
Vascular Calcification in Chronic Kidney Disease: Diversity in the Vessel Wall

 , and
, and
Abstract
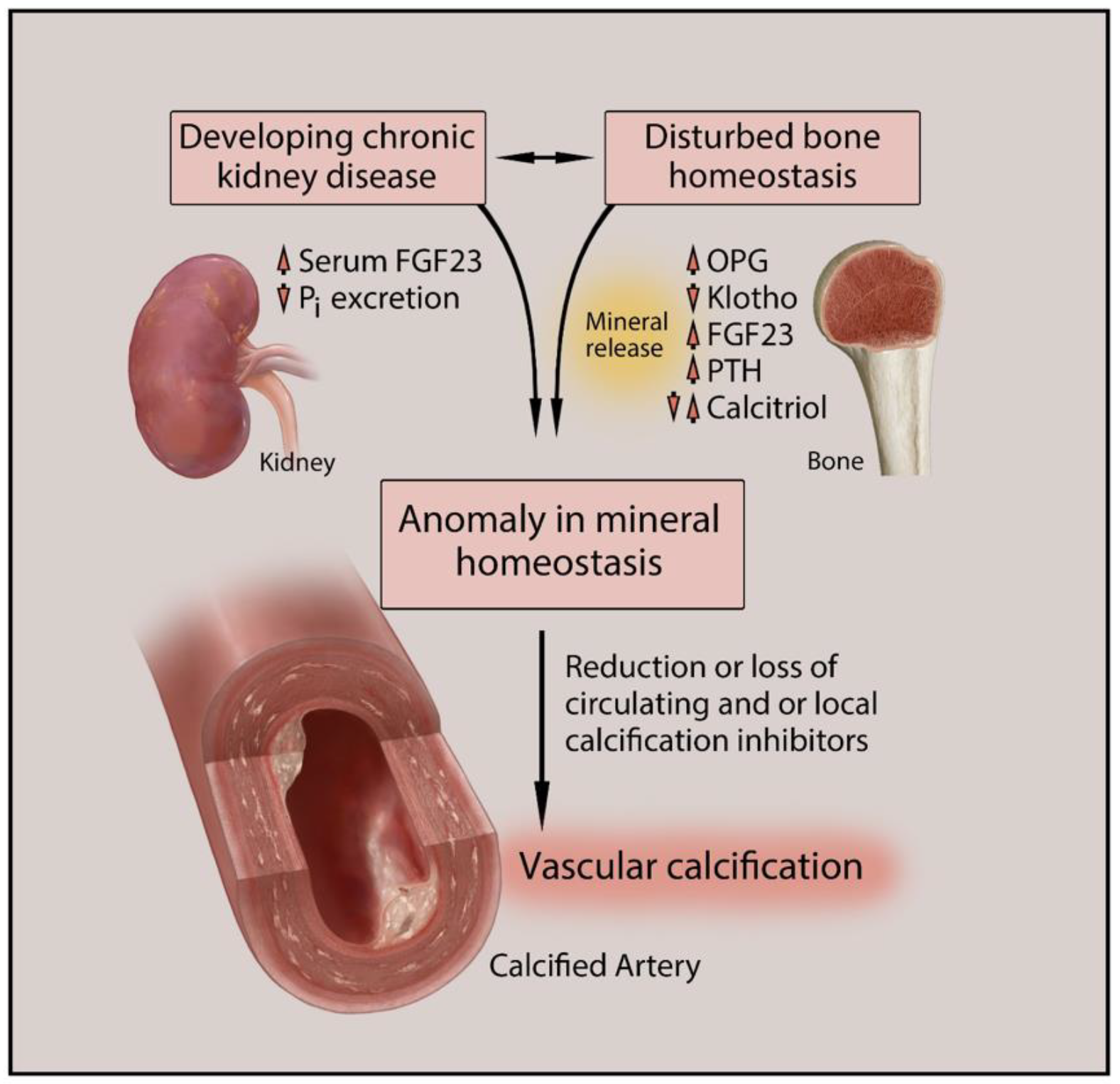
:1. Introduction
2. Types and Anatomical Presence of VC
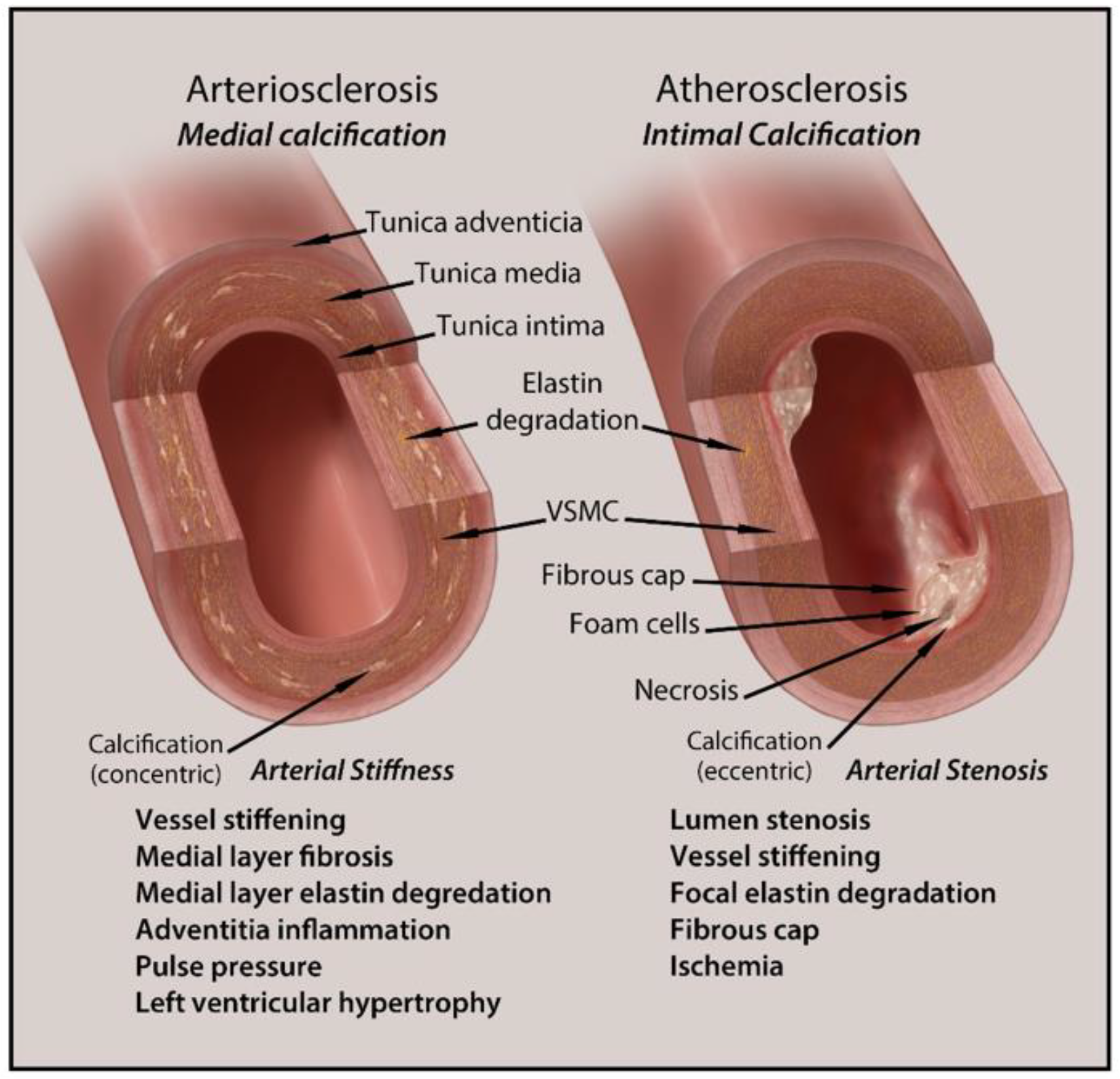
2.1. Intimal Calcification
2.2. Medial Calcification
2.3. Calciphylaxis
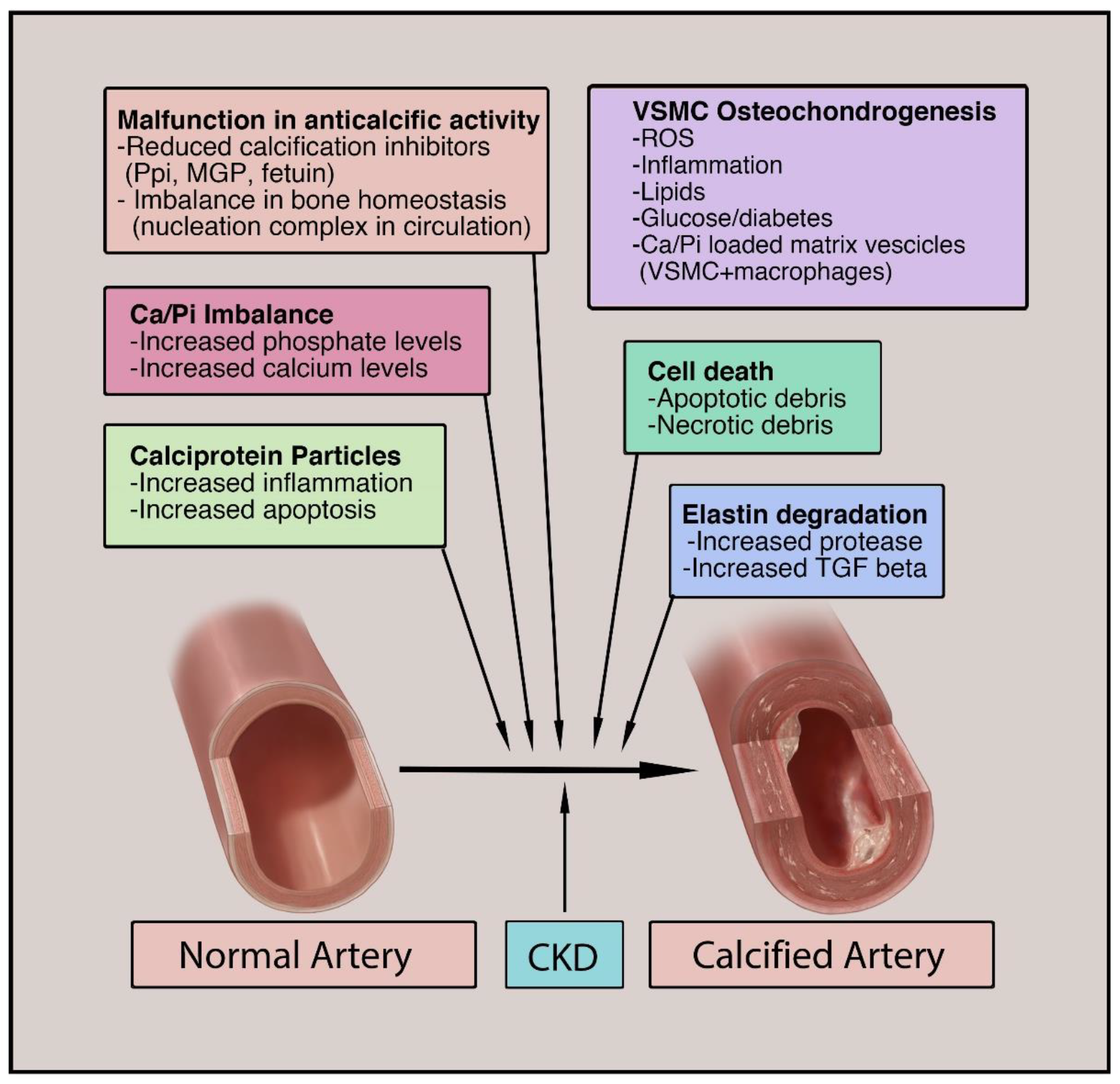
3. Major Cell Types of Vascular Calcification
3.1. Vascular Smooth Muscle Cells (VSMCs)
3.2. Macrophages
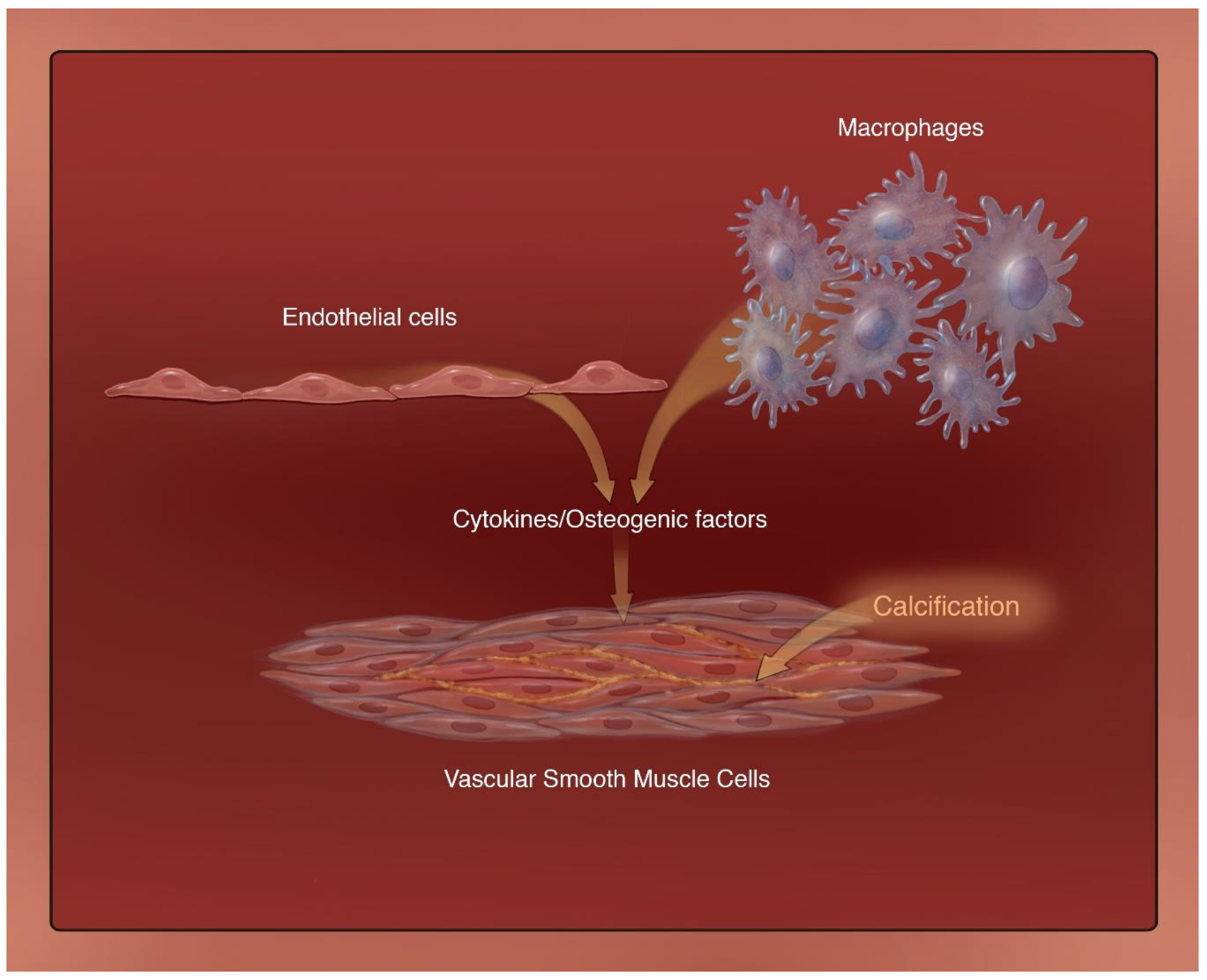
3.3. Endothelial Cells
4. Clinical Implications and Diagnosis of Vascular Calcification in CKD
5. Treatment Options for Vascular Calcification in CKD
6. Animal Models of Vascular Calcification
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gao, J.; Zhang, K.; Chen, J.; Wang, M.-H.; Wang, J.; Liu, P.; Huang, H. Roles of aldosterone in vascular calcification: An update. Eur. J. Pharmacol. 2016, 786, 186–193. [Google Scholar] [CrossRef]
- Généreux, P.; Redfors, B.; Witzenbichler, B.; Arsenault, M.-P.; Weisz, G.; Stuckey, T.D.; Rinaldi, M.J.; Neumann, F.-J.; Metzger, D.C.; Henry, T.D.; et al. Two-year outcomes after percutaneous coronary intervention of calcified lesions with drug-eluting stents. Int. J. Cardiol. 2017, 231, 61–67. [Google Scholar] [CrossRef]
- Lee, H.Y.; Park, U.J.; Kim, H.T.; Roh, Y.-N. The Effect of Severe Femoropopliteal Arterial Calcification on the Treatment Outcome of Femoropopliteal Intervention in Patients with Ischemic Tissue Loss. Vasc. Spec. Int. 2020, 36, 96–104. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Laurent, S. Mechanisms of Arterial Stiffening. Arter. Thromb. Vasc. Biol. 2020, 40, 1055–1062. [Google Scholar] [CrossRef]
- Lioufas, N.M.; Pedagogos, E.; Hawley, C.M.; Pascoe, E.M.; Elder, G.J.; Badve, S.V.; Valks, A.; Toussaint, N.D.; on behalf of the IMPROVE-CKD Investigators. Aortic Calcification and Arterial Stiffness Burden in a Chronic Kidney Disease Cohort with High Cardiovascular Risk: Baseline Characteristics of the Impact of Phosphate Reduction on Vascular End-Points in Chronic Kidney Disease Trial. Am. J. Nephrol. 2020, 51, 201–215. [Google Scholar] [CrossRef]
- Jagieła, J.; Bartnicki, P.; Rysz, J. Selected cardiovascular risk factors in early stages of chronic kidney disease. Int. Urol. Nephrol. 2020, 52, 303–314. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Vascular Calcification. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef]
- Chen, N.X.; Moe, S.M. Vascular calcification: Pathophysiology and risk factors. Curr. Hypertens. Rep. 2012, 14, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Hénaut, L.; Chillon, J.-M.; Kamel, S.; Massy, Z.A. Updates on the Mechanisms and the Care of Cardiovascular Calcification in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 233–250. [Google Scholar] [CrossRef]
- Voelkl, J.; Lang, F.; Eckardt, K.-U.; Amann, K.; Kuro-O, M.; Pasch, A.; Pieske, B.; Alesutan, I. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell. Mol. Life Sci. 2019, 76, 2077–2091. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular Calcification: An Update on Mechanisms and Challenges in Treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef]
- El Din, U.A.A.S.; Salem, M.M.; Azim, S.E.D.U.A. Vascular calcification: When should we interfere in chronic kidney disease patients and how? World J. Nephrol. 2016, 5, 398–417. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Nanami, M.; Kuragano, T. The pathogenesis of CKD complications; Attack of dysregulated iron and phosphate metabolism. Free Radic. Biol. Med. 2020, 157, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Olapoju, S.O.; Adejobi, O.I.; Le Thi, X. Fibroblast growth factor 21; review on its participation in vascular calcification pathology. Vasc. Pharmacol. 2020, 125–126, 106636. [Google Scholar] [CrossRef] [PubMed]
- Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins 2020, 12, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Sarnak, M.J. Cardiovascular complications in chronic kidney disease. Am. J. Kidney Dis. 2003, 41, 11–17. [Google Scholar] [CrossRef]
