
Copper-Associated Oxidative Stress Contributes to Cellular Inflammatory Responses in Cystic Fibrosis

 and
and
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Measurement of the cAMP-Stimulated Iodide Conductance of the Plasma Membrane
2.3. Extraction of Total RNA and Reverse Transcription
2.4. Quantitative Polymerase Chain Reaction
2.5. Cells Lysates
2.6. Electrophoresis and Immunoblotting
2.7. Cell Viability Using MTT Test
2.8. Intracellular Copper Determination
2.9. ROS Measurements
2.10. Catalase Enzyme Activity Assays
2.11. Cu/Zn- and Mn-Superoxide Dismutase (SOD) Activities
2.12. Glutathione Peroxidase (GPx) Activity
2.13. Mitochondrial Isolation
2.14. GDH and LDH Activities
2.15. Aconitase and Fumarase Activity Assays
2.16. Cytokine Secretion by Sandwich ELISA
2.17. Statistical Analysis
3. Results
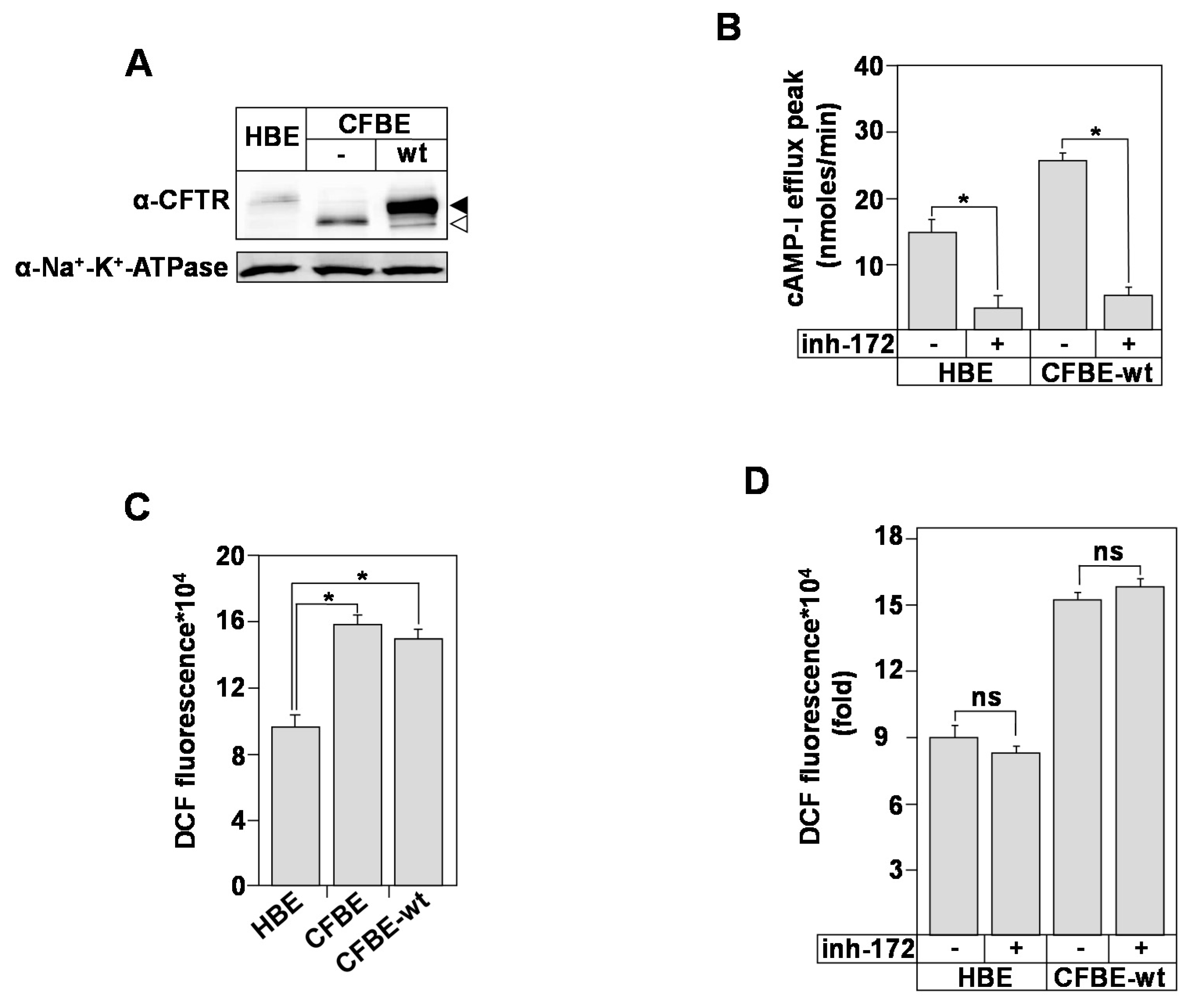
3.1. Bronchial Epithelial ROS Production Is Independent of CFTR Expression and Function
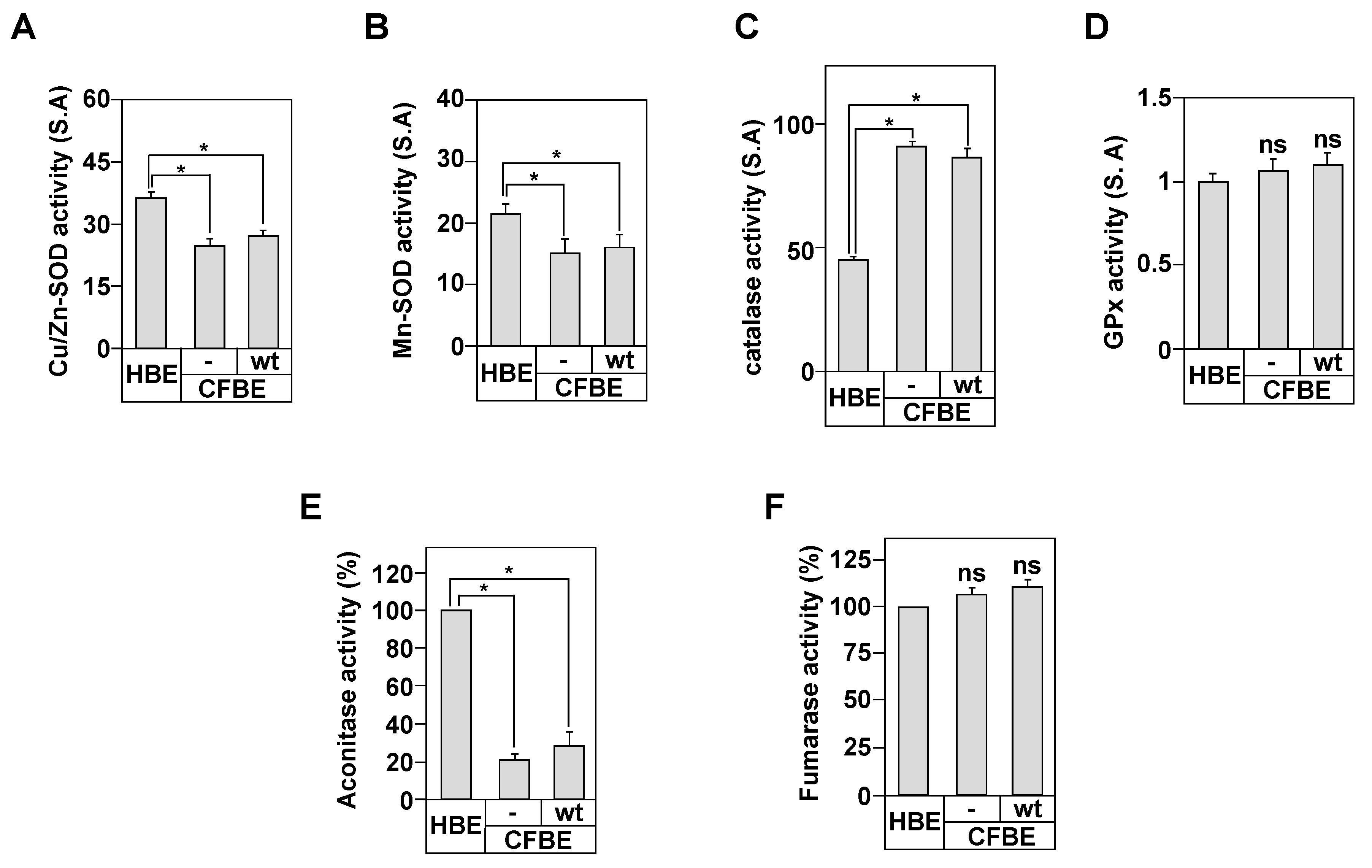
3.2. Antioxidants Enzymes Activities in the Bronchial Epithelial Cells
3.3. In Vitro Markers of Mitochondrial Oxidative Stress
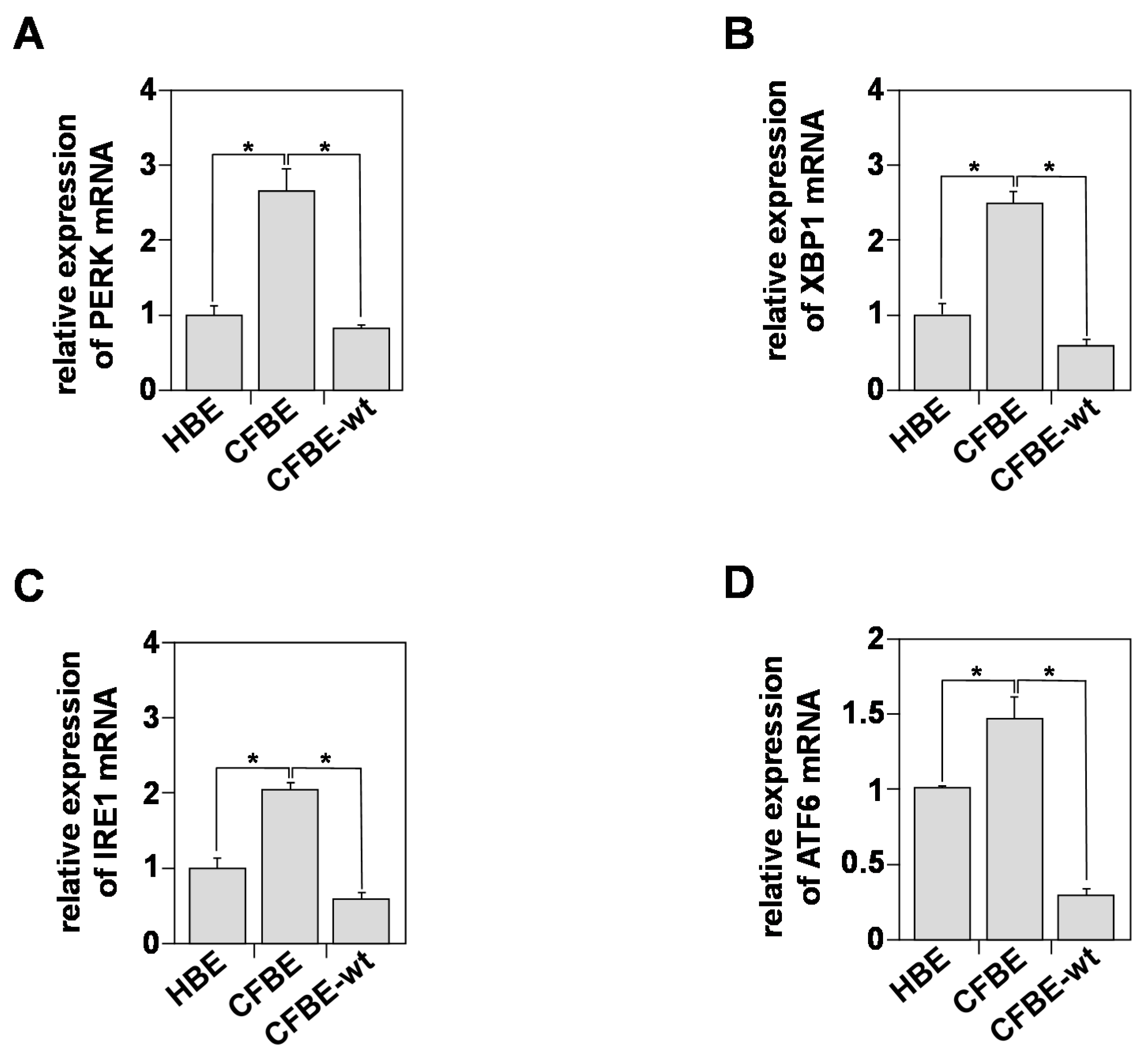
3.4. In Vitro Indicators of Endoplasmic Reticulum (ER) Stress
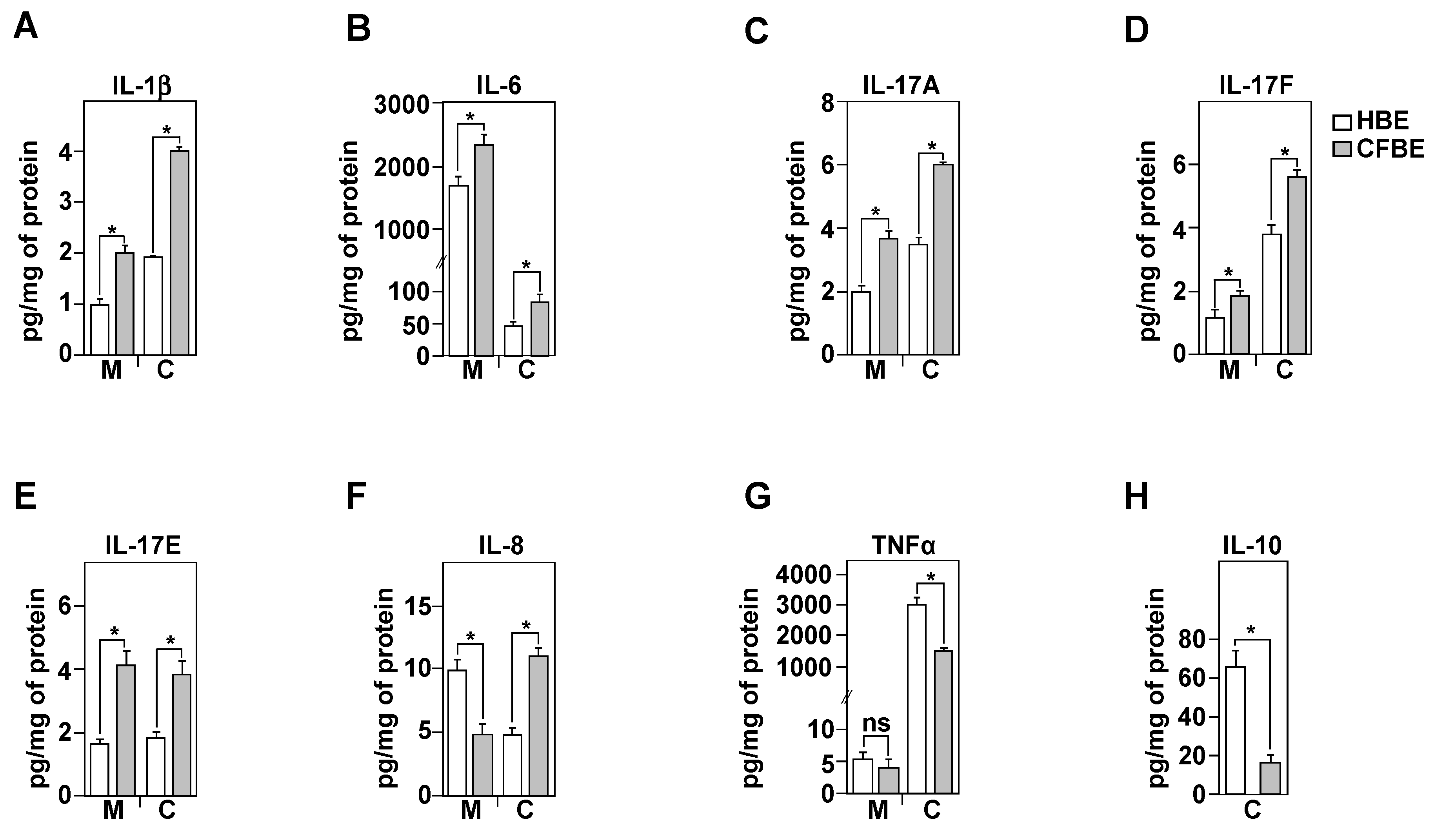
3.5. Inflammatory Profile of Healthy and CF Bronchial Epithelial Cells
3.6. IL-8 Secretion Is Associated with CFTR Function
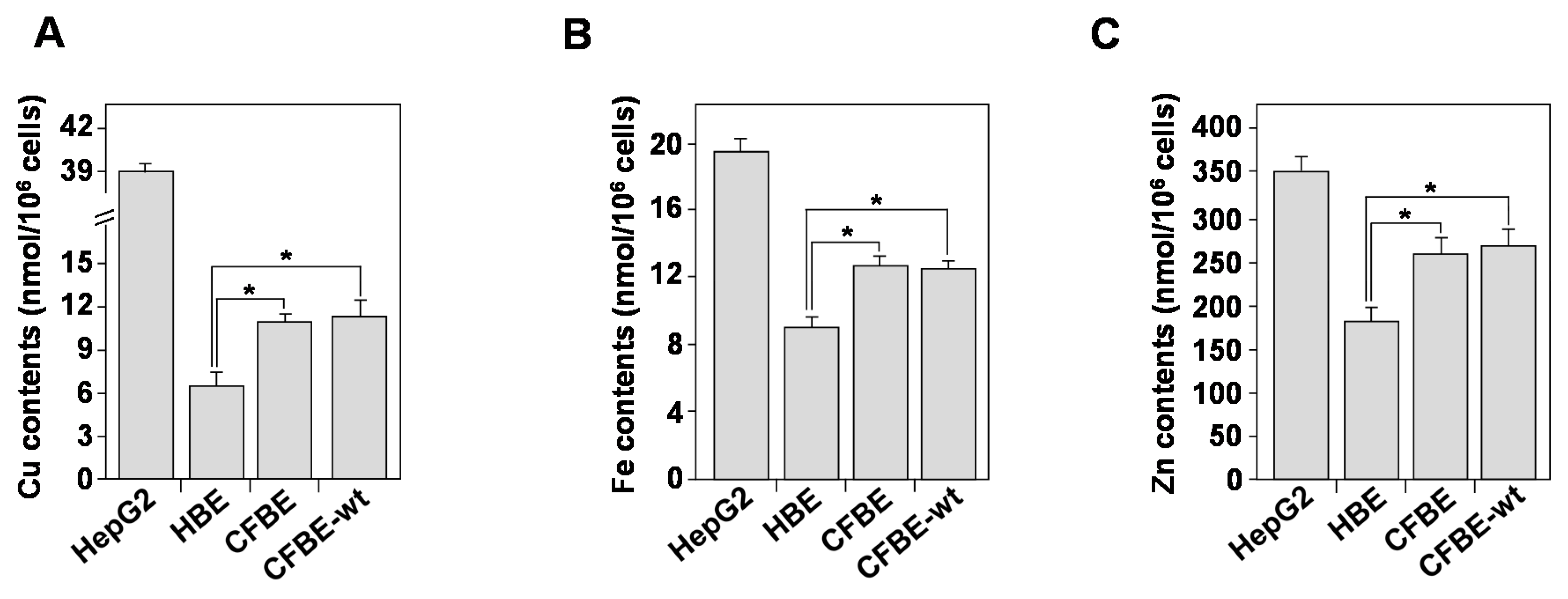
3.7. Homeostasis of Bioactive Trace Metals Is Dysregulated in CF Bronchial Epithelial Cells
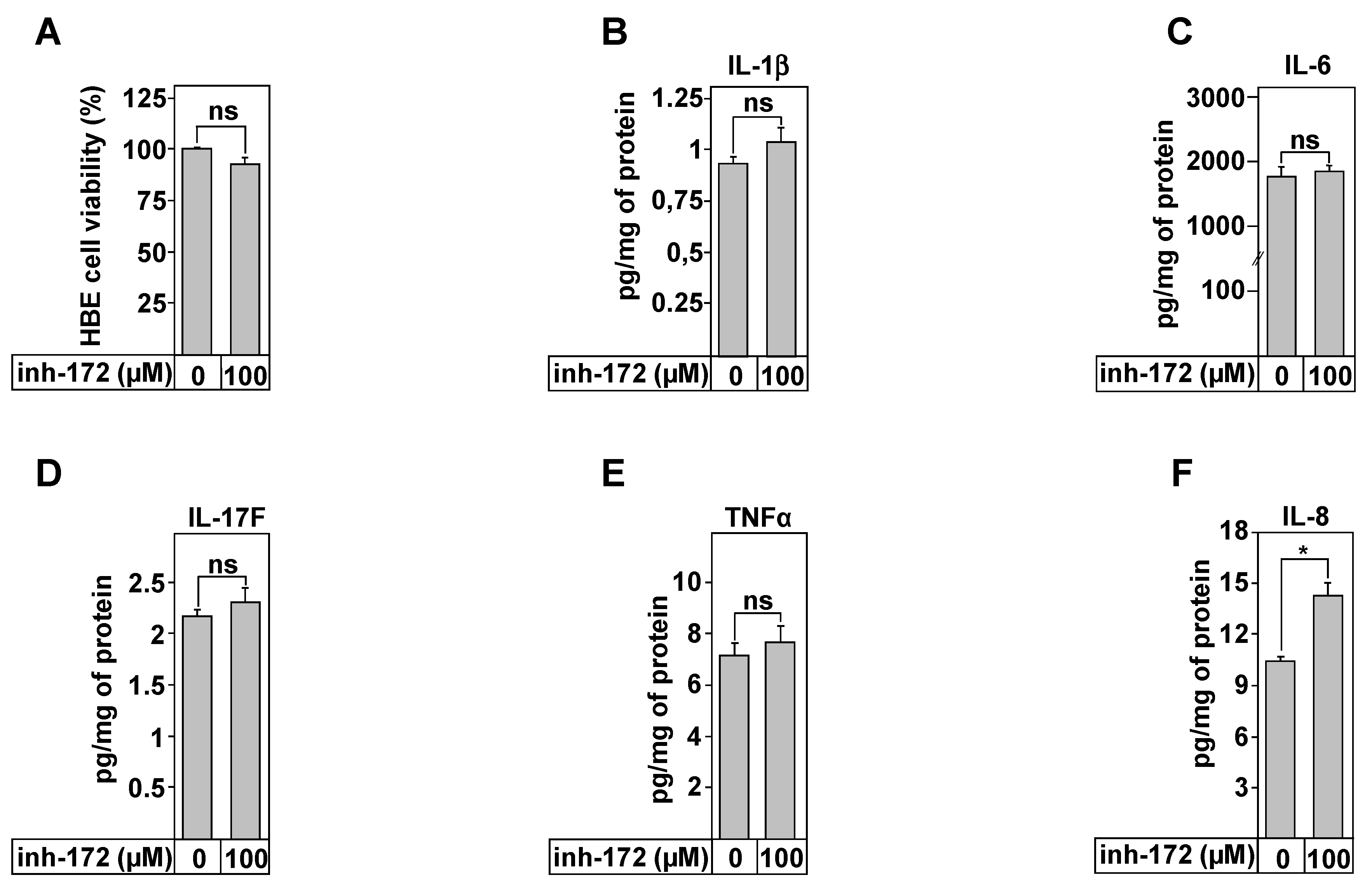
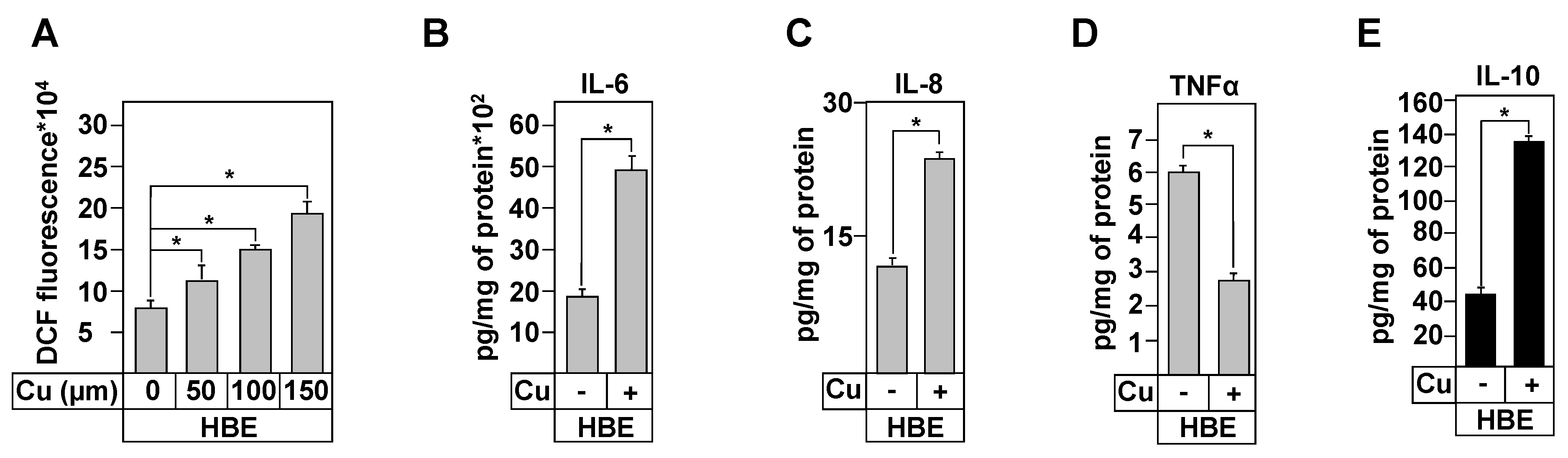
3.8. Effect of Copper Treatment on Healthy and CF Inflammation Responses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boucher, R.C. An overview of the pathogenesis of cystic fibrosis lung disease. Adv. Drug Deliv. Rev. 2002, 54, 1359–1371. [Google Scholar] [CrossRef]
