Long-Term Systemic Expression of a Novel PD-1 Blocking Nanobody from an AAV Vector Provides Antitumor Activity without Toxicity

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Animals
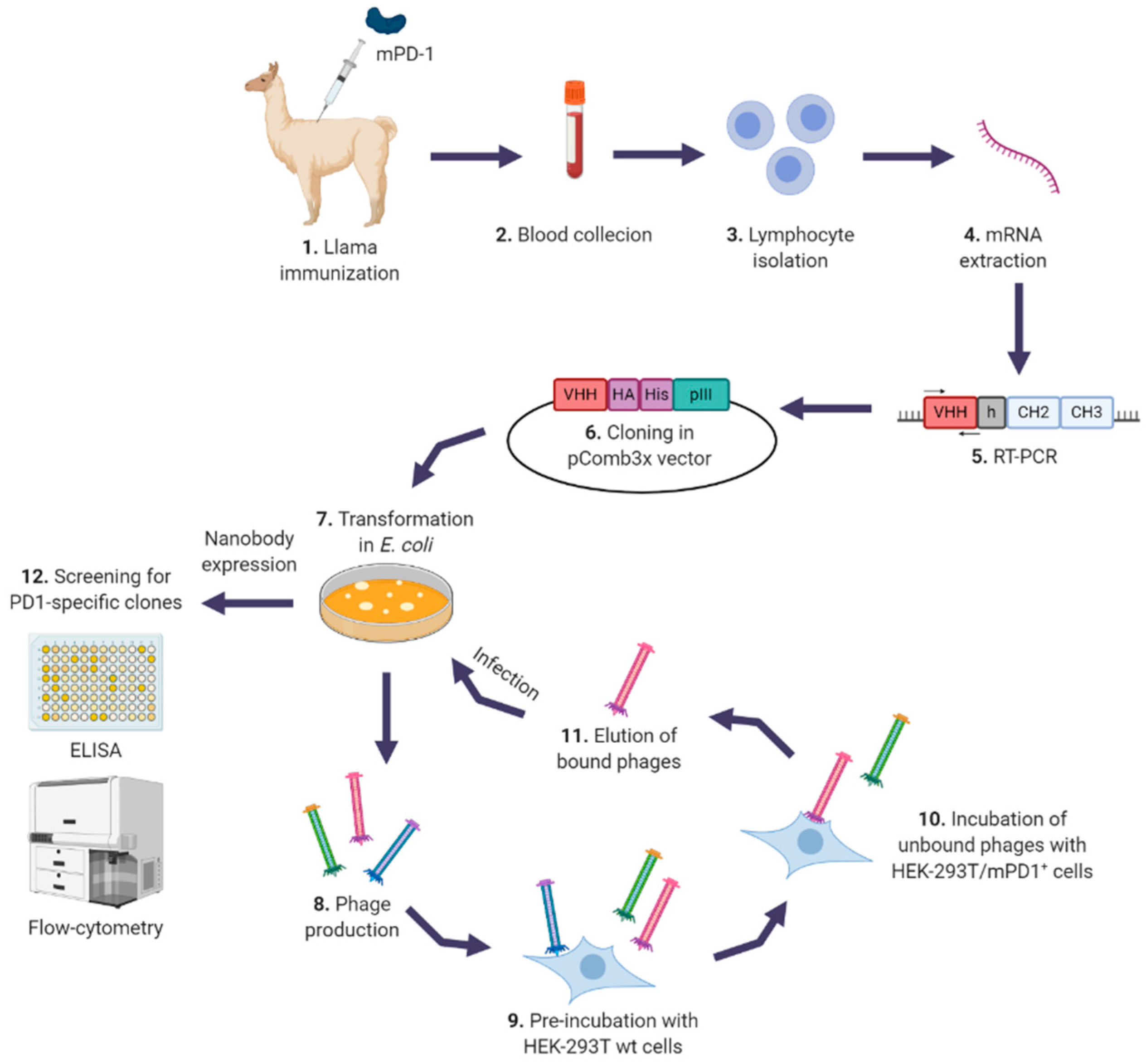
2.2. Llama Immunization and Library Construction
2.3. Selection of Specific Nanobodies
2.4. Screening for Specific Nanobodies
2.5. Large-Scale Production of Nanobodies
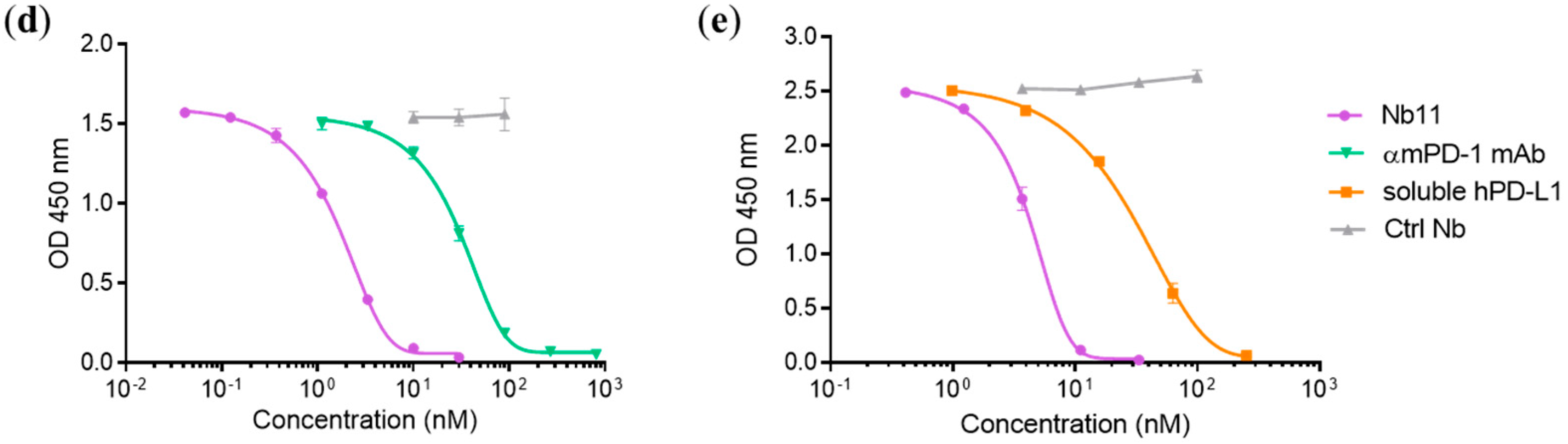
2.6. PD-1/PD-L1 Binding ELISA
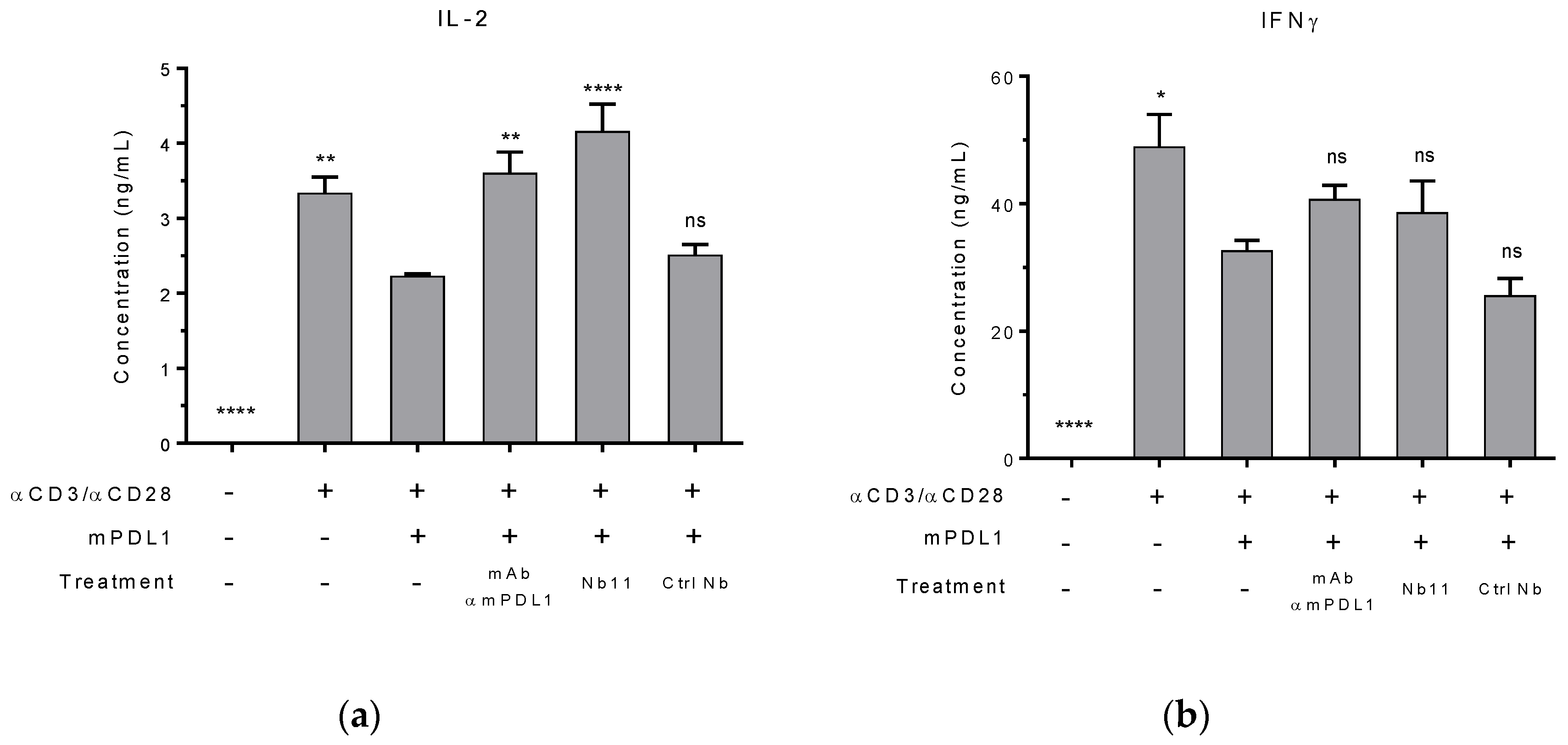
2.7. Mouse T Cell Reactivation In Vitro
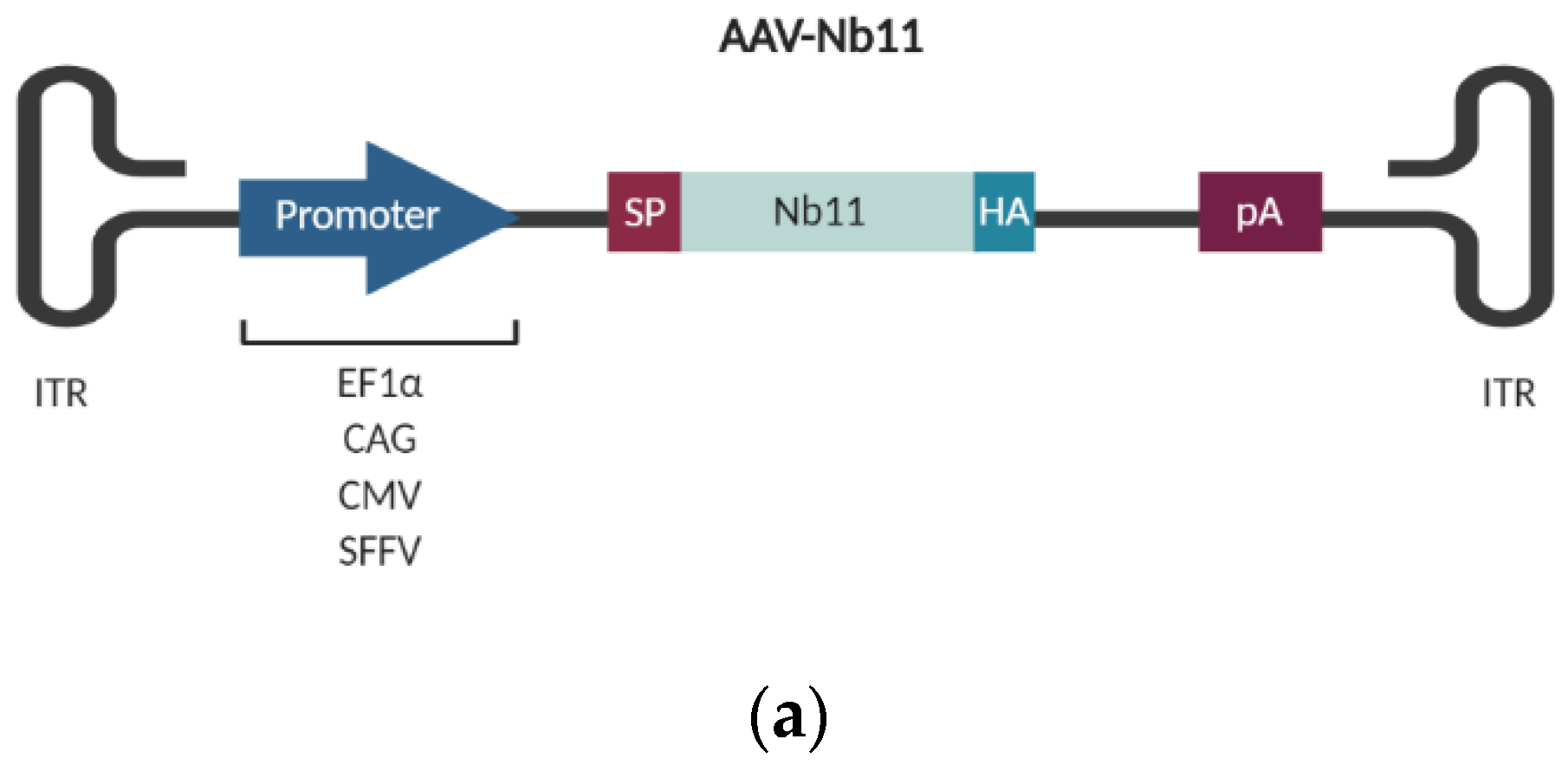
2.8. Construction of AAV Vectors Coding for Nb11
2.9. Expression of Nb11 from AAV Vectors In Vitro
2.10. Production of AAV8 Vectors
2.11. Evaluation of the Safety and Antitumor Activity of AAV8-Nb11 In Vivo
2.12. Statistical Analysis
3. Results
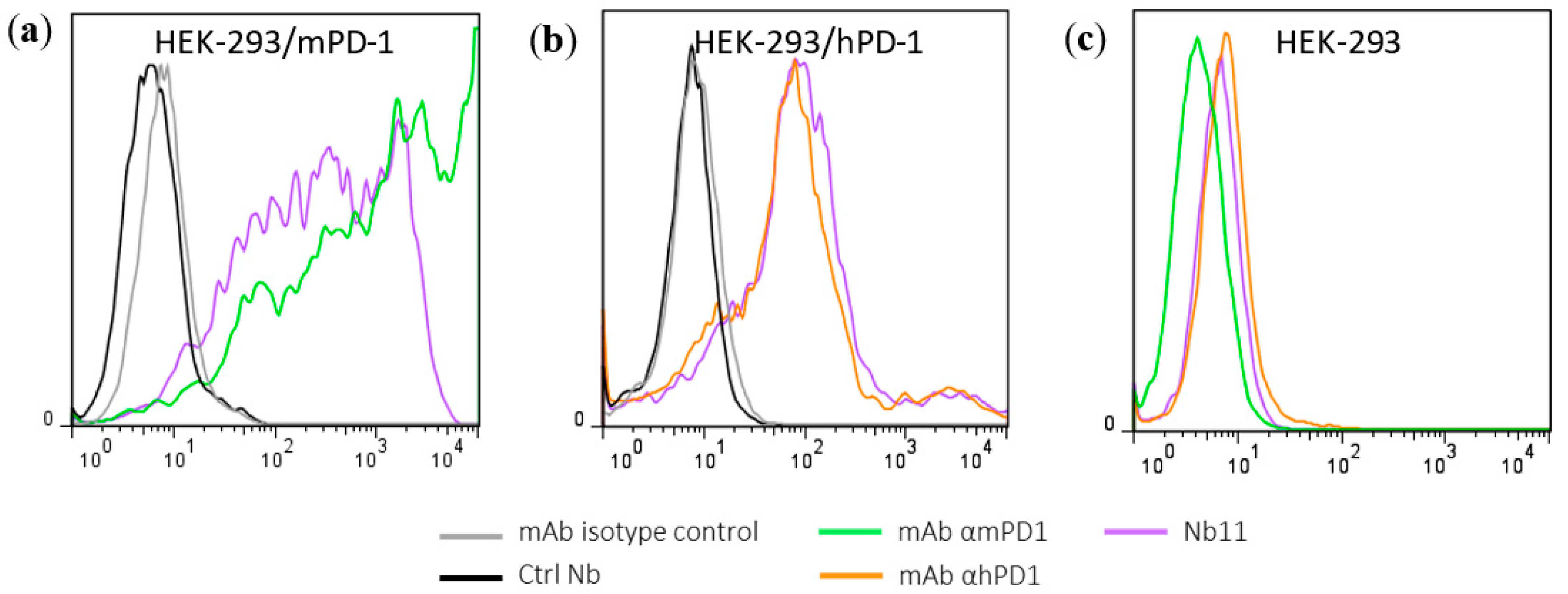
3.1. Generation and Characterization of a Nanobody that Binds to Human and Mouse PD-1
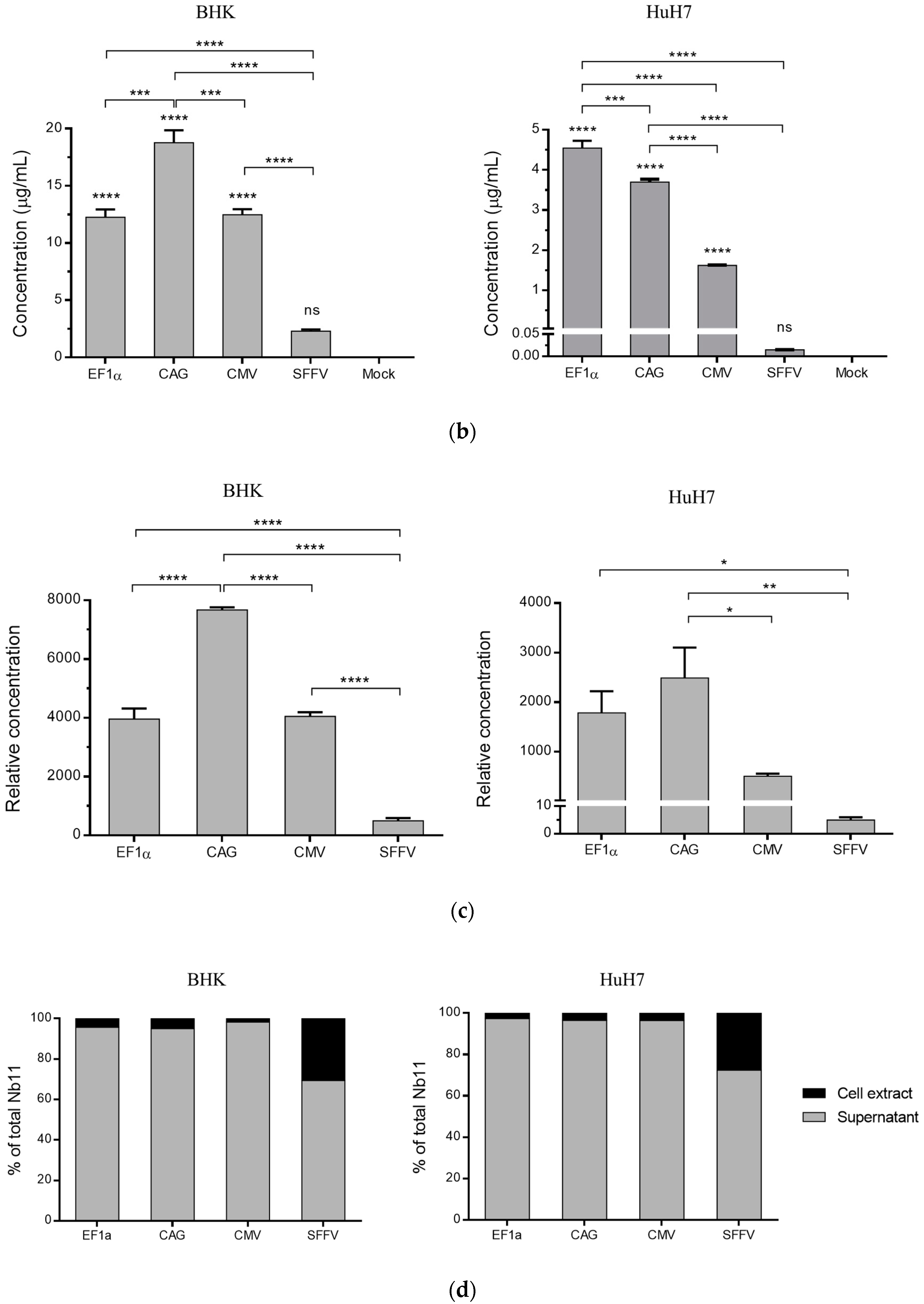
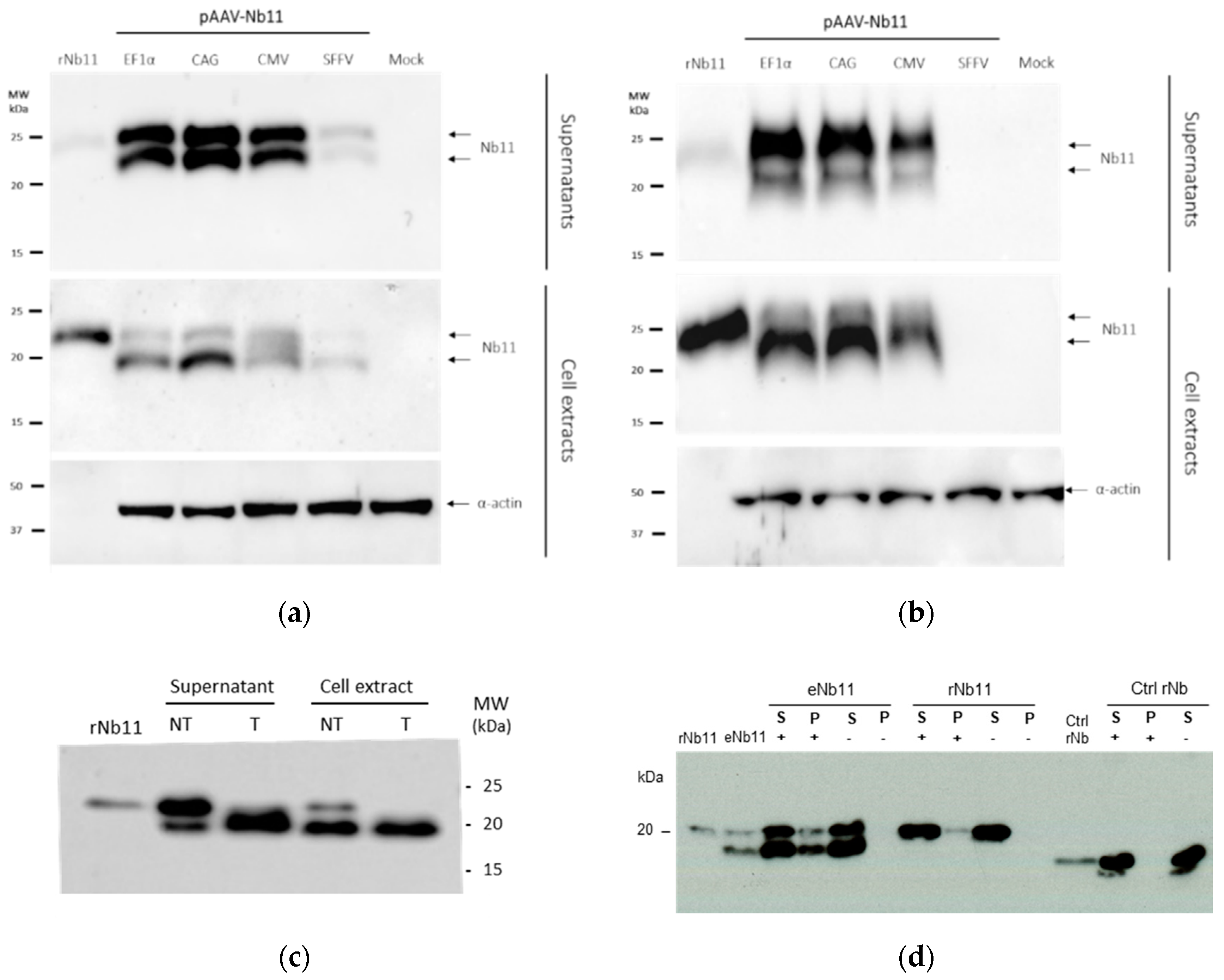
3.2. Nb11 Expression from Mammalian Cells Using AAV Vectors
3.3. AAV Vectors Can Mediate Sustained Nb11 Expression in Mice without Toxicity
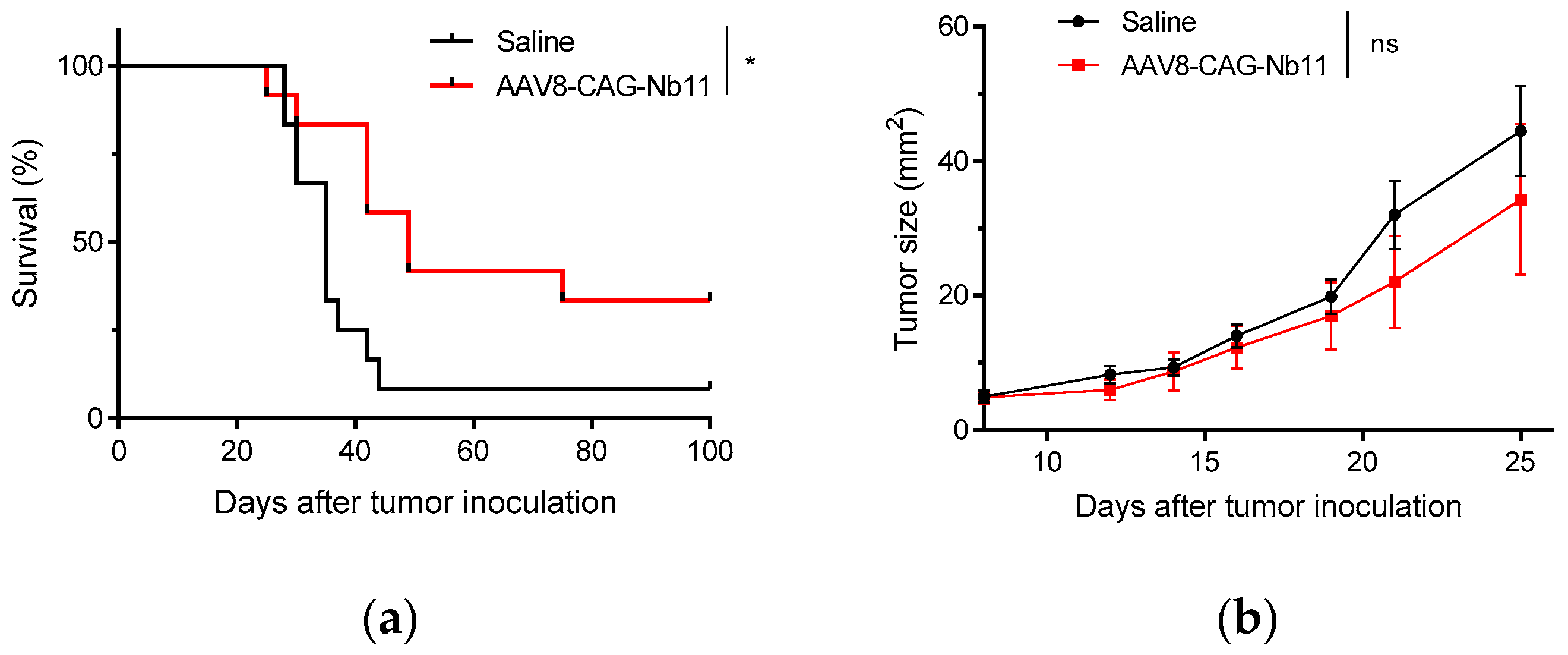