- Sheng, B.; Zhu, T.; Li, J. End-stage renal disease and pulmonary hypertension. Zhong Nan Da Xue Xue Bao Yi Xue Ban = J. Cent. South Univ. Med. Sci. 2019, 44, 1419–1422. [Google Scholar]
- Arefin, S.; Buchanan, S.; Hobson, S.; Steinmetz, J.; Alsalhi, S.; Shiels, P.G.; Kublickiene, K.; Stenvinkel, P. Nrf2 in early vascular ageing: Calcification, senescence and therapy. Clin. Chim. Acta 2020, 505, 108–118. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.-K.; Jeon, J.-H. Vascular Calcification—New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.; Towler, D.A. Arterial calcification and bone physiology: Role of the bone–vascular axis. Nat. Rev. Endocrinol. 2012, 8, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Shi, G.-P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2106–2119. [Google Scholar] [CrossRef] [Green Version]
- Cucchiari, D.; Torregrosa, J.-V. Calcifilaxis en pacientes con enfermedad renal crónica: Una enfermedad todavía desconcertante y potencialmente mortal. Nefrología 2018, 38, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Gómez, M.M.; Hernández-Betancor, I.; García-Niebla, J.; Marí-López, B.; Laynez-Cerdeña, I.; Lacalzada-Almeida, J. Valve Calcification in Aortic Stenosis: Etiology and Diagnostic Imaging Techniques. BioMed Res. Int. 2017, 2017, 5178631. [Google Scholar] [CrossRef]
- Neven, E.; Dauwe, S.; De Broe, M.E.; Haese, P.C.D.; Persy, V. Endochondral bone formation is involved in media calcification in rats and in men. Kidney Int. 2007, 72, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Qadri, S.I.; Koratala, A. Calciphylaxis with extensive arterial calcification. Clin. Case Rep. 2017, 5, 1418–1419. [Google Scholar] [CrossRef]
- Sinha, S.; Iyer, D.; Granata, A. Embryonic origins of human vascular smooth muscle cells: Implications for In Vitro modeling and clinical application. Cell. Mol. Life Sci. 2014, 71, 2271–2288. [Google Scholar] [CrossRef] [Green Version]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef]
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol. Physiol. 2014, 307, F891–F900. [Google Scholar] [CrossRef] [Green Version]
- Scholze, A.; Jankowski, J.; Pedraza-Chaverri, J.; Evenepoel, P. Oxidative Stress in Chronic Kidney Disease. Oxidative Med. Cell. Longev. 2016, 2016, 8375186. [Google Scholar] [CrossRef] [Green Version]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative Stress Induces Vascular Calcification through Modulation of the Osteogenic Transcription Factor Runx2 by AKT Signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [Green Version]
- Montezano, A.C.; Zimmerman, D.; Yusuf, H.; Burger, D.; Chignalia, A.Z.; Wadhera, V.; Van Leeuwen, F.N.; Touyz, R.M. Vascular Smooth Muscle Cell Differentiation to an Osteogenic Phenotype Involves TRPM7 Modulation by Magnesium. Hypertension 2010, 56, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, R. Emerging role for fetuin-A as contributor to morbidity and mortality in chronic kidney disease. Kidney Int. 2007, 72, 137–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial Calcification in Chronic Kidney Disease: Key Roles for Calcium and Phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlieper, G.; Schurgers, L.; Brandenburg, V.; Reutelingsperger, C.; Floege, J. Vascular calcification in chronic kidney disease: An update. Nephrol. Dial. Transplant. 2016, 31, 31–39. [Google Scholar] [CrossRef] [Green Version]
- House, S.J.; Potier, M.; Bisaillon, J.; Singer, H.A.; Trebak, M. The non-excitable smooth muscle: Calcium signaling and phenotypic switching during vascular disease. Pflug. Arch. Eur. J. Physiol. 2008, 456, 769–785. [Google Scholar] [CrossRef] [Green Version]
- Molostvov, G.; James, S.; Fletcher, S.; Bennett, J.; Lehnert, H.; Bland, R.; Zehnder, D. Extracellular calcium-sensing receptor is functionally expressed in human artery. Am. J. Physiol. Physiol. 2007, 293, F946–F955. [Google Scholar] [CrossRef]