- Riordan, J.R.; Chang, X.B. CFTR, a channel with the structure of a transporter. Biochim. Biophys. Acta 1992, 1101, 221–222. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Mohamed, A.; Kartner, N.; Chang, X.B.; Riordan, J.R.; Grinstein, S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994, 13, 6076–6086. [Google Scholar] [CrossRef]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell. Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H. Infection and inflammation in cystic fibrosis: A short review. J. Cyst. Fibros. 2005, 4 (Suppl. 2), 3–5. [Google Scholar] [CrossRef]
- Kube, D.; Sontich, U.; Fletcher, D.; Davis, P.B. Proinflammatory cytokine responses to P. aeruginosa infection in human airway epithelial cell lines. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L493–L502. [Google Scholar] [CrossRef]
- Perez, A.; Issler, A.C.; Cotton, C.U.; Kelley, T.J.; Verkman, A.S.; Davis, P.B. CFTR inhibition mimics the cystic fibrosis inflammatory profile. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L383–L395. [Google Scholar] [CrossRef]
- Saadane, A.; Soltys, J.; Berger, M. Role of IL-10 deficiency in excessive nuclear factor-kappaB activation and lung inflammation in cystic fibrosis transmembrane conductance regulator knockout mice. J. Allergy Clin. Immunol. 2005, 115, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Tabary, O.; Escotte, S.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Jacquot, J. High susceptibility for cystic fibrosis human airway gland cells to produce IL-8 through the I kappa B kinase alpha pathway in response to extracellular NaCl content. J. Immunol. 2000, 164, 3377–3384. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.S.; Stewart, P.W.; Leigh, M.W.; Noah, T.L. Quantitation of inflammatory responses to bacteria in young cystic fibrosis and control patients. Am. J. Respir. Crit. Care Med. 1999, 160, 186–191. [Google Scholar] [CrossRef]