3.4. AAV-CAG-Nb11 Treatment Can Prevent Tumor Development in Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Janik, J.E.; Abu-Eid, R.; Mkrtichyan, M.; Khleif, S.N. Programmed death-1 & its ligands: Promising targets for cancer immunotherapy. Immunotherapy 2015, 7, 777–792. [Google Scholar] [CrossRef]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Haining, W.N.; Freeman, G.J.; Sharpe, A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef]
- Noguchi, T.; Ward, J.P.; Gubin, M.M.; Arthur, C.D.; Lee, S.H.; Hundal, J.; Selby, M.J.; Graziano, R.F.; Mardis, E.R.; Korman, A.J.; et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol. Res. 2017, 5, 106–117. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, G.; Treacy, O.; Lynch, K.; Naicker, S.D.; Leonard, N.A.; Lohan, P.; Dunne, P.D.; Ritter, T.; Egan, L.J.; Ryan, A.E. Stromal cell PD-L1 inhibits CD8+ T-cell antitumor immune responses and promotes colon cancer. Cancer Immunol. Res. 2018, 6, 1426–1441. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, N.; Shah, M.; Suarez-Almazor, M.E. Adverse Events Associated with Immune Checkpoint Blockade in Patients with Cancer: A Systematic Review of Case Reports. PLoS ONE 2016, 11, e0160221. [Google Scholar] [CrossRef]
- Palmieri, D.J.; Carlino, M.S. Immune Checkpoint Inhibitor Toxicity. Curr. Oncol. Rep. 2018, 20, 72. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Arbabi Ghahroudi, M.; Desmyter, A.; Wyns, L.; Hamers, R.; Muyldermans, S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997, 414, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöl, P.; Kremer Hovinga, J.A.; Metjian, A.; De La Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Kijanka, M.; Dorresteijn, B.; Oliveira, S.; van Bergen en Henegouwen, P.M.P. Nanobody-based cancer therapy of solid tumors. Nanomedicine 2015, 10, 161–174. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, H. Expression of single-domain antibody in different systems. Appl. Microbiol. Biotechnol. 2018, 102, 539–551. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Zhang, X.; Schwartz, J.C.D.; Guo, X.; Bhatia, S.; Cao, E.; Chen, L.; Zhang, Z.Y.; Edidin, M.A.; Nathenson, S.G.; Almo, S.C. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 2004, 20, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Tabares-Da Rosa, S.; Rossotti, M.; Carleiza, C.; Carrión, F.; Pritsch, O.; Ahn, K.C.; Last, J.A.; Hammock, B.D.; González-Sapienza, G. Competitive selection from single domain antibody libraries allows isolation of high-affinity antihapten antibodies that are not favored in the llama immune response. Anal. Chem. 2011, 83, 7213–7220. [Google Scholar] [CrossRef] [Green Version]
- Delfin-Riela, T.; Rossotti, M.A.; Echaides, C.; González-Sapienza, G. A nanobody-based test for highly sensitive detection of hemoglobin in fecal samples. Anal. Bioanal. Chem. 2020, 412, 389–396. [Google Scholar] [CrossRef]