- Yamada, S.; Leaf, E.M.; Chia, J.J.; Cox, T.C.; Speer, M.Y.; Giachelli, C.M. PiT-2, a type III sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int. 2018, 94, 716–727. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-Like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neuro-Oncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikawa, E. Extracellular vesicles in cardiovascular disease: Focus on vascular calcification. J. Physiol. 2016, 594, 2877–2880. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.C.; McNair, R.; Skepper, J.N.; Figg, N.; Schurgers, L.J.; Deanfield, J.; Rees, L.; Shanahan, C.M. Chronic Mineral Dysregulation Promotes Vascular Smooth Muscle Cell Adaptation and Extracellular Matrix Calcification. J. Am. Soc. Nephrol. 2009, 21, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Zou, B.; Hou, Y.; Yan, W.; Chen, T.; Qu, S. Extracellular vesicles in vascular calcification. Clin. Chim. Acta 2019, 499, 118–122. [Google Scholar] [CrossRef]
- Coscas, R.; Bensussan, M.; Jacob, M.-P.; Louedec, L.; Massy, Z.; Sadoine, J.; Daudon, M.; Chaussain, C.; Bazin, D.; Michel, J.-B. Free DNA precipitates calcium phosphate apatite crystals in the arterial wall In Vivo. Atherosclerosis 2017, 259, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Cuervo, A.M. Autophagy in the Cellular Energetic Balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanahan, C.M. Autophagy and matrix vesicles: New partners in vascular calcification. Kidney Int. 2013, 83, 984–986. [Google Scholar] [CrossRef] [Green Version]
- Cochain, C.; Zernecke, A. Macrophages in vascular inflammation and atherosclerosis. Pflüg. Arch. Eur. J. Physiol. 2017, 469, 485–499. [Google Scholar] [CrossRef]
- Byon, C.H.; Sun, Y.; Chen, J.; Yuan, K.; Mao, X.; Heath, J.M.; Anderson, P.G.; Tintut, Y.; Demer, L.L.; Wang, D.; et al. Runx2-Upregulated Receptor Activator of Nuclear Factor κB Ligand in Calcifying Smooth Muscle Cells Promotes Migration and Osteoclastic Differentiation of Macrophages. Arter. Thromb. Vasc. Biol. 2011, 31, 1387–1396. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef]
- Adamson, S.; Leitinger, N. Phenotypic modulation of macrophages in response to plaque lipids. Curr. Opin. Lipidol. 2011, 22, 335–342. [Google Scholar] [CrossRef]
- Zhang, X.; Li, J.; Qin, J.-J.; Cheng, W.-L.; Zhu, X.; Gong, F.-H.; She, Z.; Huang, Z.; Xia, H.; Li, H. Oncostatin M receptor β deficiency attenuates atherogenesis by inhibiting JAK2/STAT3 signaling in macrophages. J. Lipid Res. 2017, 58, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, C.T.; Agarwal, S.; Ranganathan, K.; Wong, V.W.; Loder, S.; Li, J.; Delano, M.J.; Levi, B. Trauma-induced heterotopic bone formation and the role of the immune system. J. Trauma Acute Care Surg. 2016, 80, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Cai, B.; Zhang, Z.; Zhang, Y.; Wang, L.; Liu, K.; Zhang, H.; Sun, L.; Cai, H.; Lu, G.; et al. CDKN2B Methylation and Aortic Arch Calcification in Patients with Ischemic Stroke. J. Atheroscler. Thromb. 2017, 24, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Gao, C.; Liang, Y.; Wang, M.; Huang, Y.; Ma, W.; Li, T.; Jia, Y.; Yu, F.; Zhu, W.; et al. Shift of Macrophage Phenotype Due to Cartilage Oligomeric Matrix Protein Deficiency Drives Atherosclerotic Calcification. Circ. Res. 2016, 119, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C.; Riboldi, E.; Ippolito, A.; Sica, A. Molecular and epigenetic basis of macrophage polarized activation. Semin. Immunol. 2015, 27, 237–248. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Alternatively activated macrophages exhibit an anticalcifying activity dependent on extracellular ATP/pyrophosphate metabolism. Am. J. Physiol. Physiol. 2016, 310, C788–C799. [Google Scholar] [CrossRef] [Green Version]
