [50].
Several studies have demonstrated that SNCA mutated neurons have increased sensitivity to mitochondrial toxin-induced oxidative stress [49,54] which can be further aggravated by metal ion interactions [55]. Furthermore, the expression level of oxidative stress markers such as DNAJA1, HMOX2, UCHL1, and HSPB1 which have neuroprotective capacity are also significantly dysregulated [56]. In line with this endogenous antioxidant pathways are elevated through increase activity of catalase or PGC-1α as a compensatory mechanism in response to the oxidative stress [50]. SNCA-A53T iPSC-derived neurons also generate higher levels of nitrous oxide (NO) after exposure to low levels of agrichemicals, resulting in disruption of microtubules [52] (Figure 2). These data support the growing evidence that exposure to certain environmental agents can significantly contribute to the PD pathology in those with alterations in SNCA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
SNCA-mutated iPSC-derived neuronal phenotypes.
| Reference | Number of Cohorts | Type of Mutation | Cell Type | Phenotype |
|---|---|---|---|---|
| [48] | 1 PD line vs. 2 control lines | Autosomal dominant Triplication | iPSC-derived neuronal progenitor cells | 1. Elevated α-synuclein levels 2. Decreased neuronal activity 3. Increased autophagy 4. Mitochondrial dysfunction 5. Increased oxidative stress |
| [57] | 1 PD line vs. 1 control line | Autosomal dominant Triplication | iPSC-derived cortical neurons | 1. Elevated α-synuclein levels 2. Endoplasmic Reticulum stress |
| [58] | 1 PD line vs. 2 control lines | Autosomal dominant Triplication | iPSC-derived DA neurons | 1. Elevated α-synuclein levels 2. Impairment in neuronal development 3. Impairment in synaptic transmission 3. Increased autophagy |
| [47] | 1 PD line vs. 1 control line | Autosomal dominant Duplication | iPSC-derived midbrain DA and Cortical projection neurons | 1. Elevated α-synuclein levels 2. Increased α-synuclein aggregation 3. Increased phosphorylated α-synuclein 4. Increased oxidative stress |
| [56] | 1 PD line vs. 1 control line | Autosomal dominant Triplication | iPSC-derived DA neurons | 1. Increased α-synuclein aggregation 2. Increased oxidative stress |
| [55] | 1 PD line vs. 1 control line | Autosomal dominant Triplication | iPSC-derived cortical neurons | 1. Elevated α-synuclein 2. Increased oxidative stress |
| [59] | 1 PD line | Autosomal dominant Triplication | iPSC-derived DA progenitor cells | 1. Elevated α-synuclein levels 2. Increased oxidative stress 3. Increased cell death |
| [60] | 1 PD line vs. 1 control line | Autosomal dominant Triplication | iPSC-derived DA neurons | 1. Elevated α-synuclein levels 2. Altered Calcium signalling |
| [61] | 1 PD line vs. 1 control line | Autosomal dominant Triplication | iPSC-derived DA and basal forebrain cholinergic neurons | 1. Elevated α-synuclein levels 2. Increased α-synuclein aggregation 3. Increased DNA damage |
| [62] | 1 PD line vs. 3 control lines | Autosomal dominant Triplication | iPSC-derived DA neurons | 1. Elevated α-synuclein levels 2. Impairment in neuronal development 3. Increased α-synuclein phosphorylation 4. Increased cell death and apoptosis |
| [63] | 1 PD line vs. 1 control line | Autosomal dominant Triplication | iPSC-derived neuronal progenitor cells | 1. Elevated α-synuclein levels 2. Increased DNA damage |
| [51] | 1 PD line vs. 1 control line vs. 1 isogenic control line | Autosomal dominant Triplication | iPSC-derived cortical neurons | 1. Elevated α-synuclein levels 2. Mitochondrial dysfunction |
| [53] | 1 PD line vs. 2 control line | Autosomal dominant Duplication | iPSC-derived cortical forebrain glutamatergic neurons | 1. Elevated α-synuclein levels 2. Increased α-synuclein aggregation 3. Mitochondrial transport impairment |
| [49] | 1 PD line vs. 1 isogenic control line | Autosomal dominant A53T | iPSC-derived A9 DA neurons | 1. Mitochondrial dysfunction 2. Increased oxidative stress 3. Increased cell death and apoptosis 4. Neuronal maturation impairment |
| [46] | 2 PD lines vs. 1 control line | Autosomal dominant A53T and A30T | iPSC-derived neural stem cells | 1. Mitochondrial dysfunction |
| [64] | 2 PD line vs. 1 control line | Autosomal dominant A53T | iPSC-derived DA, GABAergic and glutaminergic neurons | 1. Altered synaptic activity 2. Increase α-synuclein aggregation 3. Impairment in Neuronal development |
| [52] | 1 PD line vs. 1 isogenic control line | Autosomal dominant A53T | iPSC-derived A9 DA neurons | 1. Mitochondrial transport impairment 2. Alteration in microtubules function |
| [50] | 4 PD lines vs. 3 control line | Autosomal dominant A53T and Triplication | iPSC-derived DA neurons | 1. Elevated α-synuclein levels 2. Endoplasmic Reticulum stress 3. Mitochondrial dysfunction 5. Increased autophagy 6. Increased oxidative stress |
| [65] | 1 PD line vs. 1 isogenic control line | Autosomal dominant A53T | iPSC-derived DA neurons | 1. Increased α-synuclein aggregation |
| [66] | 1 PD line vs. 1 control line | Autosomal dominant A53T | iPSC-derived DA neurons | 1. Elevated α-synuclein levels 2. Increased α-synuclein aggregation 3. Impairment in Neuronal development |
| [54] | 3 PD lines vs. 3 control lines | Autosomal dominant A53T and triplication | iPSC-derived DA neurons | 1. Elevated α-synuclein levels 2. Mitophagy impairment 3. Increased oxidative stress |
| [67] | 2 PD lines vs. 1 control line vs. 1 isogenic control line | Autosomal dominant A53T and triplication | iPSC-derived DA neurons | 1. Lysosomal dysfunction 2. Increased α-synuclein aggregation |
3.1.2. SNCA Alteration Leads to Protein Aggregation and Cellular Damage in iPSC-Derived Neurons
Lewy bodies formation as a consequence of the abnormal aggregation of α-synuclein is a major hallmark in human PD pathology and more specifically to DA neurons [62]. iPSC-derived neurons with SNCA mutations also exhibit higher levels of α-synuclein phosphorylation and increased α-synuclein aggregate formation (Figure 2; Table 2). In A53T iPSC-derived neurons reports describe posttranslational change in α-synuclein, such as Ser129 phosphorylation or ubiquitination leading to the formation of large aggregates and Lewy Bodies [64,66]. In addition, A53T mutations result in increased interactions of elevated α-synuclein levels with essential transcription factors, ribonucleoproteins, and ribosomal proteins [49,66]. This aggregation instigates a dysregulation in protein production and transcription-related mRNAs in SNCA-A53T iPSC-derived neurons [60,65,68]. This excessive accumulation of aberrant misfolded protein aggregates within the cell results in endoplasmic reticulum stress and activation of the unfolded protein response (UPR) which is also observed in both SNCA-triplication and A53T iPSC-derived neurons (Figure 2) [50,57]. A reduction in the level of a key factor in UPR pathway IREα, supports the damaging consequences of these aggregations in SNCA-triplication and A53T iPSC-derived neurons [50,57]. In the related lysosomal stress pathway, α-synuclein deactivation of the SNARE protein ykt6 leads to an impaired physiological response to lysosomal stress, in SNCA mutated iPSC-derived neurons [67]. The exposure of SNCA-triplication iPSC-derived neurons to toxins results in an elevation in cell death, caspase-3 cleavage [62] and the presence of autophagosomes [58]. This suggest that iPSC-derived neurons with SNCA mutations show higher vulnerability to toxins and undergo apoptosis. Intron 1 methylation of SNCA-triplication gene has shown to rescue this phenotype and improve cell viability [59]. Similar processes have been reported in SNCA-A53T iPSC-derived neurons by an increase in autophagy-related proteins such as p62 or the autophagosome marked LC3 after exposure to agrochemicals [49,50]. α-synuclein aggregation not only induces neuronal loss but also prevents neuronal regeneration. Significant downregulation in specific DA differentiation genes such as DLK, GABABR2, NURR1, and TH have been reported in SNCA-A53T iPSC-derived neurons [58]. Furthermore neurite growth, length are impaired with appearance of dystrophic neurite patterns in SNCA-triplication iPSC-derived neurons [66]. Neurite formation at the early stages present with many degeneration features such as swollen varicosities and spheroid inclusions leading to reduction and alteration of the number of synaptic contacts and activity respectively in SNCA-A53T iPSC-derived neurons [64]. Spontaneous Ca2+ transients with larger mean amplitude have been shown as a consequence of impairment in synaptic activity [66].
3.2. iPSC Modelling of LRKK2 and Associated Phenotypes
LRRK2 is a multi-domain protein exhibiting both kinase and GTPase functions located in the PARK8 locus on chromosome 12 [69]. LRRK2 is implicated as a significant genetic contributor to the development of autosomal dominant familial PD as well as some cases of idiopathic PD [70,71]. To date around 20 different LRRK2 mutations have been linked to PD pathophysiology [72]. LRRK2 is susceptible to several missense mutations including the LRRK2 G2019S, I2020T, Y1699C, and R1441C heterozygous mutations [73,74]. G2019S is the most commonly occurring mutation and is associated with 4% of familial PD and 1% of idiopathic PD cases [75]. LRKK2 is recognized to have pleiotropic roles across multiple domains including neurite outgrowth [76], modulation of synaptic vesicle endocytosis [77,78] and mitochondrial function and mitophagy [79,80].
3.2.1. LRKK2 Alterations Result in Mitochondrial Dysfunction in iPSC-Derived Neurons
Defective mitochondria are found to accumulate in the axons of LRRK2 mutated iPSC-derived DA neurons as a result of disruption in mitophagy [81]. In addition, there is increased level of mitochondrial DNA in LRRK2 R1441C iPSC-derived neurons compared to control neurons [82]. Mitochondrial impairments have also been observed in LRRK2 G2019S human neuroepithelial stem cells (NESCs), suggesting a defective mechanism at earlier stages of neuronal development [83]. Furthermore, LRRK2-mutated iPSC-derived neurons show higher mitochondria mobility including more bidirectional movement, suggesting that LRRK2 mutations can actively encourage mitochondrial evasion of mitophagy [84]. One study reports that defects in mitochondrial biogenesis and energetics are associated with low levels of nicotinamide adenine dinucleotide (NAD+) in LRRK2-mutated iPSC-derived neurons [85].