- Alexopoulou, A.N.; Couchman, J.R.; Whiteford, J.R. The CMV early enhancer/chicken β actin (CAG) promoter can be used to drive transgene expression during the differentiation of murine embryonic stem cells into vascular progenitors. BMC Cell Biol. 2008, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Pignataro, D.; Sucunza, D.; Vanrell, L.; Lopez-Franco, E.; Dopeso-Reyes, I.G.; Vales, A.; Hommel, M.; Rico, A.J.; Lanciego, J.L.; Gonzalez-Aseguinolaza, G. Adeno-Associated Viral Vectors Serotype 8 for Cell-Specific Delivery of Therapeutic Genes in the Central Nervous System. Front. Neuroanat. 2017, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros-Briones, M.C.; Martisova, E.; Casales, E.; Silva-Pilipich, N.; Buñuales, M.; Galindo, J.; Mancheño, U.; Gorraiz, M.; Lasarte, J.J.; Kochan, G.; et al. Short-Term Local Expression of a PD-L1 Blocking Antibody from a Self-Replicating RNA Vector Induces Potent Antitumor Responses. Mol. Ther. 2019, 27, 1892–1905. [Google Scholar] [CrossRef]
- Liechtenstein, T.; Perez-Janices, N.; Bricogne, C.; Lanna, A.; Dufait, I.; Goyvaerts, C.; Laranga, R.; Padella, A.; Arce, F.; Baratchian, M.; et al. Immune modulation by genetic modification of dendritic cells with lentiviral vectors. Virus Res. 2013, 176, 1–15. [Google Scholar] [CrossRef]