- Dakin, C.J.; Numa, A.H.; Wang, H.; Morton, J.R.; Vertzyas, C.C.; Henry, R.L. Inflammation, infection, and pulmonary function in infants and young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2002, 165, 904–910. [Google Scholar] [CrossRef]
- Bergoin, C.; Gosset, P.; Lamblin, C.; Bolard, F.; Turck, D.; Tonnel, A.B.; Wallaert, B. Cell and cytokine profile in nasal secretions in cystic fibrosis. J. Cyst. Fibros. 2002, 1, 110–115. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.; Stecenko, A.A.; King, G.; Blackwell, T.R.; Brigham, K.L.; Christman, J.W.; Blackwell, T.S. Exaggerated activation of nuclear factor-kappaB and altered IkappaB-beta processing in cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell. Mol. Biol. 2000, 23, 396–403. [Google Scholar] [CrossRef]
- Escotte, S.; Tabary, O.; Dusser, D.; Majer-Teboul, C.; Puchelle, E.; Jacquot, J. Fluticasone reduces IL-6 and IL-8 production of cystic fibrosis bronchial epithelial cells via IKK-beta kinase pathway. Eur. Respir. J. 2003, 21, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Rottner, M.; Freyssinet, J.M.; Martinez, M.C. Mechanisms of the noxious inflammatory cycle in cystic fibrosis. Respir. Res. 2009, 10, 23. [Google Scholar] [CrossRef]
- Tabary, O.; Boncoeur, E.; de Martin, R.; Pepperkok, R.; Clement, A.; Schultz, C.; Jacquot, J. Calcium-dependent regulation of NF-(kappa)B activation in cystic fibrosis airway epithelial cells. Cell. Signal. 2006, 18, 652–660. [Google Scholar] [CrossRef]
- Courtney, J.M.; Ennis, M.; Elborn, J.S. Cytokines and inflammatory mediators in cystic fibrosis. J. Cyst. Fibros. 2004, 3, 223–231. [Google Scholar] [CrossRef]