3.2.2. LRKK2 Alteration Promotes α-Synuclein Aggregation in iPSC-Derived Neurons
Data from G2019S-mutated DA neurons have indicated to a role for LRKK2 in α-synuclein pathology, resulting in increased endogenous α-synuclein aggregation [86,87], accelerating neuronal loss [88]. Accumulation of α-synuclein additionally affects LRRK2 gain of function mutations [79] suggesting that LRRK2 mutations may confer increased susceptibility to PD through SNCA [89]. In addition, iPSC-derived astrocytes with LRRK2 mutations display increased α-synuclein aggregation, leading to cell death [90]. Chemical amelioration of chaperone mediated autophagy has been shown to rescue astrocytes and DA neurons via the clearance of α-synuclein [90].
iPSC-derived DA neurons with LRRK2 G2019S and R1441C mutations have impaired development and differentiation capability [91,92,93,94,95,96,97,98]. LRRK2 mutated DA neurons have additionally altered neurite outgrowth and aggregation of microtubules as well as altered calcium dynamics in vitro [99]. Studies have continually implicated endo-lysosomal system dysfunction in PD pathogenesis with the serine/threonine kinase activity of lark2 as a key factor in endocytosis of synaptic vesicles [100]. iPSC ventral midbrain neurons with G2019S mutations have impaired endocytosis [101]. Consistent with this, central endocytosis proteins including dynamin-1, and various Rab proteins are significantly downregulated in iPSC-derived neurons [101,102]. Downregulation of these proteins is central to the endocytosis pathways, leading to defective clathrin-mediated synaptic vesicle endocytosis that may confer PD pathogenesis by dysregulation in these pathways [102].
Table 2.
LRRK2-mutated iPSC-derived neuronal phenotypes.
| Reference | Number of Cohorts | Type of Mutation | Cell Type | Phenotype |
|---|---|---|---|---|
| [88] | 1 PD and isogenic KO line vs. isogenic controls | LG2019S | iPSC-derived cortical neurons | 1. Increased neuronal degeneration 2. Degeneration-associated neuroinflammation |
| [83] | 3 PD and 2 isogenic KO lines vs. 4 controls and isogenic lines | G2019S | iPSC-derived neural stem cells | 1. Deficient dopaminergic differentiation 2. Mitochondrial dysfunction 3. Increased cell death |
| [91] | 3 PD lines vs. 3 control lines | G2019S | iPSC-derived DA neurons | 1. Impairment in neuronal development |
| [102] | 8 PD lines vs. 4 control lines | G2019S and R1441C | iPSC-derived DA neurons | 1. Decreased Endocytosis |
| [84] | 2 PD lines vs. 2 control lines | G2019S, R1441C | iPSC-derived DA neurons | 1. Mitochondrial dysfunction. |
| [62] | 6 PD lines vs. 3 control lines | G2019S | iPSC-derived DA neurons | 1. Increased oxidative stress 2. Increased neuronal degeneration 3. Increased α-synuclein aggregation |
| [90] | 2 PD lines vs. 3 control lines vs. 1 isogenic control line | G2019S, | iPSC-derived DA neurons | 1. Increased neuronal degeneration |
| [96] | 4 PD lines vs. 4 control lines | G2019S | iPSC-derived DA neurons | 1. Increased neuronal degeneration 2. Increased α-synuclein aggregation 3. Increased autophagy |
| [81] | 6 PD lines vs. 3 control lines | G2019S | iPSC-derived DA neurons | 1. Decreased neuronal development 2. Increased neuronal degeneration |
| [88] | 3 PD lines vs. 3 control lines | G2019S | iPSC-derived DA organoids | 1. Mitophagy impairment |
| [103] | 1 PD line | G2019S | iPSC-derived neural stem cells | 1. Increased α-synuclein aggregation |
| [92] | 8 PD lines vs. 5 control lines vs. 4 gene edited controls | G2019S | iPSC-derived DA neurons | 1. Increased mitochondrial dysfunction 2. Increased oxidative Stress |
| [97] | 2 PD lines vs. 2 control and H1, H9 lines | G2019S | iPSC-derived neural stem cells | 1. Altered calcium signalling 2. Impaired neuronal development |
| [93] | 4 PD lines vs. 4 control line | G2019S | iPSC-derived DA neurons | 1. Nuclear envelope impairment 2. Increased proteasome stress |
| [94] | 3 PD lines vs. 1 control line | G2019S | iPSC-derived DA neurons | 1. Impaired neuronal development 2. Decreased mitophagy |
| [87] | 3 PD lines vs. 4 control lines | G2019S | iPSC-derived DA neurons | 1. Impaired neuronal Development |
| [104] | 4 PD lines vs. 7 control lines | G2019S | iPSC-derived DA neurons | 1. Increased α-synuclein aggregation 2. Increased oxidative stress |
| [95] | 1 PD and 1 isogenic KO line vs. 1 control and 1 isogenic control line | G2019S | iPSC-derived DA neurons | 1. Increased apoptosis and neuronal cell death 2. Decreased mitosis |
| [80] | 2 PD lines vs. 4 control lines | G2019S | iPSC-derived DA neurons | 1. Impaired Neuronal development 2. Decreased α-synuclein phosphorylation |
| [82] | 12 PD lines vs. 3 control lines | G2019S, R1441C | iPSC-derived DA neurons | 1. Increased Tau and α-synuclein aggregation. 2. Impaired Neuronal development |
| [99] | 3 PD lines vs. 3 control lines | G2019S | iPSC-derived sensory and DA neurons | 1. Mitochondrial dysfunction |
| [85] | 3 PD lines vs. 3 control lines | G2019S | iPSC-derived sensory, Glutamatergic and DA neurons | 1. Large microtubule-containing neurite aggregations 2. Altered calcium signalling |
| [83] | 3 PD and 2 isogenic KO lines vs. 4 control lines and 2 isogenic control | G2019S | iPSC-derived neural stem cells | 1. Mitochondrial Dysfunction |
| [105] | 2 PD lines vs. 2 control lines | G2019S | iPSC-derived DA neurons | 1. Mitochondrial Dysfunction 2. Altered mitophagy |
3.3. iPSC Modelling of PARK2 Mutations and Associated Phenotypes
Parkin is an E3 ubiquitin ligase, residing in the cytosol, that functions in the ubiquitin proteasome pathway [106,107]. PARK2 which is located on the 6q25.2–27 chromosome encodes parkin and is the most frequent gene mutation associated with autosomal recessive early onset familial PD [107]. Fifty percent of all PD cases under the age of 45 are associated with parkin mutations [108] (Table 3). Mutations in parkin range from small deletions and base pair substitution to large deletions spanning hundreds of nucleotides [109]. Parkin has been primarily shown to be important in maintaining normal mitochondrial function and integrity [110].
PARKIN Mutations Result in Mitochondrial Dysfunction and Oxidative Stress in iPSC-Derived Neurons
Mitochondrial dysfunction, abnormal morphology, and impaired mitochondrial homeostasis are some of the key features displayed by parkin iPSC-derived DA neurons. These neurons exhibit swollen cristae and a highly condensed matrix in the inner mitochondrial membrane (IMM), with abnormal mitochondrial morphology directly affecting function [111] as well as an elevation in the number of enlarged mitochondria [112,113]. Pyruvate kinase M and 14-3-3 epsilon are among the most dysregulated mitochondrial proteins associated with parkin mutated neurons, and this pair have also been consistently shown to be changed in post mortem brain tissues of PD patients [113,114,115].
The culmination of mitochondrial dysfunction is thought to be an increase in oxidative stress, leading to dopamine oxidation [116]. Under normal physiological conditions the transcription of mitochondrial enzymes such as monoamine oxidases (MAO) A and B are limited, as parkin suppresses dopamine-induced oxidative stress [117,118]. However, the levels of MAO-A and B were found to be significantly increased in parkin mutated iPSC-derived DA neurons, suggesting an escalation in dopamine-induced oxidative stress [118]. Increased oxidative stress is a consistent factor observed in numerous independent studies of parkin mutations [111,112,119]. Furthermore, anti-oxidative proteins are significantly reduced in parkin mutated iPSC-derived DA neurons [120]. Conversely, Nrf2, a protein promoting antioxidant gene expression is significantly enhanced in parkin mutated iPSC-derived neurons [111]. Related to this oxidative stress inducing environment, there is a marked decrease in dopamine uptake and increased levels of spontaneous dopamine release in iPSC-derived DA neurons with parkin mutations [121]. This increased spontaneous leak of dopamine is observed in both heterozygous and homozygous forms of parkin neurons, independent of intracellular Ca2+ [121] (Figure 3). Additionally, the number of correctly folded and trafficked dopamine transporters (DAT) are significantly decreased [121]. Thus, parkin is presumed to mitigate dopamine oxidation and control the transmission of dopamine. A further study has demonstrated that the activation of dopamine D1-class receptors in parkin neurons leads to large rhythmic outbursts of spontaneous excitatory postsynaptic currents (sEPSCs) [122] (Figure 3). These rhythmic outburst of sEPSCs resemble oscillatory activities observed within basal ganglia neurons in PD. Overexpression of parkin in this same study resulted in a significant rescue of iPSC-derived neurons, returning oscillatory activities to normal levels [122]. These data show that parkin mutations enhance abnormal dopaminergic modulation and release in neurons.
Table 3.
PARK2-mutated iPSC-derived neuronal phenotypes.
| Reference | Number of Cohorts | Type of Mutation | Cell Type | Phenotype |
|---|---|---|---|---|
| [119] | 6 PD patient lines vs. 3 control lines | Exon 2–4 or 6–7 deletions | iPSC-derived DA neurons | 1. Increased Oxidative stress 2. Mitophagy impairment |
| [122] | 3 PD patient lines vs. 3 control lines | Exon 3–5 or R42P deletions | iPSC-derived DA neurons | 1. Dopamine dysregulation |
| [111] | 2 PD patient lines vs. 2 control lines | Exon 2–4 or Exon 6–7 deletions | iPSC-derived DA neurons | 1. Increased oxidative stress 2. Mitochondrial dysfunction 3. Increase α-synuclein aggregation |
| [123] | 4 PD patient lines with 1 control line | Exon 3–4, R275W or R42P deletions | iPSC-derived DA neurons | 1. Mitochondrial dysfunction 2. Increase α-synuclein aggregation |
| [124] | 1 PD patient line vs. 1 control line | Del202-203AG and IVS1+1G/A | iPSC-derived DA neurons | 1. Increased cell death |
| [113] | 2 Isogenic mutated PD lines vs. 1 control line | Exon 2 deletion | iPSC-derived DA neurons | 1. Mitochondrial dysfunction |
| [118] | 2 PD lines vs. 2 control lines | Exon 4 deletion | iPSC-derived DA neurons | 1. Dopamine dysregulation 2. Increased oxidative stress |
| [125] | 2 PD lines vs. 2 control lines | Exon 2–4 deletion | iPSC-derived DA neurons | 1. Mitochondrial dysfunction |
| [126] | 2 Isogenic mutated iPSC lines vs. 1 control line | Exon 2 deletion | iPSC-derived DA neurons | 1. Lysosomal dysfunction |
| [120] | 1 PD line vs. 1 control line | Exon 5 deletion | iPSC-derived DA neurons | 1. Increase α-synuclein aggregation 2. Reduced level of anti-oxidative proteins |
| [127] | 3 PD lines vs. 3 control lines | Exon 7 deletion, c.1072delT or Exon 1 deletion and c.924C>Tor c.1072delT | iPSC-derived DA neurons | 1. Mitochondrial dysfunction |
| [112] | 2 PD lines vs. 2 control lines | c.1366C.T and c.1072Tdel | iPSC-derived DA neurons | 1. Dopamine dysregulation 2. Increase cell death 3. Increase α-synuclein aggregation |
3.4. iPSC Modelling of PINK1 Mutations and Associated Phenotypes
Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) mutations are the second most frequent cause of early-onset PD and is involved in an autosomal recessive familial form of PD [128]. PINK1-related PD usually appears in the third or fourth decade of life, and like other recessive early onset forms, presents as a slow progression of the disease with a consistent response to levodopa treatment [129]. Forty-two different mutations have been found within the exons of the PINK1 in both heterozygous and homozygous states with Q456X is the most prevalent form of PINK1-related PD mutation [130,131] (Table 4). PINK1 consists of a C-terminal kinase domain and a mitochondrial targeting sequence at the N-terminus. Cytosolic PINK1 is released by truncation of the N-terminal portion of the gene in a proteasome-dependent manner [132,133]. PINK1 has been shown to have an essential role in mitochondrial function, calcium homeostasis, autophagy/mitophagy, protection from stress, and protein misfold [134,135,136].