- Murillo, O.; Luqui, D.M.; Gazquez, C.; Martinez-Espartosa, D.; Navarro-Blasco, I.; Monreal, J.I.; Guembe, L.; Moreno-Cermeño, A.; Corrales, F.J.; Prieto, J.; et al. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J. Hepatol. 2016, 64, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Broos, K.; Keyaerts, M.; Lecocq, Q.; Renmans, D.; Nguyen, T.; Escors, D.; Liston, A.; Raes, G.; Breckpot, K.; Devoogdt, N. Non-invasive assessment of murine PD-L1 levels in syngeneic tumor models by nuclear imaging with nanobody tracers. Oncotarget 2017, 8, 41932–41946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Wei, H.; Wang, X.; Bai, Y.; Wang, P.; Wu, J.; Jiang, X.; Wang, Y.; Cai, H.; Xu, T.; et al. Structural basis of a novel PD-L1 nanobody for immune checkpoint blockade. Cell Discov. 2017, 3, 17004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Jiang, K.; Wang, T.; Zhang, W.; Shi, M.; Chen, B.; Hua, Z. Nanobody against PDL1. Biotechnol. Lett. 2020, 42, 727–736. [Google Scholar] [CrossRef]
- Wen, B.; Zhao, L.; Wang, Y.; Qiu, C.; Xu, Z.; Huang, K.; Zhu, H.; Li, Z.; Li, H. Nanobodies targeting the interaction interface of programmed death receptor 1 (PD-1)/PD-1 ligand 1 (PD-1/PD-L1). Prep. Biochem. Biotechnol. 2020, 50, 252–259. [Google Scholar] [CrossRef]
- Xian, Z.; Ma, L.; Zhu, M.; Li, G.; Gai, J.; Chang, Q.; Huang, Y.; Ju, D.; Wan, Y. Blocking the PD-1-PD-L1 axis by a novel PD-1 specific nanobody expressed in yeast as a potential therapeutic for immunotherapy. Biochem. Biophys. Res. Commun. 2019, 519, 267–273. [Google Scholar] [CrossRef]