- Villa-Bellosta, R.; Sorribas, V. Prevention of Vascular Calcification by Polyphosphates and Nucleotides. Circ. J. 2013, 77, 2145–2151. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.; Polewski, M.D.; Van Etten, D.; Terkeltaub, R.A. Chondrogenesis Mediated by PP i Depletion Promotes Spontaneous Aortic Calcification in NPP1−/− Mice. Arter. Thromb. Vasc. Biol. 2005, 25, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, Z.; Zhang, L.; Yan, J.; Shao, C.; Jing, L.; Li, L.; Wang, Z. Role of Macrophages in the Progression and Regression of Vascular Calcification. Front. Pharmacol. 2020, 11, 661. [Google Scholar] [CrossRef]
- Dube, P.R.; Birnbaumer, L.; Vazquez, G. Evidence for constitutive bone morphogenetic protein-2 secretion by M1 macrophages: Constitutive auto/paracrine osteogenic signaling by BMP-2 in M1 macrophages. Biochem. Biophys. Res. Commun. 2017, 491, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Dube, P.R.; Chikkamenahalli, L.L.; Birnbaumer, L.; Vazquez, G. Reduced calcification and osteogenic features in advanced atherosclerotic plaques of mice with macrophage-specific loss of TRPC3. Atherosclerosis 2018, 270, 199–204. [Google Scholar] [CrossRef]
- Solanki, S.; Dube, P.R.; Tano, J.-Y.; Birnbaumer, L.; Vazquez, G. Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages. Am. J. Physiol. Physiol. 2014, 307, C521–C531. [Google Scholar] [CrossRef] [Green Version]
- Solanki, S.; Dube, P.R.; Birnbaumer, L.; Vazquez, G. Reduced Necrosis and Content of Apoptotic M1 Macrophages in Advanced Atherosclerotic Plaques of Mice with Macrophage-Specific Loss of Trpc3. Sci. Rep. 2017, 7, 42526. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Chatrou, M.L.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.M.; Alvarez-Hernandez, D.; et al. Vascular Smooth Muscle Cell Calcification Is Mediated by Regulated Exosome Secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Ciceri, P.; Galassi, A.; Mangano, M.; Carugo, S.; Capelli, I.; Cianciolo, G. The Key Role of Phosphate on Vascular Calcification. Toxins 2019, 11, 213. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wei, G.; Liu, B.; Zhou, X.; Xiao, H.; Dong, N.; Li, F. The Role of High Mobility Group Box 1 Protein in Interleukin-18-Induced Myofibroblastic Transition of Valvular Interstitial Cells. Cardiology 2016, 135, 168–178. [Google Scholar] [CrossRef]
- Kwon, D.-H.; Kim, Y.-K.; Kook, H. New Aspects of Vascular Calcification: Histone Deacetylases and Beyond. J. Korean Med. Sci. 2017, 32, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Hénaut, L.; Candellier, A.; Boudot, C.; Grissi, M.; Mentaverri, R.; Choukroun, G.; Brazier, M.; Kamel, S.; Massy, Z.A. New Insights into the Roles of Monocytes/Macrophages in Cardiovascular Calcification Associated with Chronic Kidney Disease. Toxins 2019, 11, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, P.V.L.N.S.; Gouroju, S.; Bitla, A.R.; Vinapamula, K.S.; Manohar, S.M.; Vishnubhotla, S. Role of gut-derived uremic toxins on oxidative stress and inflammation in patients with chronic kidney disease. Indian J. Nephrol. 2017, 27, 359–364. [Google Scholar] [CrossRef]
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Mazière, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Novel phosphate-activated macrophages prevent ectopic calcification by increasing extracellular ATP and pyrophosphate. PLoS ONE 2017, 12, e0174998. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.J.; Ni, J.W.; Ding, F.H.; Fang, Y.H.; Wang, X.Q.; Wang, H.B.; Chen, X.N.; Chen, N.; Zhan, W.W.; Lu, L.; et al. p-Cresyl sulfate is associated with carotid arteriosclerosis in hemodialysis patients and promotes atherogenesis in apoE−/− mice. Kidney Int. 2016, 89, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Hahn, K.; Kanbay, M.; Lanaspa, M.A.; Johnson, R.J.; Ejaz, A.A. Serum uric acid and acute kidney injury: A mini review. J. Adv. Res. 2017, 8, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraci, G.; Mule’, G.; Morreale, M.; Cusumano, C.; Castiglia, A.; Gervasi, F.; D’Ignoto, F.; Mogavero, M.; Geraci, C.; Cottone, S. Association between uric acid and renal function in hypertensive patients: Which role for systemic vascular involvement? J. Am. Soc. Hypertens. 