PINK1 Mutations Result in Loss of Mitochondrial Function and Increases Reactive Oxygen Species Generation in iPSC-Derived Neurons
PINK1 mutations are thought to impair mitochondrial function due to a loss of function, based on the upregulation of PGC-1α and an increase in mitochondrial DNA (mtDNA) copy number (Figure 3) [137]. Under normal physiological conditions, mitochondrial damage activates PINK1 kinase activity, and activated PINK1 phosphorylates ubiquitin at a conserved residue of Ser65 [138]. Parkin cooperates with PINK1 in the phosphorylation process, preparing the damaged mitochondria for lysosomal and proteasomal targeted degradation [139]. Pathogenic mtDNA mutations are found widely in individuals with PD, resulting in mitochondrial dysfunction. Siebler et al., demonstrated that while the level of mtDNA is decreased in wild-type neurons, it remained unchanged in PINK1−/− iPSC-derived neurons upon mitochondrial depolarization, suggesting an increase accumulation of mitochondria DNA dues to loss of PINK function [137]. PINK1+/− iPSC-derived neurons show a significant number of cells with fragmented mitochondria suggestion an alteration in mitochondrial cycling dynamics towards increased organelle fission [140] (Figure 3).
In addition, iPSC-derived neurons with PINK1 mutations show a significant reduction in the level of endogenous parkin levels and are unable to initiate mitophagy due to dysfunction in ubiquitination pathways [141]. Furthermore, the level of Phosphorylated-Ser65-Ub signals are significantly reduced within iPSC-derived TH+ neurons with PINK1 p.G411S mutation [125,142]. Consistent with this, the recruitment of parkin to mitochondria is impaired upon depolarization of mitochondria in PINK1 mutated iPSC-derived DA neurons [137]. Overexpression of wild type PINK1 in these DA neurons restored the translocation of parkin to mitochondria [137]. These studies highlighted the vital role of PINK1 in mitochondrial function and pathogenesis of PD.
One of the major indicators of mitochondrial dysfunction is the generation of reactive oxygen species (ROS) which in turn leads to cell damage due to oxidative stress [143]. Significant damage to lipids, proteins, and nucleic acids have been identified in fibroblast of s carrying PINK mutation [144]. Further study demonstrated that PINK1 deficiency results in an increased basal ROS in both the mitochondria (Figure 3) and cytoplasm, leading to increased oxidative stress in iPSC-derived DA neurons [145]. One of the key mechanisms in detoxification and prevention of ROS associated mitochondrial damage in the cytoplasm is the oxidation of glutathione (GSH). iPSC-derived neurons with PINK1 Q456X mutation display reduced GSH levels and showed increase vulnerability after exposure to low concentrations of valinomycin, concanamycin A, MPP+ and hydrogen peroxide (all promotors of oxidative stress) in comparison to control neurons [84].
Table 4.
PINK1-mutated iPSC-derived neuronal phenotypes.
| Reference | Number of Cohorts | Type of Mutation | Cell Type | Phenotype |
|---|---|---|---|---|
| [137] | 3 PD lines vs. 1 control line | c.1366C>T, c.509T>G | iPSC-derived DA neurons | 1. Mitochondrial dysfunction |
| [84] | 5 PD lines vs. 2 control lines | Q456X, R1441C | iPSC-derived DA neurons | 1. Increase in oxidative stress 2. Mitochondrial dysfunction |
| [141] | 1 PD line vs. 1 control line | V170G | iPSC-derived DA neurons | 1. Impairement in mitophagy |
| [140] | 7 PD lines vs. 5 control lines | Exon 4 or 7 deletion | iPSC-derived DA neurons | 1. Dysregulation of LRKK2 levels 2. Mitochondrial dysfunction |
3.5. iPSC Modelling of GBA Mutation and Associated Phenotypes
The GBA gene is located on chromosome 1 (1q21) and encodes for a lysosomal glucocerebrosidase enzyme (GCase), hydrolysing the glucosylceramide (GlcCer) into ceramide and glucose [146]. GBA mutation was first associated with PD approximately 14 years ago as a result of a PD like phenotype in PD with Gaucher disease [147]. The onset of PD with GBA mutations have been reported to be 30% at 80 years, with 9.1% of GBA carriers develop PD [148]. The main GBA mutations are p.N370S and p.L444P, enhancing the Lewy bodies formation, leading to PD and dementia [149]. Both mutations exhibit a reduced GCase activity that trigger an abnormal accumulation of α-synuclein [146]. Moreover, GBA has a key role in mitochondrial function and autophagy [150].
3.5.1. GBA Mutations Result in Disrupted Mitochondrial Function in iPSC-Derived Neurons
Mutations in all pN370S, pL444P, and RecNcil GBA iPSC-derived neurons have altered mitochondria morphology and function (Table 5). Morphological assessment using transmission electronic microscope (TEM) has demonstrated that mitochondrial have larger diameters and altered cristae in iPSCs-derived DA neurons in comparison to controls. Alterations have been additionally observed as a result of reduced oxygen consume rate (OCR) and complex I activity (CI) in GBA mutated neurons, associating with increased mtROS levels [151]. The ratio between long and short isoforms of fusion protein OPA1 are elevated in GBA-mutated iPSC-derived neurons, suggesting disruptions in mitophagy process, and subsequently in mitochondrial dynamics [151]. Furthermore, expression of mitophagy adaptor proteins such as BNIP3L/NIX have been significantly reduced in GBA mutated iPSC-derived neurons [151].
Table 5.
GBA-mutated iPSC-derived neuronal phenotype.
| Reference | Number of Cohorts | Type of Mutation | Cell Type | Phenotype |
|---|---|---|---|---|
| [150] | 3 PD lines vs. 3 control lines | Heterozygous N370S | iPSC-derived DA neurons | 1. Lysosomal dysfunction 2. Autophagy dysfunction 3. Endoplasmic Reticulum stress |
| [152] | 1 PD line vs. 1 control line | Heterozygous N370S | iPSC-derived DA neurons | 1. α- synuclein aggregation 2. Lipid dyshomeostasis |
| [151] | 4 PD lines vs. 2 isogenic control lines | Heterozygous N370S, L444P and RecNciI | iPSC-derived DA neurons | 1. Mitochondrial dysfunction 3. Lipid dyshomeostasis 4. Alteration in mitochondria and lysosome Colocalization 5. Endoplasmic reticulum stress |
| [153] | Homozygous N370S | iPSC-derived DA neurons | 1.Dopamine dysregulation | |
| [154] | 1 PD line vs. 3 controls lines | Heterozygous N3070S | iPSC-derived DA neurons | 1. Dopamine dysregulation 2. Elevated α-synuclein levels. |
| [155] | 7 PD lines vs. 3 control lines | N370S | iPSC-derived DA neurons | 1. Autophagic and autophagosome dysfunction. 2. Elevated α-synuclein levels |
| [156] | 2 PD lines vs. 2 isogenic control lines | null GBA (CRISPR-Cas) | iPSC-derived cortical neurons | 1. Reduction in Gcase activity 2. Lysosomal dysfunction |
| [157] | 3 PD lines vs. 3 control lines | N370S | iPSC-derived DA neurons | 1. Lysosomal dysfunction 2. Elevated α-synuclein levels |
3.5.2. GBA Mutations Result in ES Stress in iPSC-Derived Neurons
iPSC derived DA neurons with GBA mutations are vulnerable to increased endoplasmic reticulum (ER) stress, autophagic/lysosomal dysfunction, and eventually enlargement of lysosomal compartments [150]. This results from the accumulation of misfolded GBA in the ER, leading to the activation of the UPR, supported by an upregulation of Bip/GRP78, calreticulin and additional UPR-mediators such as PDI, calnexin and IRE1-alpha [150]. These findings suggest that accumulation of misfolded proteins can result in autophagy disturbances. In line with this, GBA-pN370S iPSCs-derived DA neurons display elevated level of LC3B-II, the lipidated form of the autophagosome marker, reflecting an increase in level of autophagosomes [150]. This was further confirmed in a study by Yang et al., in which the levels of LC3 and p62 were decreased, suggesting an impairment in autophagosome formation [155].
Impairments in lysosomal number and degradation processes are also associated with the reduced GCase activity in GBA-mutated cortical and DA neurons [150,156]. The level of cathepsin D, a lysosomal protease that interacts with GCase products is decreased, promoting a dysregulation in α-synuclein levels, disturbing lysosomal function in GBA-pN370S mutated iPSC-derived DA neurons [157]. The observed lysosome enlargement was found to be associated with an impairment of cargo degradation accompanied by elevated level of lysosomal markers LAMP1 and LAMP2 in iPSC-derived DA neurons with GBA-pN370S mutation [150]. Consistent with these findings, the electron microscopy images showed an accumulation of dense debris in lysosomes, suggesting abnormal lysosome clearance [150].
3.5.3. GBA Mutations Result in α-Synuclein Aggregation in iPSC-Derived Neurons
The observed defects in autophagic/lysosome pathways enhance α-synuclein aggregation, impairing its release, as seen in GBA iPSCs-derived DA neurons [158]. Reduction of GCase activity by the accumulation of GlcCer enhance the formation of soluble toxic α-synuclein [152]. mRNA levels of α-synuclein in iPSCs-derived DA neurons with GBA mutation showed no significant difference compared to control, suggesting that GBA mutation only disrupt α-synuclein processing and not its transcription [154]. Accumulation of α-synuclein observed, impaired the degradation dopamine in GBA-pN370S heterozygous null neurons [156].
Degradation of DA neurons are controlled by monoamine oxidases (MAO). iPSC-derived DA neurons affected by GBA-pN370S have demonstrated an elevated activity of MAO [154]. These results are supported by an increase in MAO mRNA levels and protein, suggesting MAO upregulation in PD-affected patients [154]. Additionally, proteins implicated in dopamine level such as dopamine transporter DAT and VMAT2, were decreased in mRNA expression in iPSCs- derived DA neurons of GD with parkinsonism, against iPSCs-derived DA neurons of GD without parkinsonism [153]. Dopamine absorption studies in iPSCs-derived DA neurons shows that the level of dopamine in DA neurons with Gaucher Disease with parkinsonism is reduced [153].