- De Vlieger, D.; Ballegeer, M.; Rossey, I.; Schepens, B.; Saelens, X. Single-Domain Antibodies and Their Formatting to Combat Viral Infections. Antibodies 2019, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Sheoran, A.S.; Dmitriev, I.P.; Kashentseva, E.A.; Cohen, O.; Mukherjee, J.; Debatis, M.; Shearer, J.; Tremblay, J.M.; Beamer, G.; Curiel, D.T.; et al. Adenovirus vector expressing Stx1/Stx2-neutralizing agent protects piglets infected with Escherichia coli O157: H7 against fatal systemic intoxication. Infect. Immun. 2015, 83, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Burmistrova, D.A.; Tillib, S.V.; Shcheblyakov, D.V.; Dolzhikova, I.V.; Shcherbinin, D.N.; Zubkova, O.V.; Ivanova, T.I.; Tukhvatulin, A.I.; Shmarov, M.M.; Logunov, D.Y.; et al. Genetic passive immunization with adenoviral vector expressing chimeric nanobody-Fc molecules as therapy for genital infection caused by mycoplasma hominis. PLoS ONE 2016, 11, e0150958. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Shi, L.; Yu, H.; Zhang, Y.; Chen, K.; Saint Fleur, A.; Bai, G.; Feng, H. Intravenous adenovirus expressing a multi-specific, single-domain antibody neutralizing TcdA and TcdB protects mice from Clostridium difficile infection. Pathog. Dis. 2016, 74, ftw078. [Google Scholar] [CrossRef] [Green Version]