2016, 10, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Andrés, M.; Quintanilla, M.-A.; Sivera, F.; Sánchez-Payá, J.; Pascual, E.; Vela, P.; Ruiz-Nodar, J.-M. Silent Monosodium Urate Crystal Deposits Are Associated with Severe Coronary Calcification in Asymptomatic Hyperuricemia: An Exploratory Study. Arthritis Rheumatol. 2016, 68, 1531–1539. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, S.-H.; Choi, A.R.; Kim, S.; Choi, H.Y.; Kim, H.J.; Park, H.-C. Asymptomatic hyperuricemia is independently associated with coronary artery calcification in the absence of overt coronary artery disease. Medicine 2017, 96, e6565. [Google Scholar] [CrossRef]
- Kiss, L.Z.; Bagyura, Z.; Csobay-Novák, C.; Lux, Á.; Polgár, L.; Jermendy, Á.; Soós, P.; Szelid, Z.; Maurovich-Horvat, P.; Becker, D.; et al. Serum Uric Acid Is Independently Associated with Coronary Calcification in an Asymptomatic Population. J. Cardiovasc. Transl. Res. 2019, 12, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Aroor, A.R.; Jia, G.; Habibi, J.; Sun, Z.; Ramirez-Perez, F.I.; Brady, B.; Chen, D.; Martinez-Lemus, L.A.; Manrique, C.; Nistala, R.; et al. Uric acid promotes vascular stiffness, maladaptive inflammatory responses and proteinuria in western diet fed mice. Metab. Clin. Exp. 2017, 74, 32–40. [Google Scholar] [CrossRef]
- Andrews, E.S.; Perrenoud, L.; Nowak, K.L.; You, Z.; Pasch, A.; Chonchol, M.; Kendrick, J.; Jalal, D. Examining the effects of uric acid-lowering on markers vascular of calcification and CKD-MBD; A post-hoc analysis of a randomized clinical trial. PLoS ONE 2018, 13, e0205831. [Google Scholar] [CrossRef] [PubMed]
- Rogacev, K.S.; Seiler, S.; Zawada, A.M.; Reichart, B.; Herath, E.; Roth, D.; Ulrich, C.; Fliser, D.; Heine, G.H. CD14++CD16+ monocytes and cardiovascular outcome in patients with chronic kidney disease. Eur. Heart J. 2010, 32, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizobuchi, M.; Towler, D.; Slatopolsky, E. Vascular Calcification: The Killer of Patients with Chronic Kidney Disease. J. Am. Soc. Nephrol. 2009, 20, 1453–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergenreider, E.; Heydt, S.; Tréguer, K.; Boettger, T.; Horrevoets, A.J.G.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012, 14, 249–256. [Google Scholar] [CrossRef]
- Shin, V.; Zebboudj, A.F.; Boström, K. Endothelial Cells Modulate Osteogenesis in Calcifying Vascular Cells. J. Vasc. Res. 2004, 41, 193–201. [Google Scholar] [CrossRef]
- Yao, Y.; Jumabay, M.; Ly, A.; Radparvar, M.; Cubberly, M.R.; Boström, K.I. A Role for the Endothelium in Vascular Calcification. Circ. Res. 2013, 113, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Carmona, A.; Triviño, F.; Rodriguez, M.; Alvarez-Benito, M.; Martín-Malo, A.; Alvarez-Lara, M.-A.; Ramírez, R.; Aljama, P.; Carracedo, J. Endothelial damage and vascular calcification in patients with chronic kidney disease. Am. J. Physiol. Physiol. 2014, 307, F1302–F1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Guihard, P.J.; Blazquez-Medela, A.M.; Guo, Y.; Moon, J.H.; Jumabay, M.; Boström, K.I.; Yao, Y. Serine Protease Activation Essential for Endothelial–Mesenchymal Transition in Vascular Calcification. Circ. Res. 2015, 117, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Boström, K.I.; Yao, J.; Guihard, P.J.; Blazquez-Medela, A.M.; Yao, Y. Endothelial-mesenchymal transition in atherosclerotic lesion calcification. Atherosclerosis 2016, 253, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Hjortnaes, J.; Shapero, K.; Goettsch, C.; Hutcheson, J.D.; Keegan, J.; Kluin, J.; Mayer, J.E.; Bischoff, J.; Aikawa, E. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis 2015, 242, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Tang, R.-N.; Liu, H.; Pan, M.-M.; Liu, B.-C. Cinacalcet ameliorates aortic calcification in uremic rats via suppression of endothelial-to-mesenchymal transition. Acta Pharmacol. Sin. 2016, 37, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Rutkovskiy, A.; Lund, M.; Siamansour, T.S.; Reine, T.M.; Kolset, S.O.; Sand, K.L.; Ignatieva, E.; Gordeev, M.L.; Stensløkken, K.-O.; Valen, G.; et al. Mechanical stress alters the expression of calcification-related genes in vascular interstitial and endothelial cells. Interact. Cardiovasc. Thorac. Surg. 2019, 28, 803–811. [Google Scholar] [CrossRef]