- van der Vliet, A.; Eiserich, J.P.; Marelich, G.P.; Halliwell, B.; Cross, C.E. Oxidative stress in cystic fibrosis: Does it occur and does it matter? Adv. Pharmacol. 1997, 38, 491–513. [Google Scholar] [CrossRef]
- Brown, R.K.; Kelly, F.J. Evidence for increased oxidative damage in patients with cystic fibrosis. Pediatr. Res. 1994, 36, 487–493. [Google Scholar] [CrossRef]
- Haleng, J.; Pincemail, J.; Defraigne, J.O.; Charlier, C.; Chapelle, J.P. Oxidative stress. Rev. Med. Liege 2007, 62, 628–638. [Google Scholar]
- Galli, F.; Battistoni, A.; Gambari, R.; Pompella, A.; Bragonzi, A.; Pilolli, F.; Iuliano, L.; Piroddi, M.; Dechecchi, M.C.; Cabrini, G.; et al. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim. Biophys. Acta 2012, 1822, 690–713. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; White, T.B.; Cross, C.E.; Forman, H.J.; Sokol, R.J.; Borowitz, D. Antioxidants in cystic fibrosis. Conclusions from the CF antioxidant workshop, Bethesda, Maryland, November 11–12, 2003. Free Radic. Biol. Med. 2007, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.; Lubamba, B.A. Role of IRE1alpha/XBP-1 in Cystic Fibrosis Airway Inflammation. Int. J. Mol. Sci. 2017, 18, 118. [Google Scholar] [CrossRef]
- Day, B.J.; van Heeckeren, A.M.; Min, E.; Velsor, L.W. Role for cystic fibrosis transmembrane conductance regulator protein in a glutathione response to bronchopulmonary pseudomonas infection. Infect. Immun. 2004, 72, 2045–2051. [Google Scholar] [CrossRef]
- Back, E.I.; Frindt, C.; Nohr, D.; Frank, J.; Ziebach, R.; Stern, M.; Ranke, M.; Biesalski, H.K. Antioxidant deficiency in cystic fibrosis: When is the right time to take action? Am. J. Clin. Nutr. 2004, 80, 374–384. [Google Scholar] [CrossRef]