3.6. iPSC Modelling of DJ-1 Mutation and Associated Phenotypes
Dj-1 is a small protein with 189 amino acid residues, usually forming homodimers having a key role in anti-oxidant activities as well as directly inhibiting α-synuclein aggregation [159]. Mutations in DJ-1 have shown to cause early onset, autosomal recessive PD, either due to a base-pair deletion or a homozygous point mutation (L166P) [160,161]. The effects of mutations in DJ-1 on the development of PD has not been extensively studied in iPSC-derived DA neurons (Table 6). Increased dopamine oxidation and oxidative stress have been observed in iPSC-derived DA neurons, triggering mitochondrial oxidative stress, leading to the inactivation of glucocerebrosidaes [160]. This inactivation in turn inhibits lysosomal functions, elevating the level of α-synuclein, a known phenotype observed in iPSC- derived neurons from PD [160]. A more recent study utilized hiPSC-derived DA neurons carrying DJ-1 mutation and demonstrated a dysregulation in lysosomal proteins and activity [81,162,163].
4. Evidence That iPSC Models Confirm the Key Phenotypes Found in Human Pathology
DA degeneration is a key element in the development of PD, and increasingly the evidence for the direct or indirect involvement of mitochondrial dysfunction is growing. Numerous studies show that samples from PD patients show dysregulation in mitochondrial protein expression, glucose metabolism, and reduction in complex 1 activity [164,165,166]. The data to date shows that the DA neurons from the substantia nigra have a high energy demand to support their long, arborized axons and transmitter release sites, making these cells vulnerable to degeneration. This is supported by the evidence from iPSC-derived neurons with mutations in LRRK2, PARK2, and SNCA, where these differentiated neurons have shorter outgrowths and reduced numbers of neurites [58,167,168].
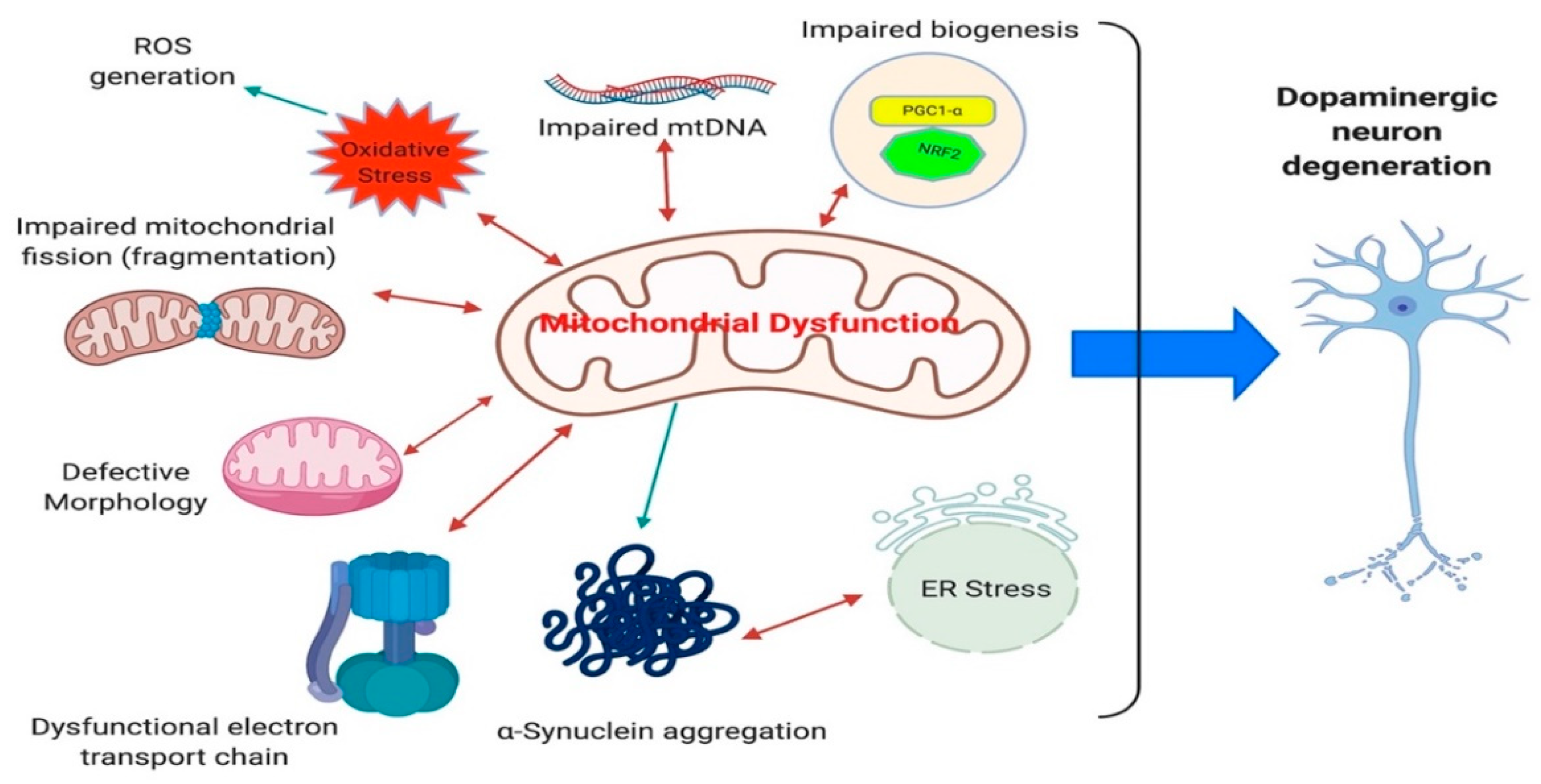
Alterations in both the functional and morphological aspects of mitochondria in iPSC-derived neurons have been demonstrated (Table 1, Table 2, Table 3, Table 4, Table 5 and Table 6). PINK1, PARK2, LRRK2 G2019S, and SNCA mutations recapitulated the disease state very well, neurons present with an increase in fragmented mitochondria and an increase in mitochondrial content [48,57,82,111,140] (Figure 4). This suggests an overall reduction in the number of active functional mitochondria within iPSC-derived DA neurons and supports the energy drain as a causative agent in DA degeneration observed in patients. Furthermore, the morphology of mitochondria are shown to be swollen and disorganized in iPSC-derived DA neurons [48,151] (Figure 4). These alterations in mitochondrial content are in line with the idea that the neurons capacity to produce energy is compromised. As evidenced by the reduction in nicotinamide adenine dinucleotide (NAD+), in LRRK2 G2019S iPSC-derived DA neurons [85]. Additionally, GBA mutations result in a reduction the NAD+/NADH ratio and treatment with NAD+ precursor nicotinamide riboside rescued the respiratory capacity in these neurons [151]. In the same domain, basal and maximal respiration capacity, ATP-linked respiration (Figure 4) and ATP production is significantly decreased in SNCA, GBA, PINK1 and LRRK2 G2019S iPSC-derived neurons [48,85,151,169]. All these data are consistent with the reduced levels of ATP synthase observed in substantia nigra of PD patients and re-enforces the quality of iPSC model systems to model the disease state as regards mitochondrial function [165].
Mitochondrial DNA (mtDNA) damage and accumulation of mtDNA deletions have been observed in substantia nigra of PD patients, which correlate with the cellular respiratory defects [170,171]. Moreover, a reduction in mtDNA copy number has been detected in cerebrospinal fluid of PD patients and in other patients with different neurodegenerative disorders [172,173]. Damage to mtDNA can lead to the breakdown of double-stranded DNA, causing the deletion and loss of several kilobases of mtDNA. This connection between double-strand breaks and mtDNA deletions has been studied in mouse models, developing PD-related behavioral phenotypes and degeneration of nigrostrial brain regions [174]. Increased mtDNA has been confirmed in neurons with PINK1 and LRRK2 G2019S mutations [82,137]. The mtDNA damage can explain the reported complex I deficiency observed in PD patients since the genes found in mtDNA encode components of the electron transport chain [175,176] (Figure 4). Studies in iPSC-derived neurons from different genetic background all confirm their association with the pathogenesis of PD. While these results suggest that mitochondrial alterations increase neuronal vulnerability and neuronal loss as seen in PD, it is still unclear whether these defects are causative agents or more downstream event in PD pathology.
An additional effect of accumulating somatic mtDNA deletions and reduction in ATP synthase is an increase in oxidative stress within DA neurons. Oxidative stress is one of the main factors involved in pathogenesis of PD and has been identified in post-mortem studies [177,178,179,180]. Increased reactive oxygen species generation (Figure 4) is evident due to an increase in respiration and metabolic demand, leading to an elevation in electron flux. In addition, reduced level of antioxidants has been shown in the brains of PD patients [181,182]. While these phenotypes showing increased oxidative stress could relate to mitochondrial dysfunction, an additional source could be the processing of dopamine by oxidases [183].
Oxidative stress is also one of the major phenotypes observed in many of the iPSC-derived DA neurons from different genetic backgrounds (Table 1, Table 2, Table 3, Table 4, Table 5 and Table 6). Increases in the level of carbonylated proteins are associated with both LRRK2 and parkin mutations [118,184]. Additionally, the levels of proteins involved in dopamine oxidation (MAO-A and MAO-B) are significantly elevated in parkin, SNCA triplication, GBA, and LRRK2 G2019S iPSC-derived DA neurons [56,79,118,154]. This phenotype can be rescued with the expression of MAO-A and -B, while the opposite occurs when parkin is overexpressed, further supporting the relationship between oxidative stress and PD pathology and the utility of iPSC models [118]. Consistent with this, the level of anti-oxidative genes Nrf2, NQO1 are upregulated in parkin mutated iPSC-derived neurons [111]. Furthermore, increased levels of ER stress and activation of unfolded protein response (UPR) have been demonstrated in substantia nigra of PD patients. ER stress is one of the least explored phenotypes which has been confirmed in GBA, LRRK2 G2019S, and SNCA A53T iPSC-derived DA and cortical neurons [57,150,151,185]. The relationship between protein aggregation and cellular stress has been widely studied in post-mortem PD brains [177]. The utility of iPSC neurons makes them an ideal platform to better understand ER stress and study protein aggregation i.e., α-synuclein and eventual DA neuronal loss, which is central to PD pathology (Figure 4).
Oxidative stress is associated with increased uptake and accumulation of α-synuclein [186] as evidenced by elevated dendritic mitochondrial stress in DA neurons [187,188,189]. α-synuclein is the main protein expressed both in Lewy bodies and Lewy neurites; hence, it is important to understand the potential toxic effects of these aggregations in DA neurons. α-synuclein levels are increased in LRRK2 G2019S, parkin, PINK1, GBA, SNCA triplication, and DJ-1 iPSC-derived DA and cortical neurons (Table 1, Table 2, Table 3, Table 4, Table 5 and Table 6). Abnormal expression of α-synuclein [190] and its phosphorylation at serin 129 (pS129) are the most abundant form which is found in Lewy bodies [51,62,64,112,123]. Abnormal accumulation of the phosphorylated form of α-synuclein has also been observed in LRRK2 G2019S, SNCA triplication, and parkin mutated iPSC-derived DA neurons [51,62,64,112,123]. While these data suggest a strong association of α-synuclein alterations in iPSC-derived DA neurons, it is still unclear if α-synuclein directly induces DA neuron toxicity.