- Chiu, J.-J.; Usami, S.; Chien, S. Vascular endothelial responses to altered shear stress: Pathologic implications for atherosclerosis. Ann. Med. 2009, 41, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Urs, S.; Venkatesh, D.; Tang, Y.; Henderson, T.; Yang, X.; Friesel, R.E.; Rosen, C.J.; Liaw, L. Sprouty1 is a critical regulatory switch of mesenchymal stem cell lineage allocation. FASEB J. 2010, 24, 3264–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merle, B.; Garnero, P. The multiple facets of periostin in bone metabolism. Osteoporos. Int. 2012, 23, 1199–1212. [Google Scholar] [CrossRef] [PubMed]
- Canfield, A.E.; Farrington, C.; Dziobon, M.D.; Boot-Handford, R.P.; Heagerty, A.M.; Kumar, S.N.; Roberts, I.S.D. The involvement of matrix glycoproteins in vascular calcification and fibrosis: An immunohistochemical study. J. Pathol. 2001, 196, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Van Thienen, J.V.; Fledderus, J.O.; Dekker, R.J.; Rohlena, J.; Van Ijzendoorn, G.A.; Kootstra, N.A.; Pannekoek, H.; Horrevoets, A.J.G. Shear stress sustains atheroprotective endothelial KLF2 expression more potently than statins through mRNA stabilization. Cardiovasc. Res. 2006, 72, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of Endothelial Shear Stress in the Natural History of Coronary Atherosclerosis and Vascular Remodeling. J. Am. Coll. Cardiol. 2007, 49, 2379–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, S. Mechanotransduction and endothelial cell homeostasis: The wisdom of the cell. Am. J. Physiol. Circ. Physiol. 2007, 292, H1209–H1224. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.-C.; Liu, W.-C.; Zheng, C.-M.; Zheng, J.-Q.; Yen, T.-H.; Lu, K.-C. Role of Vitamin D in Uremic Vascular Calcification. BioMed Res. Int. 2017, 2017, 2803579. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-J.; Hsu, S.-C.; Huang, S.-M.; Lee, H.-S.; Lin, S.-H.; Tsai, C.-S.; Shih, C.-C.; Lin, C.-Y. Hyperphosphatemia induces protective autophagy in endothelial cells through the inhibition of Akt/mTOR signaling. J. Vasc. Surg. 2015, 62, 210–221.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.-Y.; Zhao, M.-M.; Cai, Y.; Guan, Q.-C.; Zhao, Y.; Guan, Y.; Kong, W.; Zhu, W.-G.; Xu, M.-J.; Wang, X. Phosphate-induced autophagy counteracts vascular calcification by reducing matrix vesicle release. Kidney Int. 2013, 83, 1042–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol. Dial. Transplant. 2019, 34, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Zhao, Y.; Wang, B.; Li, B.; Sheng, Y.; Liu, M.; Li, H.; Xiu, R. Endothelial Cells Promote Calcification in Aortic Smooth Muscle Cells from Spontaneously Hypertensive Rats. Cell. Physiol. Biochem. 2018, 49, 2371–2381. [Google Scholar] [CrossRef]
- Cianciolo, G.; Galassi, A.; Capelli, I.; Schillaci, R.; La Manna, G.; Cozzolino, M. Klotho-FGF23, Cardiovascular Disease, and Vascular Calcification: Black or White? Curr. Vasc. Pharmacol. 2018, 16, 143–156. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium Regulates Key Components of Vascular Smooth Muscle Cell–Derived Matrix Vesicles to Enhance Mineralization. Circ. Res. 2011, 109, e1–e12. [Google Scholar] [CrossRef] [Green Version]
- Houben, E.; Neradova, A.; Schurgers, L.J.; Vervloet, M. The influence of phosphate, calcium and magnesium on matrix Gla-protein and vascular calcification: A systematic review. G. Ital. Nefrol. 2017, 33, 1724–5590. [Google Scholar]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2009, 113, S1–S130. [Google Scholar]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human Vascular Smooth Muscle Cells Undergo Vesicle-Mediated Calcification in Response to Changes in Extracellular Calcium and Phosphate Concentrations: A Potential Mechanism for Accelerated Vascular Calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [Green Version]