- Velsor, L.W.; Kariya, C.; Kachadourian, R.; Day, B.J. Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Respir. Cell. Mol. Biol. 2006, 35, 579–586. [Google Scholar] [CrossRef]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuca, K.; Musilek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef]
- Kouadri, A.; El Khatib, M.; Cormenier, J.; Chauvet, S.; Zeinyeh, W.; El Khoury, M.; Macari, L.; Richaud, P.; Coraux, C.; Michaud-Soret, I.; et al. Involvement of the Prion Protein in the Protection of the Human Bronchial Epithelial Barrier against Oxidative Stress. Antioxid. Redox Signal. 2019, 31, 59–74. [Google Scholar] [CrossRef]
- Ma, T.; Thiagarajah, J.R.; Yang, H.; Sonawane, N.D.; Folli, C.; Galietta, L.J.; Verkman, A.S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Investig. 2002, 110, 1651–1658. [Google Scholar] [CrossRef]
- Benharouga, M.; Haardt, M.; Kartner, N.; Lukacs, G.L. COOH-terminal truncations promote proteasome-dependent degradation of mature cystic fibrosis transmembrane conductance regulator from post-Golgi compartments. J. Cell. Biol. 2001, 153, 957–970. [Google Scholar] [CrossRef]
- Alfaidy, N.; Chauvet, S.; Donadio-Andrei, S.; Salomon, A.; Saoudi, Y.; Richaud, P.; Aude-Garcia, C.; Hoffmann, P.; Andrieux, A.; Moulis, J.M.; et al. Prion protein expression and functional importance in developmental angiogenesis: Role in oxidative stress and copper homeostasis. Antioxid. Redox Signal. 2013, 18, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Beers, R.F., Jr.; Sizer, I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef]
- Marklund, S.; Marklund, G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Flohe, L.; Gunzler, W.A. Assays of glutathione peroxidase. Methods Enzymol. 1984, 105, 114–121. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Lopez-Perez, M.J.; Enriquez, J.A. Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods 2002, 26, 292–297. [Google Scholar] [CrossRef]