The Potential and Limitations of iPSC-Derived Neurons
The development of iPSC model systems provides immense potential in the realm of patient-specific disease modelling. This is particularly true in the case of Parkinson‘s disease, through the generation of DA neurons from PD patients. These cells facilitate the study of gene specific alterations, enabling researchers to characterize different functional and morphological deficiencies in patients own cells and should in the future allow more personalized, targeted therapies. The iPSC-derived neurons carrying gene specific mutations have been shown to display characteristics commonly found in human PD pathology which supports the further exploration of iPSC models. The next phase of this approach to disease modelling and therapy development is to fully characterize these cells and maximize the opportunity.
Many of the pathways highlighted here are directly associated with DA neuron vulnerability and intersect at the controlled generation and release of dopamine. iPSC-derived neurons are ideal for mechanistic-based drug development trials targeting the suppression of oxidative stress, maintenance of the DA morphology and reduction in aggregation/accumulation of α-synuclein. iPSC-derived neurons provide a platform for screening many drugs in order to ameliorate the detrimental phenotypes observed in PD patient derived neurons. For example, high throughput screening of a library of compounds revealed the ability of Isoxazole to specifically target MEFC2-PCG1α pathway, preventing neuronal damage [49]. Similarly, potential targets which are identified through screening can be validated using iPSC-derived neurons. In a study by Soldner et al. they validated a SNCA enhancer variant that is associated with PD with use of CRISPR/Cas9 edited iPSC-derived neurons [191]. iPSCs can be a potential model to bridge the gap between the pre-clinical and clinical trials studies, increasing the translation prospects of potential drug candidates, providing valuable opportunities which can eventually be extrapolated for therapeutic interventions for PD.
However, despite the many advantages, there are limitations in the use of iPSC neurons for modeling PD pathology in a dish. PD is associated with aging and it is a limitation in iPSC technology to generate an age matched model to more fully recapitulate the PD phenotypes. DA degeneration is a hallmark of PD which has not been observed in iPSC- derived DA neurons. A study by Miller et al., reported that the addition of progerin to parkin and PINK1 iPSC-derived DA neurons can enhance apoptosis and shortening of the neurites, accelerating an aging process [192]. Progerin treatments in animal models have additionally shown reduction in survival of DA neurons [192]. Overexpression of progerin can improve the modelling of the late-onset PD. Telomerase inhibitor 2-[(E)-3-naphthalen-2-yl-but-2-enoylamino]-benzoic acid (BIBR1532) also exhibited a reduction in neurite branching increase in mitochondrial stress and DNA break in parkin iPSC-derived DA neurons [193]. While addition of these factors are beneficial in representing true DA neurons in iPSC modelling for PD phenotypes, it is, as yet difficult to distinguish the phenotypes observed from the factors-derived or normal DA neurons.
Another pitfall in iPSC modelling is the efficiency of different differentiation protocols and two-dimensional monolayer systems, which may not truly represent the complex neuronal signalling in vivo or in 3D in vitro scenarios. Protocols have been established for the rapid generation of organoids containing DA neurons, astrocytes and oligodendrocytes from iPSC-derived from PD patients [194,195]. Jo et al., have demonstrated electrically active mature DA neurons with the capability of dopamine production in 3D organoids [194]. Diminishing the genetic backgrounds has also been achieved with the use of CRISPR genome editing technology and use of isogenic controls. This not only eliminates the effect of genetic background but also minimizes the heterogeneity within iPSC lines. This can potentially provide new insights into understanding the complexity of DA degeneration process in PD. Furthermore, this may also help us to investigate and recapitulate other aspects of PD pathology such as neuroinflammation or Lewy bodies formation which as yet have not been established in iPSC-derived neurons. Very few studies using iPSC modelling have been used to study the contribution of astrocytes or microglia, which is a clear feature seen in PD.
5. Conclusions
Human PD-derived iPSCs are a powerful tool for improving our understanding of PD pathology and the underlying mechanism for DA degeneration and loss. There are still challenges in using iPSCs for the accurate recapitulation of PD phenotypes in humans. New 3D differentiation protocols, CRISPR genome editing technologies can advance the field for improving these challenges, paving the way towards a potential therapeutic target. iPSC technology can provide an exceptional opportunity not only to understand the PD pathology but additionally to understand the development of the disease, gaining invaluable information to facilitate the translation of research in a dish to clinical treatment.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The emerging evidence of the parkinson pandemic. J. Parkinsons. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.; Gilbert, R.M. Epidemiology of parkinson disease. Neurol. Clin. 2016, 34, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Ahlskog, J.E.; Mielke, M.M.; Rocca, W.A. Incidence and time trends of drug-induced parkinsonism: A 30-year population-based study. Mov. Disord. 2017, 32, 227–234. [Google Scholar] [CrossRef]
- Moisan, F.; Kab, S.; Mohamed, F.; Canonico, M.; Le Guern, M.; Quintin, C.; Carcaillon, L.; Nicolau, J.; Duport, N.; Singh-Manoux, A.; et al. Parkinson disease male-to-female ratios increase with age: French nationwide study and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 952–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginis, P.; Nackaerts, E.; Nieuwboer, A.; Heremans, E. Cueing for people with Parkinson’s disease with freezing of gait: A narrative review of the state-of-the-art and novel perspectives. Ann Phys Rehabil Med 2018, 61, 407–413. [Google Scholar] [CrossRef]
- Lim, I.; van Wegen, E.; de Goede, C.; Deutekom, M.; Nieuwboer, A.; Willems, A.; Jones, D.; Rochester, L.; Kwakkel, G. Effects of external rhythmical cueing on gait in patients with Parkinson’s disease: A systematic review. Clin. Rehabil. 2005, 19, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Gasser, T.; Hardy, J.; Mizuno, Y. Milestones in PD genetics. Mov. Disord. 2011, 26, 1042–1048. [Google Scholar] [CrossRef]
- Perez-Roca, L.; Adame-Castillo, C.; Campdelacreu, J.; Ispierto, L.; Vilas, D.; Rene, R.; Alvarez, R.; Gascon-Bayarri, J.; Serrano-Munoz, M.A.; Ariza, A.; et al. Glucocerebrosidase mRNA is Diminished in Brain of Lewy Body Diseases and Changes with Disease Progression in Blood. Aging Dis 2018, 9, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Schiesling, C.; Kieper, N.; Seidel, K.; Krüger, R. Review: Familial Parkinson’s disease--genetics, clinical phenotype and neuropathology in relation to the common sporadic form of the disease. Neuropathol Appl Neurobiol 2008, 34, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Ann. N. Y. Acad. Sci. 2008, 1147, 93–104. [Google Scholar] [CrossRef]
- Zhang, L.; Hao, J.; Zheng, Y.; Su, R.; Liao, Y.; Gong, X.; Liu, L.; Wang, X. Fucoidan Protects Dopaminergic Neurons by Enhancing the Mitochondrial Function in a Rotenone-induced Rat Model of Parkinson’s Disease. Aging Dis 2018, 9, 590–604. [Google Scholar] [CrossRef] [Green Version]
- Potashkin, J.A.; Blume, S.R.; Runkle, N.K. Limitations of animal models of Parkinson’s disease. Parkinsons. Dis. 2010, 2011, 658083. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Experimental models of Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, R.; Sun, M.; Wang, W.; Zhang, J.; Zhang, L.; Zhen, J.; Qian, Y.; Zheng, Y.; Wang, X. A Novel Immunosuppressor, (5R)-5-Hydroxytriptolide, Alleviates Movement Disorder and Neuroinflammation in a 6-OHDA Hemiparkinsonian Rat Model. Aging Dis 2017, 8, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Chiba, K.; Trevor, A.; Castagnoli, N. Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem. Biophys. Res. Commun. 1984, 120, 574–578. [Google Scholar] [CrossRef]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic animal models of Parkinson’s disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell. 2007, 1, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, S. Induction of pluripotent stem cells from mouse fibroblasts by four transcription factors. Cell Prolif. 2008, 41 (Suppl. S1), 51–56. [Google Scholar] [CrossRef]
- Grealish, S.; Diguet, E.; Kirkeby, A.; Mattsson, B.; Heuer, A.; Bramoulle, Y.; Van Camp, N.; Perrier, A.L.; Hantraye, P.; Björklund, A.; et al. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell 2014, 15, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Jiang, H.; Zhang, B.; Feng, J. Modeling Parkinson’s Disease Using Patient-specific Induced Pluripotent Stem Cells. J. Parkinsons. Dis. 2018, 8, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Mao, C.; Fan, L.; Luo, H.; Hu, Z.; Zhang, S.; Yang, Z.; Zheng, H.; Sun, H.; Fan, Y.; et al. Modeling parkinson’s disease using induced pluripotent stem cells. Stem Cells Int. 2020, 2020, 1061470. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marton, R.M.; Ioannidis, J.P.A. A Comprehensive Analysis of Protocols for Deriving Dopaminergic Neurons from Human Pluripotent Stem Cells. Stem Cells Transl. Med. 2019, 8, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Ling, K.-H.; Tan, J.J.; Lu, C.-B. Development and differentiation of midbrain dopaminergic neuron: From bench to bedside. Cells 2020, 9, 1489. [Google Scholar] [CrossRef] [PubMed]
- Theka, I.; Caiazzo, M.; Dvoretskova, E.; Leo, D.; Ungaro, F.; Curreli, S.; Managò, F.; Dell’Anno, M.T.; Pezzoli, G.; Gainetdinov, R.R.; et al. Rapid generation of functional dopaminergic neurons from human induced pluripotent stem cells through a single-step procedure using cell lineage transcription factors. Stem Cells Transl. Med. 2013, 2, 473–479. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Fasano, C.A.; Chambers, S.M.; Lee, G.; Tomishima, M.J.; Studer, L. Efficient derivation of functional floor plate tissue from human embryonic stem cells. Cell Stem Cell 2010, 6, 336–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajani, S.; Raina, A.; Fokken, C.; Kügler, S.; Bähr, M. Homogenous generation of dopaminergic neurons from multiple hiPSC lines by transient expression of transcription factors. Cell Death Dis. 2019, 10, 898. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.; Anastacio, H.; Bardy, C. Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. npj Parkinsons Disease 2020, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankratz, N.; Foroud, T. Genetics of Parkinson disease. Genet. Med. 2007, 9, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, M.; Massano, J. An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 2017, 135, 273–284. [Google Scholar] [CrossRef]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Miraglia, F.; Ricci, A.; Rota, L.; Colla, E. Subcellular localization of alpha-synuclein aggregates and their interaction with membranes. Neural Regen. Res. 2018, 13, 1136–1144. [Google Scholar]