- Hastie, E.; Samulski, R.J. Adeno-Associated Virus at 50: A Golden Anniversary of Discovery, Research, and Gene Therapy Success—A Personal Perspective. Hum. Gene Ther. 2015, 26, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Vanrell, L.; Di Scala, M.; Blanco, L.; Otano, I.; Gil-Farina, I.; Baldim, V.; Paneda, A.; Berraondo, P.; Beattie, S.G.; Chtarto, A.; et al. Development of a liver-specific tet-on inducible system for AAV vectors and its application in the treatment of liver cancer. Mol. Ther. 2011, 19, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Liu, J.Q.; Shi, M.; Cheng, X.; Ding, M.; Zhang, J.C.; Davis, J.P.; Varikuti, S.; Satoskar, A.R.; Lu, L.; et al. IL-27 gene therapy induces depletion of Tregs and enhances the efficacy of cancer immunotherapy. JCI Insight 2018, 3, e98745. [Google Scholar] [CrossRef] [Green Version]
- Verhelle, A.; Nair, N.; Everaert, I.; Van Overbeke, W.; Supply, L.; Zwaenepoel, O.; Peleman, C.; Van Dorpe, J.; Lahoutte, T.; Devoogdt, N.; et al. AAV9 delivered bispecific nanobody attenuates amyloid burden in the gelsolin amyloidosis mouse model. Hum. Mol. Genet. 2017, 26, 1353–1364. [Google Scholar] [CrossRef] [Green Version]
- Del Rosario, J.M.M.; Smith, M.; Zaki, K.; Risley, P.; Temperton, N.; Engelhardt, O.G.; Collins, M.; Takeuchi, Y.; Hufton, S.E. Protection From Influenza by Intramuscular Gene Vector Delivery of a Broadly Neutralizing Nanobody Does Not Depend on Antibody Dependent Cellular Cytotoxicity. Front. Immunol. 2020, 11, 627. [Google Scholar] [CrossRef]