- Andersson, B.S.; Jones, D.P. Use of digitonin fractionation to determine mitochondrial transmembrane ion distribution in cells during anoxia. Anal. Biochem. 1985, 146, 164–172. [Google Scholar] [CrossRef]
- Schmidt, E.; Schmidt, F.W. Methods and value of determination of glutamic acid dehydrogenase activity in the serum. A contribution to the importance of examination of enzyme relations in the serum. Klin. Wochenschr. 1962, 40, 962–969. [Google Scholar] [CrossRef]
- Moskwa, P.; Lorentzen, D.; Excoffon, K.J.; Zabner, J.; McCray, P.B., Jr.; Nauseef, W.M.; Dupuy, C.; Banfi, B. A novel host defense system of airways is defective in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 174–183. [Google Scholar] [CrossRef]
- Boncoeur, E.; Criq, V.S.; Bonvin, E.; Roque, T.; Henrion-Caude, A.; Gruenert, D.C.; Clement, A.; Jacquot, J.; Tabary, O. Oxidative stress induces extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase in cystic fibrosis lung epithelial cells: Potential mechanism for excessive IL-8 expression. Int. J. Biochem. Cell. Biol. 2008, 40, 432–446. [Google Scholar] [CrossRef]
- Ambrosio, G.; Zweier, J.L.; Duilio, C.; Kuppusamy, P.; Santoro, G.; Elia, P.P.; Tritto, I.; Cirillo, P.; Condorelli, M.; Chiariello, M.; et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J. Biol. Chem. 1993, 268, 18532–18541. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Martino, M.E.; Olsen, J.C.; Fulcher, N.B.; Wolfgang, M.C.; O’Neal, W.K.; Ribeiro, C.M. Airway epithelial inflammation-induced endoplasmic reticulum Ca2+ store expansion is mediated by X-box binding protein-1. J. Biol. Chem. 2009, 284, 14904–14913. [Google Scholar] [CrossRef]