- Wersinger, C.; Sidhu, A. Attenuation of dopamine transporter activity by α-synuclein. Neurosci. Lett. 2003, 340, 189–192. [Google Scholar] [CrossRef]
- Carnwath, T.; Mohammed, R.; Tsiang, D. The direct and indirect effects of α-synuclein on microtubule stability in the pathogenesis of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2018, 14, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, R.; Kuhn, W.; Leenders, K.L.; Sprengelmeyer, R.; Müller, T.; Woitalla, D.; Portman, A.T.; Maguire, R.P.; Veenma, L.; Schröder, U.; et al. Familial parkinsonism with synuclein pathology: Clinical and PET studies of A30P mutation carriers. Neurology 2001, 56, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, S.; Paschalis, C.; Athanassiadou, A.; Papadimitriou, A.; Ellul, J.; Polymeropoulos, M.H.; Papapetropoulos, T. Clinical phenotype in patients with alpha-synuclein Parkinson’s disease living in Greece in comparison with patients with sporadic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2001, 70, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.; Goldman, J.; Zabetian, C.; Mata, I.; Leverenz, J. SNCA G51D Missense Mutation Causing Juvenile Onset Parkinson’s Disease (P5. 8-026). Neurology 2019, 92. [Google Scholar]
- Arias-Fuenzalida, J.; Jarazo, J.; Qing, X.; Walter, J.; Gomez-Giro, G.; Nickels, S.L.; Zaehres, H.; Schöler, H.R.; Schwamborn, J.C. FACS-Assisted CRISPR-Cas9 Genome Editing Facilitates Parkinson’s Disease Modeling. Stem Cell Rep. 2017, 9, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Brazdis, R.-M.; Alecu, J.E.; Marsch, D.; Dahms, A.; Simmnacher, K.; Lörentz, S.; Brendler, A.; Schneider, Y.; Marxreiter, F.; Roybon, L.; et al. Demonstration of brain region-specific neuronal vulnerability in human iPSC-based model of familial Parkinson’s disease. Hum. Mol. Genet. 2020, 29, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Flierl, A.; Oliveira, L.M.A.; Falomir-Lockhart, L.J.; Mak, S.K.; Hesley, J.; Soldner, F.; Arndt-Jovin, D.J.; Jaenisch, R.; Langston, J.W.; Jovin, T.M.; et al. Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS ONE 2014, 9, e112413. [Google Scholar] [CrossRef] [Green Version]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [Green Version]
- Zambon, F.; Cherubini, M.; Fernandes, H.J.R.; Lang, C.; Ryan, B.J.; Volpato, V.; Bengoa-Vergniory, N.; Vingill, S.; Attar, M.; Booth, H.D.E.; et al. Cellular α-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 2019, 28, 2001–2013. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stykel, M.G.; Humphries, K.; Kirby, M.P.; Czaniecki, C.; Wang, T.; Ryan, T.; Bamm, V.; Ryan, S.D. Nitration of microtubules blocks axonal mitochondrial transport in a human pluripotent stem cell model of Parkinson’s disease. FASEB J. 2018, 32, 5350–5364. [Google Scholar] [CrossRef] [Green Version]
- Prots, I.; Grosch, J.; Brazdis, R.-M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [Green Version]
- Shaltouki, A.; Hsieh, C.-H.; Kim, M.J.; Wang, X. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 2018, 136, 607–620. [Google Scholar] [CrossRef]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schüle, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef] [PubMed]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, L.M.A.; Falomir-Lockhart, L.J.; Botelho, M.G.; Lin, K.H.; Wales, P.; Koch, J.C.; Gerhardt, E.; Taschenberger, H.; Outeiro, T.F.; Lingor, P.; et al. Elevated α-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 2015, 6, e1994. [Google Scholar] [CrossRef] [Green Version]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA expression by targeted editing of DNA methylation: A potential strategy for precision therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef] [Green Version]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Tagliafierro, L.; Zamora, M.E.; Chiba-Falek, O. Multiplication of the SNCA locus exacerbates neuronal nuclear aging. Hum. Mol. Genet. 2019, 28, 407–421. [Google Scholar] [CrossRef]
- Lin, L.; Göke, J.; Cukuroglu, E.; Dranias, M.R.; VanDongen, A.M.J.; Stanton, L.W. Molecular Features Underlying Neurodegeneration Identified through In Vitro Modeling of Genetically Diverse Parkinson’s Disease Patients. Cell Rep. 2016, 15, 2411–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, V.; Mitra, J.; Hegde, P.M.; Pandey, A.; Sengupta, S.; Mitra, S.; Rao, K.S.; Hegde, M.L. Chromatin-Bound Oxidized α-Synuclein Causes Strand Breaks in Neuronal Genomes in in vitro Models of Parkinson’s Disease. J. Alzheimers Dis. 2017, 60, S133–S150. [Google Scholar] [CrossRef]
- Kouroupi, G.; Taoufik, E.; Vlachos, I.S.; Tsioras, K.; Antoniou, N.; Papastefanaki, F.; Chroni-Tzartou, D.; Wrasidlo, W.; Bohl, D.; Stellas, D.; et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3679–E3688. [Google Scholar] [CrossRef] [Green Version]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef] [Green Version]
- Zygogianni, O.; Antoniou, N.; Kalomoiri, M.; Kouroupi, G.; Taoufik, E.; Matsas, R. In Vivo Phenotyping of Familial Parkinson’s Disease with Human Induced Pluripotent Stem Cells: A Proof-of-Concept Study. Neurochem. Res. 2019, 44, 1475–1493. [Google Scholar] [CrossRef] [PubMed]
- Cuddy, L.K.; Wani, W.Y.; Morella, M.L.; Pitcairn, C.; Tsutsumi, K.; Fredriksen, K.; Justman, C.J.; Grammatopoulos, T.N.; Belur, N.R.; Zunke, F.; et al. Stress-Induced Cellular Clearance Is Mediated by the SNARE Protein ykt6 and Disrupted by α-Synuclein. Neuron 2019, 104, 869–884.e11. [Google Scholar] [CrossRef] [PubMed]
- Khurana, V.; Peng, J.; Chung, C.Y.; Auluck, P.K.; Fanning, S.; Tardiff, D.F.; Bartels, T.; Koeva, M.; Eichhorn, S.W.; Benyamini, H.; et al. Genome-Scale Networks Link Neurodegenerative Disease Genes to α-Synuclein through Specific Molecular Pathways. Cell Syst. 2017, 4, 157–170.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Tan, Y.-C.; Poulose, S.; Olanow, C.W.; Huang, X.-Y.; Yue, Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J. Neurochem. 2007, 103, 238–247. [Google Scholar]
- Roosen, D.A.; Cookson, M.R. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 2016, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Gómez-Garre, P.; Díaz-Corrales, F.J.; Carrillo, F.; Carballo, M.; Palomino, A.; Díaz-Martín, J.; Mejías, R.; Vime, P.J.; López-Barneo, J.; et al. Prevalence and clinical features of LRRK2 mutations in patients with Parkinson’s disease in southern Spain. Eur. J. Neurol. 2009, 16, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Sammler, E. LRRK2 kinase in Parkinson’s disease. Science 2018, 360, 36–37. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-W.; Cannon, J.R. LRRK2 mutations and neurotoxicant susceptibility. Exp. Biol. Med. 2015, 240, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Di Fonzo, A.; Tassorelli, C.; De Mari, M.; Chien, H.F.; Ferreira, J.; Rohé, C.F.; Riboldazzi, G.; Antonini, A.; Albani, G.; Mauro, A.; et al. Italian Parkinson’s Genetics Network Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson’s disease. Eur. J. Hum. Genet. 2006, 14, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Marder, K.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Tang, M.-X.; Lee, A.; Raymond, D.; Mirelman, A.; Saunders-Pullman, R.; Clark, L.; et al. LRRK2 Ashkenazi Jewish Consortium Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015, 85, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, D.; Dowman, J.; Hammond, R.; Leete, T.; Inoue, K.; Abeliovich, A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 2006, 52, 587–593. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.; Jeong, H.; Kwon, J.; Heo, H.Y.; Kwon, J.J.; Yun, H.J.; Kim, C.-H.; Han, B.S.; Tong, Y.; Shen, J.; et al. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 2008, 314, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Suaga, P.; Rivero-Ríos, P.; Fdez, E.; Blanca Ramírez, M.; Ferrer, I.; Aiastui, A.; López De Munain, A.; Hilfiker, S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 2014, 23, 6779–6796. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schüle, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic parkinson’s disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, L.H.; Laganière, J.; Cooper, O.; Mak, S.K.; Vu, B.J.; Huang, Y.A.; Paschon, D.E.; Vangipuram, M.; Sundararajan, R.; Urnov, F.D.; et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: Reversal by gene correction. Neurobiol. Dis. 2014, 62, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, J.; Bolognin, S.; Antony, P.M.A.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X.; et al. Neural stem cells of parkinson’s disease patients exhibit aberrant mitochondrial morphology and functionality. Stem Cell Rep. 2019, 12, 878–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.J.; Sison, S.L.; Meade, M.R.; Broniowska, K.A.; Corbett, J.A.; Ebert, A.D. Decreased Sirtuin Deacetylase Activity in LRRK2 G2019S iPSC-Derived Dopaminergic Neurons. Stem Cell Rep. 2017, 9, 1839–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; Krainc, D. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 5576–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schüle, B.; et al. LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef] [Green Version]
- Clark, L.N.; Wang, Y.; Karlins, E.; Saito, L.; Mejia-Santana, H.; Harris, J.; Louis, E.D.; Cote, L.J.; Andrews, H.; Fahn, S.; et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology 2006, 67, 1786–1791. [Google Scholar] [CrossRef]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Borgs, L.; Peyre, E.; Alix, P.; Hanon, K.; Grobarczyk, B.; Godin, J.D.; Purnelle, A.; Krusy, N.; Maquet, P.; Lefebvre, P.; et al. Dopaminergic neurons differentiating from LRRK2 G2019S induced pluripotent stem cells show early neuritic branching defects. Sci. Rep. 2016, 6, 33377. [Google Scholar] [CrossRef]
- Korecka, J.A.; Talbot, S.; Osborn, T.M.; de Leeuw, S.M.; Levy, S.A.; Ferrari, E.J.; Moskites, A.; Atkinson, E.; Jodelka, F.M.; Hinrich, A.J.; et al. Neurite Collapse and Altered ER Ca2+ Control in Human Parkinson Disease Patient iPSC-Derived Neurons with LRRK2 G2019S Mutation. Stem Cell Rep. 2019, 12, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Marrone, L.; Bus, C.; Schöndorf, D.; Fitzgerald, J.C.; Kübler, M.; Schmid, B.; Reinhardt, P.; Reinhardt, L.; Deleidi, M.; Levin, T.; et al. Generation of iPSCs carrying a common LRRK2 risk allele for in vitro modeling of idiopathic Parkinson’s disease. PLoS ONE 2018, 13, e0192497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momcilovic, O.; Sivapatham, R.; Oron, T.R.; Meyer, M.; Mooney, S.; Rao, M.S.; Zeng, X. Derivation, Characterization, and Neural Differentiation of Integration-Free Induced Pluripotent Stem Cell Lines from Parkinson’s Disease Patients Carrying SNCA, LRRK2, PARK2, and GBA Mutations. PLoS ONE 2016, 11, e0154890. [Google Scholar] [CrossRef] [Green Version]