- Disthabanchong, S. Vascular calcification in chronic kidney disease: Pathogenesis and clinical implication. World J. Nephrol. 2012, 1, 43–53. [Google Scholar] [CrossRef]
- Merjanian, R.; Budoff, M.; Adler, S.; Berman, N.; Mehrotra, R. Coronary artery, aortic wall, and valvular calcification in nondialyzed individuals with type 2 diabetes and renal disease. Kidney Int. 2003, 64, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamprea-Montealegre, J.A.; McClelland, R.L.; Astor, B.C.; Matsushita, K.; Shlipak, M.; de Boer, I.H.; Szklo, M. Chronic kidney disease, plasma lipoproteins, and coronary artery calcium incidence: The Multi-Ethnic Study of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, H.; Toto, R.; Peshock, R.; Cooper, R.; Victor, R. Association between Chronic Kidney Disease and Coronary Artery Calcification: The Dallas Heart Study. J. Am. Soc. Nephrol. 2004, 16, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Qunibi, W.Y.; AbouZahr, F.; Mizani, M.R.; Nolan, C.R.; Arya, R.; Hunt, K.J. Cardiovascular calcification in Hispanic Americans (HA) with chronic kidney disease (CKD) due to type 2 diabetes. Kidney Int. 2005, 68, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Russo, D.; Palmiero, G.; De Blasio, A.P.; Balletta, M.M.; Andreucci, V.E. Coronary artery calcification in patients with CRF not undergoing dialysis. Am. J. Kidney Dis. 2004, 44, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-W.; Adler, S.G.; Budoff, M.J.; Takasu, J.; Ashai, J.; Mehrotra, R. Coronary artery calcification and mortality in diabetic patients with proteinuria. Kidney Int. 2010, 77, 1107–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellasi, A.; Raggi, P. Vascular calcification in patients with kidney disease: Techniques and Technologies to Assess Vascular Calcification. Semin. Dial. 2007, 20, 129–133. [Google Scholar] [CrossRef]
- Uhlig, K. There Is No Practical Utility in Routinely Screening Dialysis Patients for Vascular Calcification. Semin. Dial. 2010, 23, 277–279. [Google Scholar] [CrossRef]
- Beto, J.; Bhatt, N.; Gerbeling, T.; Patel, C.; Drayer, D. Overview of the 2017 KDIGO CKD-MBD Update: Practice Implications for Adult Hemodialysis Patients. J. Ren. Nutr. 2019, 29, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Ix, J.H.; Katz, R.; Kestenbaum, B.; Fried, L.F.; Kramer, H.; Stehman-Breen, C.; Shlipak, M.G. Association of Mild to Moderate Kidney Dysfunction and Coronary Calcification. J. Am. Soc. Nephrol. 2008, 19, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Jun, M.; Lv, J.; Perkovic, V.; Jardine, M.J. Managing cardiovascular risk in people with chronic kidney disease: A review of the evidence from randomized controlled trials. Ther. Adv. Chronic Dis. 2011, 2, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Himmelsbach, A.; Ciliox, C.; Goettsch, C. Cardiovascular Calcification in Chronic Kidney Disease—Therapeutic Opportunities. Toxins 2020, 12, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal, S.A.; Vandermeer, B.; Raggi, P.; Mendelssohn, D.C.; Chatterley, T.; Dorgan, M.; Lok, C.E.; Fitchett, D.; Tsuyuki, R.T. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: An updated systematic review and meta-analysis. Lancet 2013, 382, 1268–1277. [Google Scholar] [CrossRef]
- Block, G.A.; Bushinsky, D.A.; Cheng, S.; Cunningham, J.; Dehmel, B.; Drueke, T.B.; Ketteler, M.; KewalRamani, R.; Martin, K.J.; Moe, S.M.; et al. Effect of Etelcalcetide vs. Cinacalcet on Serum Parathyroid Hormone in Patients Receiving Hemodialysis with Secondary Hyperparathyroidism. JAMA 2017, 317, 156–164. [Google Scholar] [CrossRef]
- Chitalia, N.; Recio-Mayoral, A.; Kaski, J.C.; Banerjee, D. Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis 2012, 220, 265–268. [Google Scholar] [CrossRef]
- Perazella, M.A.; Markowitz, G.S. Bisphosphonate nephrotoxicity. Kidney Int. 2008, 74, 1385–1393. [Google Scholar] [CrossRef] [Green Version]