- Kelly, M.; Trudel, S.; Brouillard, F.; Bouillaud, F.; Colas, J.; Nguyen-Khoa, T.; Ollero, M.; Edelman, A.; Fritsch, J. Cystic fibrosis transmembrane regulator inhibitors CFTR(inh)-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition. J. Pharmacol. Exp. Ther. 2010, 333, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Mackey, M.M.; Diaz, A.A.; Cox, D.P. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: Implications for diseases associated with iron accumulation. Redox Rep. 2009, 14, 102–108. [Google Scholar] [CrossRef]
- Zwolak, I. The Role of Selenium in Arsenic and Cadmium Toxicity: An Updated Review of Scientific Literature. Biol. Trace Elem. Res. 2020, 193, 44–63. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Anderson, G.J.; Bell, S.C.; Reid, D.W. Elevated metal concentrations in the CF airway correlate with cellular injury and disease severity. J. Cyst. Fibros. 2014, 13, 289–295. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef]
- Voisin, G.; Bouvet, G.F.; Legendre, P.; Dagenais, A.; Masse, C.; Berthiaume, Y. Oxidative stress modulates the expression of genes involved in cell survival in DeltaF508 cystic fibrosis airway epithelial cells. Physiol. Genom. 2014, 46, 634–646. [Google Scholar] [CrossRef]
- Valdivieso, A.G.; Santa-Coloma, T.A. CFTR activity and mitochondrial function. Redox Biol. 2013, 1, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Rab, A.; Jurkuvenaite, A.; Mazur, M.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Activation of the unfolded protein response by deltaF508 CFTR. Am. J. Respir. Cell. Mol. Biol. 2008, 39, 448–457. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; Boucher, R.C. Role of endoplasmic reticulum stress in cystic fibrosis-related airway inflammatory responses. Proc. Am. Thorac. Soc. 2010, 7, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Tabary, O.; Zahm, J.M.; Hinnrasky, J.; Couetil, J.P.; Cornillet, P.; Guenounou, M.; Gaillard, D.; Puchelle, E.; Jacquot, J. Selective up-regulation of chemokine IL-8 expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am. J. Pathol. 1998, 153, 921–930. [Google Scholar] [CrossRef]