- Qing, X.; Walter, J.; Jarazo, J.; Arias-Fuenzalida, J.; Hillje, A.-L.; Schwamborn, J.C. CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and α-Synuclein modulation in dopaminergic neurons. Stem Cell Res. 2017, 24, 44–50. [Google Scholar] [CrossRef]
- Fernández-Santiago, R.; Carballo-Carbajal, I.; Castellano, G.; Torrent, R.; Richaud, Y.; Sánchez-Danés, A.; Vilarrasa-Blasi, R.; Sánchez-Pla, A.; Mosquera, J.L.; Soriano, J.; et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol. Med. 2015, 7, 1529–1546. [Google Scholar] [CrossRef]
- Liu, G.-H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahnassawy, L.; Nicklas, S.; Palm, T.; Menzl, I.; Birzele, F.; Gillardon, F.; Schwamborn, J.C. The parkinson’s disease-associated LRRK2 mutation R1441G inhibits neuronal differentiation of neural stem cells. Stem Cells Dev. 2013, 22, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.J.; Ebert, A.D. Neurite Aggregation and Calcium Dysfunction in iPSC-Derived Sensory Neurons with Parkinson’s Disease-Related LRRK2 G2019S Mutation. Stem Cell Rep. 2015, 5, 1039–1052. [Google Scholar] [CrossRef] [Green Version]
- Kett, L.R.; Dauer, W.T. Endolysosomal dysfunction in Parkinson’s disease: Recent developments and future challenges. Mov. Disord. 2016, 31, 1433–1443. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.-Y.; Li, X.; Wang, J.; Powell, J.; Wang, Q.; Zhang, Y.; Chen, Z.; Wicinski, B.; Hof, P.; Ryan, T.A.; et al. Parkinson’s Disease-Associated LRRK2 Hyperactive Kinase Mutant Disrupts Synaptic Vesicle Trafficking in Ventral Midbrain Neurons. J. Neurosci. 2017, 37, 11366–11376. [Google Scholar] [CrossRef] [PubMed]
- Connor-Robson, N.; Booth, H.; Martin, J.G.; Gao, B.; Li, K.; Doig, N.; Vowles, J.; Browne, C.; Klinger, L.; Juhasz, P.; et al. An integrated transcriptomics and proteomics analysis reveals functional endocytic dysregulation caused by mutations in LRRK2. Neurobiol. Dis. 2019, 127, 512–526. [Google Scholar] [CrossRef]
- Kim, J.; Daadi, M.M. Non-cell autonomous mechanism of Parkinson’s disease pathology caused by G2019S LRRK2 mutation in Ashkenazi Jewish patient: Single cell analysis. Brain Res. 2019, 1722, 146342. [Google Scholar] [CrossRef] [PubMed]
- Nickels, S.L.; Walter, J.; Bolognin, S.; Gérard, D.; Jaeger, C.; Qing, X.; Tisserand, J.; Jarazo, J.; Hemmer, K.; Harms, A.; et al. Impaired serine metabolism complements LRRK2-G2019S pathogenicity in PD patients. Parkinsonism Relat. Disord. 2019, 67, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.-H.; Mo, J.-S.; Kim, M.-Y.; Ann, E.-J.; Ahn, J.-S.; Jo, E.-H.; Lee, H.-J.; Lee, Y.C.; Seol, W.; Yarmoluk, S.M.; et al. LRRK2 functions as a scaffolding kinase of ASK1-mediated neuronal cell death. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2356–2368. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. French Parkinson’s Disease Genetics Study Group; European Consortium on Genetic Susceptibility in Parkinson’s Disease Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef]
- West, A.B.; Maidment, N.T. Genetics of parkin-linked disease. Hum. Genet. 2004, 114, 327–336. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and α-Synuclein Accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [Green Version]
- Bogetofte, H.; Jensen, P.; Ryding, M.; Schmidt, S.I.; Okarmus, J.; Ritter, L.; Worm, C.S.; Hohnholt, M.C.; Azevedo, C.; Roybon, L.; et al. PARK2 Mutation Causes Metabolic Disturbances and Impaired Survival of Human iPSC-Derived Neurons. Front. Cell Neurosci. 2019, 13, 297. [Google Scholar] [CrossRef] [Green Version]
- Aroso, M.; Ferreira, R.; Freitas, A.; Vitorino, R.; Gomez-Lazaro, M. New insights on the mitochondrial proteome plasticity in Parkinson’s disease. Proteomics Clin Appl 2016, 10, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Hulette, C.; Wang, Y.; Zhang, T.; Pan, C.; Wadhwa, R.; Zhang, J. Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: Relevance to Parkinson disease. Mol. Cell Proteomics 2006, 5, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Hwang, O. Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol. 2013, 22, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Ren, Y.; Zhao, J.; Feng, J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum. Mol. Genet. 2004, 13, 1745–1754. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Jiang, Q.; Liu, W.; Feng, J. Parkin suppresses the expression of monoamine oxidases. J. Biol. Chem. 2006, 281, 8591–8599. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Akamatsu, W.; Kisa, F.; Sone, T.; Ishikawa, K.-I.; Kuzumaki, N.; Katayama, H.; Miyawaki, A.; Hattori, N.; Okano, H. Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem. Biophys. Res. Commun. 2017, 483, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-H.; Lee-Chen, G.-J.; Wu, Y.-R.; Chen, Y.-J.; Lin, J.-L.; Li, M.; Chen, I.-C.; Lo, Y.-S.; Wu, H.-C.; Chen, C.-M. Impairment of proteasome and anti-oxidative pathways in the induced pluripotent stem cell model for sporadic Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 24, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ren, Y.; Yuen, E.Y.; Zhong, P.; Ghaedi, M.; Hu, Z.; Azabdaftari, G.; Nakaso, K.; Yan, Z.; Feng, J. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 2012, 3, 668. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Hu, Z.; Jiang, H.; Yan, Z.; Feng, J. Dopamine Induces Oscillatory Activities in Human Midbrain Neurons with Parkin Mutations. Cell Rep. 2017, 19, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Shaltouki, A.; Sivapatham, R.; Pei, Y.; Gerencser, A.A.; Momčilović, O.; Rao, M.S.; Zeng, X. Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Rep. 2015, 4, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Konovalova, E.V.; Lopacheva, O.M.; Grivennikov, I.A.; Lebedeva, O.S.; Dashinimaev, E.B.; Khaspekov, L.G.; Fedotova, E.Y.; Illarioshkin, S.N. Mutations in the Parkinson’s Disease-Associated PARK2 Gene Are Accompanied by Imbalance in Programmed Cell Death Systems. Acta Naturae 2015, 7, 146–149. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Ishikawa, K.-I.; Inoshita, T.; Izawa, N.; Takanashi, M.; Sato, S.; Onodera, O.; Akamatsu, W.; Okano, H.; Imai, Y.; et al. Evidence that phosphorylated ubiquitin signaling is involved in the etiology of Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 3172–3185. [Google Scholar] [CrossRef]
- Okarmus, J.; Bogetofte, H.; Schmidt, S.I.; Ryding, M.; García-López, S.; Ryan, B.J.; Martínez-Serrano, A.; Hyttel, P.; Meyer, M. Lysosomal perturbations in human dopaminergic neurons derived from induced pluripotent stem cells with PARK2 mutation. Sci. Rep. 2020, 10, 10278. [Google Scholar] [CrossRef]
- Zanon, A.; Kalvakuri, S.; Rakovic, A.; Foco, L.; Guida, M.; Schwienbacher, C.; Serafin, A.; Rudolph, F.; Trilck, M.; Grünewald, A.; et al. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum. Mol. Genet. 2017, 26, 2412–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Albanese, A.; Valente, E.M.; Romito, L.M.; Bellacchio, E.; Elia, A.E.; Dallapiccola, B. The PINK1 phenotype can be indistinguishable from idiopathic Parkinson disease. Neurology 2005, 64, 1958–1960. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, P.; Lesage, S.; Lohmann, E.; Thobois, S.; De Michele, G.; Borg, M.; Agid, Y.; Dürr, A.; Brice, A. French Parkinson’s Disease Genetics Study Group Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain 2006, 129, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Bonifati, V.; Rohé, C.F.; Breedveld, G.J.; Fabrizio, E.; De Mari, M.; Tassorelli, C.; Tavella, A.; Marconi, R.; Nicholl, D.J.; Chien, H.F.; et al. Italian Parkinson Genetics Network Early-onset parkinsonism associated with PINK1 mutations: Frequency, genotypes, and phenotypes. Neurology 2005, 65, 87–95. [Google Scholar] [CrossRef]
- Lin, W.; Kang, U.J. Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 2008, 106, 464–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Arena, G.; Valente, E.M. PINK1 in the limelight: Multiple functions of an eclectic protein in human health and disease. J. Pathol. 2017, 241, 251–263. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Azkona, G.; López de Maturana, R.; Del Rio, P.; Sousa, A.; Vazquez, N.; Zubiarrain, A.; Jimenez-Blasco, D.; Bolaños, J.P.; Morales, B.; Auburger, G.; et al. LRRK2 Expression Is Deregulated in Fibroblasts and Neurons from Parkinson Patients with Mutations in PINK1. Mol. Neurobiol. 2018, 55, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Rakovic, A.; Shurkewitsch, K.; Seibler, P.; Grünewald, A.; Zanon, A.; Hagenah, J.; Krainc, D.; Klein, C. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: Study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 2013, 288, 2223–2237. [Google Scholar] [CrossRef] [Green Version]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Bogaerts, V.; Theuns, J.; van Broeckhoven, C. Genetic findings in Parkinson’s disease and translation into treatment: A leading role for mitochondria? Genes Brain Behav. 2008, 7, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Hoepken, H.-H.; Gispert, S.; Morales, B.; Wingerter, O.; Del Turco, D.; Mülsch, A.; Nussbaum, R.L.; Müller, K.; Dröse, S.; Brandt, U.; et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis. 2007, 25, 401–411. [Google Scholar] [CrossRef]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.Y.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; Deas, E.; et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Straniero, L.; Rimoldi, V.; Samarani, M.; Goldwurm, S.; Di Fonzo, A.; Krüger, R.; Deleidi, M.; Aureli, M.; Soldà, G.; Duga, S.; et al. The GBAP1 pseudogene acts as a ceRNA for the glucocerebrosidase gene GBA by sponging miR-22-3p. Sci. Rep. 2017, 7, 12702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaïti, B.; Bonaïti-Pellié, C.; Brice, A. French Parkinson Disease Genetic Group Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Velayati, A.; Yu, W.H.; Sidransky, E. The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep 2010, 10, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, H.; et al. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Woodard, C.M.; Campos, B.A.; Kuo, S.-H.; Nirenberg, M.J.; Nestor, M.W.; Zimmer, M.; Mosharov, E.V.; Sulzer, D.; Zhou, H.; Paull, D.; et al. iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson’s disease. Cell Rep. 2014, 9, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.-Y.; Beavan, M.; Chau, K.-Y.; Taanman, J.-W.; Schapira, A.H.V. A Human Neural Crest Stem Cell-Derived Dopaminergic Neuronal Model Recapitulates Biochemical Abnormalities in GBA1 Mutation Carriers. Stem Cell Rep. 2017, 8, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Novis, H.S.; Gasser, E.; Lin, S.; LaVoie, M.J. LRRK2 Kinase Inhibition Rescues Deficits in Lysosome Function Due to Heterozygous GBA1 Expression in Human iPSC-Derived Neurons. Front. Neurosci. 2020, 14, 442. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-Y.; Gegg, M.; Chau, D.; Schapira, A. Glucocerebrosidase activity, cathepsin D and monomeric α-synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol. Dis. 2020, 134, 104620. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.A. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid. Redox Signal. 2011, 15, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahfeldt, T.; Ordureau, A.; Bell, C.; Sarrafha, L.; Sun, C.; Piccinotti, S.; Grass, T.; Parfitt, G.M.; Paulo, J.A.; Yanagawa, F.; et al. Pathogenic Pathways in Early-Onset Autosomal Recessive Parkinson’s Disease Discovered Using Isogenic Human Dopaminergic Neurons. Stem Cell Rep. 2020, 14, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.-Y.; Kang, W.-Y.; Chen, Y.-M.; Jiang, T.-F.; Zhang, J.; Zhang, L.-N.; Ding, J.-Q.; Liu, J.; Chen, S.-D. DJ-1 Inhibits α-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef] [Green Version]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef]