- Bergner, R.; Diel, I.J.; Henrich, D.; Hoffmann, M.; Uppenkamp, M. Differences in Nephrotoxicity of Intravenous Bisphosphonates for the Treatment of Malignancy Related Bone Disease. Oncol. Res. Treat. 2006, 29, 534–540. [Google Scholar] [CrossRef]
- Louvet, L.; Büchel, J.; Steppan, S.; Passlick-Deetjen, J.; Massy, Z.A. Magnesium prevents phosphate-induced calcification in human aortic vascular smooth muscle cells. Nephrol. Dial. Transplant. 2012, 28, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Sharples, E.J.; Pereira, D.; Summers, S.; Cunningham, J.; Rubens, M.; Goldsmith, D.; Yaqoob, M.M. Coronary artery calcification measured with electron-beam computerized tomography correlates poorly with coronary artery angiography in dialysis patients. Am. J. Kidney Dis. 2004, 43, 313–319. [Google Scholar] [CrossRef]
- Maniscalco, B.S.; Taylor, K.A. Calcification in coronary artery disease can be reversed by EDTA–tetracycline long-term chemotherapy. Pathophysiology 2004, 11, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Krieger, N.S.; Frick, K.K.; Bushinsky, D.A. Mechanism of acid-induced bone resorption. Curr. Opin. Nephrol. Hypertens. 2004, 13, 423–436. [Google Scholar] [CrossRef]
- Mendoza, F.J.; Lopez, I.; Montes de Oca, A.; Perez, J.; Rodriguez, M.; Aguilera-Tejero, E. Metabolicacidosis inhibits soft tissue calcification in uremic rats. Kidney Int. 2008, 73, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Simpson, C.L.; Lindley, S.; Eisenberg, C.; Basalyga, D.M.; Starcher, B.C.; Simionescu, D.T.; Vyavahare, N.R. Toward cell therapy for vascular calcification: Osteoclast-mediated demineralization of calcifiedelastin. Cardiovasc. Pathol. 2007, 16, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Neven, E.; D’Haese, P.C. Vascular Calcification in Chronic Renal Failure. Circ. Res. 2011, 108, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Shobeiri, N.; Adams, M.; Holden, R. Vascular Calcification in Animal Models of CKD: A Review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef]
- Herrmann, J.; Babic, M.; Tölle, M.; Van Der Giet, M.; Schuchardt, M. Research Models for Studying Vascular Calcification. Int. J. Mol. Sci. 2020, 21, 2204. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Eddington, H.; Kalra, P.A. Vascular calcification: Lessons from scientific models. J. Ren. Care 2009, 35, 51–56. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Promoters | Inhibitors |
|---|---|
| Bone morphogenetic protein (BMP) 2, 4, and 6 Osteocalcin Alkaline phosphatase (Sex determining region Y)-box 9 (SOX9) Osterix Matrix metalloproteinase (MMP) 2, 3, and 7 Runt-related transcription factor 2 (Runx2) Calcium Phosphate Glucose Advanced glycation end products Oxidized low-density lipoproteins Collagen I Receptor activator of nuclear factor-kB ligand (RANKL) | Matrix Gla protein (MGP) Osteopontin Osteoprotegerin Vitamin K Magnesium Bone morphogenetic protein 7 (BMP7) Fetuin Klotho Parathyroid hormone (PTH) Pyrophosphate Carbonic anhydrase Collagen IV Inorganic pyrophosphate (PPi) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dube, P.; DeRiso, A.; Patel, M.; Battepati, D.; Khatib-Shahidi, B.; Sharma, H.; Gupta, R.; Malhotra, D.; Dworkin, L.; Haller, S.; et al. Vascular Calcification in Chronic Kidney Disease: Diversity in the Vessel Wall. Biomedicines 2021, 9, 404. https://doi.org/10.3390/biomedicines9040404
Dube P, DeRiso A, Patel M, Battepati D, Khatib-Shahidi B, Sharma H, Gupta R, Malhotra D, Dworkin L, Haller S, et al. Vascular Calcification in Chronic Kidney Disease: Diversity in the Vessel Wall. Biomedicines. 2021; 9(4):404. https://doi.org/10.3390/biomedicines9040404
Chicago/Turabian StyleDube, Prabhatchandra, Armelle DeRiso, Mitra Patel, Dhanushya Battepati, Bella Khatib-Shahidi, Himani Sharma, Rajesh Gupta, Deepak Malhotra, Lance Dworkin, Steven Haller, and et al. 2021. "Vascular Calcification in Chronic Kidney Disease: Diversity in the Vessel Wall" Biomedicines 9, no. 4: 404. https://doi.org/10.3390/biomedicines9040404