- Bargagli, E.; Lavorini, F.; Pistolesi, M.; Rosi, E.; Prasse, A.; Rota, E.; Voltolini, L. Trace metals in fluids lining the respiratory system of patients with idiopathic pulmonary fibrosis and diffuse lung diseases. J. Trace Elem. Med. Biol. 2017, 42, 39–44. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence 5′→3′ | Size |
|---|---|---|
| PERK | FW: TCTGTTCAGCTCTGGGTTG TBW: CCGAAGTTCAAAGTGGCCAA | 158 bp |
| XBP-1 | FW: TGTCACCCCTCCAGAACATC BW: AAGGGAGGCTGGTAAGGAAC | 196 bp |
| IRE1 | FW: AGCAAGAGGACAGGCTCAAT BW: CATCTGAACTTCGGCATGGG | 205 pb |
| ATF6 | FW: GTGTCAGAGAACCAGAGGCT BW: GGTGCCTCCTTTGATTTGCA | 166 bp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouadri, A.; Cormenier, J.; Gemy, K.; Macari, L.; Charbonnier, P.; Richaud, P.; Michaud-Soret, I.; Alfaidy, N.; Benharouga, M. Copper-Associated Oxidative Stress Contributes to Cellular Inflammatory Responses in Cystic Fibrosis. Biomedicines 2021, 9, 329. https://doi.org/10.3390/biomedicines9040329
Kouadri A, Cormenier J, Gemy K, Macari L, Charbonnier P, Richaud P, Michaud-Soret I, Alfaidy N, Benharouga M. Copper-Associated Oxidative Stress Contributes to Cellular Inflammatory Responses in Cystic Fibrosis. Biomedicines. 2021; 9(4):329. https://doi.org/10.3390/biomedicines9040329
Chicago/Turabian StyleKouadri, Amal, Johanna Cormenier, Kevin Gemy, Laurence Macari, Peggy Charbonnier, Pierre Richaud, Isabelle Michaud-Soret, Nadia Alfaidy, and Mohamed Benharouga. 2021. "Copper-Associated Oxidative Stress Contributes to Cellular Inflammatory Responses in Cystic Fibrosis" Biomedicines 9, no. 4: 329. https://doi.org/10.3390/biomedicines9040329