- Ferrer, I.; Perez, E.; Dalfó, E.; Barrachina, M. Abnormal levels of prohibitin and ATP synthase in the substantia nigra and frontal cortex in Parkinson’s disease. Neurosci. Lett. 2007, 415, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.; Allen, G.F.; Mamais, A.; Ling, H.; Li, A.; Duberley, K.E.; Hargreaves, I.P.; Pope, S.; Holton, J.L.; Lees, A.; et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson’s disease. Neurobiol. Aging 2014, 35, 1111–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skibinski, G.; Nakamura, K.; Cookson, M.R.; Finkbeiner, S. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J. Neurosci. 2014, 34, 418–433. [Google Scholar] [CrossRef]
- Ren, Y.; Jiang, H.; Hu, Z.; Fan, K.; Wang, J.; Janoschka, S.; Wang, X.; Ge, S.; Feng, J. Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 2015, 33, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.-B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Brennan, R.; Kurzawa-Akanbi, M.; Yarnall, A.; Thouin, A.; Mollenhauer, B.; Burn, D.; Chinnery, P.F.; Hudson, G. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann. Neurol. 2015, 78, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Lowes, H.; Pyle, A.; Duddy, M.; Hudson, G. Cell-free mitochondrial DNA in progressive multiple sclerosis. Mitochondrion 2019, 46, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Pinto, M.; Hida, A.; Moraes, C.T. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci. 2011, 31, 17649–17658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, W.D.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [Green Version]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. et Biophys. Acta (BBA) Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Toulorge, D.; Schapira, A.H.V.; Hajj, R. Molecular changes in the postmortem parkinsonian brain. J. Neurochem. 2016, 139 (Suppl. S1), 27–58. [Google Scholar] [CrossRef]
- Yoritaka, A.; Hattori, N.; Uchida, K.; Tanaka, M.; Stadtman, E.R.; Mizuno, Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mythri, R.B.; Venkateshappa, C.; Harish, G.; Mahadevan, A.; Muthane, U.B.; Yasha, T.C.; Srinivas Bharath, M.M.; Shankar, S.K. Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochem. Res. 2011, 36, 1452–1463. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Navarro, A.; Boveris, A.; Bández, M.J.; Sánchez-Pino, M.J.; Gómez, C.; Muntané, G.; Ferrer, I. Human brain cortex: Mitochondrial oxidative damage and adaptive response in Parkinson disease and in dementia with Lewy bodies. Free Radic. Biol. Med. 2009, 46, 1574–1580. [Google Scholar] [CrossRef]
- van Muiswinkel, F.L.; de Vos, R.A.I.; Bol, J.G.J.M.; Andringa, G.; Jansen Steur, E.N.H.; Ross, D.; Siegel, D.; Drukarch, B. Expression of NAD(P)H:quinone oxidoreductase in the normal and Parkinsonian substantia nigra. Neurobiol. Aging 2004, 25, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology (Basel) 2019, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Ohta, E.; Nihira, T.; Uchino, A.; Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Takahashi, K.; Hayakawa, H.; Nagai, M.; Ohyama, M.; et al. I2020T mutant LRRK2 iPSC-derived neurons in the Sagamihara family exhibit increased Tau phosphorylation through the AKT/GSK-3β signaling pathway. Hum. Mol. Genet. 2015, 24, 4879–4900. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, W.; Schlachetzki, J.C.M.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schäffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.-M. Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013, 54, 71–83. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef]
- Dryanovski, D.I.; Guzman, J.N.; Xie, Z.; Galteri, D.J.; Volpicelli-Daley, L.A.; Lee, V.M.-Y.; Miller, R.J.; Schumacker, P.T.; Surmeier, D.J. Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 2013, 33, 10154–10164. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vera, E.; Bosco, N.; Studer, L. Generating Late-Onset Human iPSC-Based Disease Models by Inducing Neuronal Age-Related Phenotypes through Telomerase Manipulation. Cell Rep. 2016, 17, 1184–1192. [Google Scholar] [CrossRef] [Green Version]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.-D.; Göke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.-P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzel, A.S.; Smits, L.M.; Hemmer, K.; Hachi, S.; Moreno, E.L.; van Wuellen, T.; Jarazo, J.; Walter, J.; Brüggemann, I.; Boussaad, I.; et al. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem Cell Rep. 2017, 8, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Parkinson’s disease (PD) modelling. PD have been modelled and studied using post-mortem (A) tissues derived from PD. (B) Animal models have been vastly used both by knocking down specific gene or with use of chemicals. This help researchers to investigate the associated phenotypes such as mitochondrial dysfunction, neuronal degeneration and protein folding and aggregation for creating an efficient animal model and use in drug discovery and toxicity. (C) Novel induced pluripotent stem cells have been derived from somatic cells of a PD or a healthy individual, leading to generate a disease model in a dish where different phenotypes can be investigated and pave the way towards drug discovery, toxicity testing and cell therapy interventions.
Figure 1.
Parkinson’s disease (PD) modelling. PD have been modelled and studied using post-mortem (A) tissues derived from PD. (B) Animal models have been vastly used both by knocking down specific gene or with use of chemicals. This help researchers to investigate the associated phenotypes such as mitochondrial dysfunction, neuronal degeneration and protein folding and aggregation for creating an efficient animal model and use in drug discovery and toxicity. (C) Novel induced pluripotent stem cells have been derived from somatic cells of a PD or a healthy individual, leading to generate a disease model in a dish where different phenotypes can be investigated and pave the way towards drug discovery, toxicity testing and cell therapy interventions.

Figure 2.
Induced pluripotent stem cell (iPSC)-derived dopaminergic neurons modelling the role of mutant SNCA. The illustration shows the impact of the mutated α-synuclein in different cellular processes within the cell.
Figure 2.
Induced pluripotent stem cell (iPSC)-derived dopaminergic neurons modelling the role of mutant SNCA. The illustration shows the impact of the mutated α-synuclein in different cellular processes within the cell.

Figure 3.
Effect of PINK1 and parkin mutation in IPSC-derived neurons. Parkin mutation in iPSC-derived neurons showed impairment in their requitement to PINK1, alteration in spontaneous postsynaptic current activity, reduction in dopamine receptor and release. PINK1 mutation in iPSC-derived neurons displayed fragmented mitochondria with alteration their DNA level, ATP, membrane potential and oxidative stress. Additionally, there is an increase in apoptotic cell death and α-synuclein aggregation and accumulation.
Figure 3.
Effect of PINK1 and parkin mutation in IPSC-derived neurons. Parkin mutation in iPSC-derived neurons showed impairment in their requitement to PINK1, alteration in spontaneous postsynaptic current activity, reduction in dopamine receptor and release. PINK1 mutation in iPSC-derived neurons displayed fragmented mitochondria with alteration their DNA level, ATP, membrane potential and oxidative stress. Additionally, there is an increase in apoptotic cell death and α-synuclein aggregation and accumulation.

Figure 4.
Pathways of mitochondrial dysfunction, a major cellular and clinical phenotype in PD. Mitochondrial dysfunction can result from impairment in mitochondrial fission, change in mitochondrial morphology, electron transport chain, increase in mtDNA, elevation in oxidative stress leading to reactive oxygen species generation, alteration in mitochondrial biogenesis and electron transport dysfunction. These can lead and associated with protein aggregation and eventual endoplasmic (ER) stress that ultimately results in degeneration of dopaminergic neurons that underlines the PD pathogenesis.
Figure 4.
Pathways of mitochondrial dysfunction, a major cellular and clinical phenotype in PD. Mitochondrial dysfunction can result from impairment in mitochondrial fission, change in mitochondrial morphology, electron transport chain, increase in mtDNA, elevation in oxidative stress leading to reactive oxygen species generation, alteration in mitochondrial biogenesis and electron transport dysfunction. These can lead and associated with protein aggregation and eventual endoplasmic (ER) stress that ultimately results in degeneration of dopaminergic neurons that underlines the PD pathogenesis.

Table 6.
DJ-1-mutated iPSC-derived neuronal phenotype.
| Reference | Number of Cohorts | Type of Mutation | Cell Type | Phenotype |
|---|---|---|---|---|
| [162] | 1 PD isogonic lines vs. control lines | DJ-1 | iPSC-derived DA neurons | 1. Increased oxidative stress 2. Mitochondrial dysfunction 3. Lysosomal dysfunction |
| [160] | 3 PD lines vs. 2 control lines | c.192G>C | iPSC-derived DA neurons | 1. Increased oxidative stress 2. lysosomal dysfunction 3. α -synuclein aggregation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Avazzadeh, S.; Baena, J.M.; Keighron, C.; Feller-Sanchez, Y.; Quinlan, L.R. Modelling Parkinson’s Disease: iPSCs towards Better Understanding of Human Pathology. Brain Sci. 2021, 11, 373. https://doi.org/10.3390/brainsci11030373
AMA Style
Avazzadeh S, Baena JM, Keighron C, Feller-Sanchez Y, Quinlan LR. Modelling Parkinson’s Disease: iPSCs towards Better Understanding of Human Pathology. Brain Sciences. 2021; 11(3):373. https://doi.org/10.3390/brainsci11030373
Chicago/Turabian StyleAvazzadeh, Sahar, Jara Maria Baena, Cameron Keighron, Yajaira Feller-Sanchez, and Leo R. Quinlan. 2021. "Modelling Parkinson’s Disease: iPSCs towards Better Understanding of Human Pathology" Brain Sciences 11, no. 3: 373. https://doi.org/10.3390/brainsci11030373
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.