- Winiarska, M.; Nowis, D.; Firczuk, M.; Zagozdzon, A.; Gabrysiak, M.; Sadowski, R.; Barankiewicz, J.; Dwojak, M.; Golab, J. Selection of an optimal promoter for gene transfer in normal B cells. Mol. Med. Rep. 2017, 16, 3041–3048. [Google Scholar] [CrossRef] [Green Version]
- Reul, J.; Frisch, J.; Engeland, C.E.; Thalheimer, F.B.; Hartmann, J.; Ungerechts, G.; Buchholz, C.J. Tumor-specific delivery of immune checkpoint inhibitors by engineered AAV vectors. Front. Oncol. 2019, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, H.; Bell, P.; McCarter, R.J.; He, J.; Calcedo, R.; Vandenberghe, L.H.; Morizono, H.; Batshaw, M.L.; Wilson, J.M. Systematic evaluation of AAV vectors for liver directed gene transfer in murine models. Mol. Ther. 2010, 18, 118–125. [Google Scholar] [CrossRef]
- Affolter, T.; Llewellyn, H.P.; Bartlett, D.W.; Zong, Q.; Xia, S.; Torti, V.; Ji, C. Inhibition of immune checkpoints PD-1, CTLA-4, and IDO1 coordinately induces immune-mediated liver injury in mice. PLoS ONE 2019, 14, e0217276. [Google Scholar] [CrossRef]
- Marabelle, A.; Tselikas, L.; de Baere, T.; Houot, R. Intratumoral immunotherapy: Using the tumor as the remedy. Ann. Oncol. 2017, 28, xii33–xii43. [Google Scholar] [CrossRef]
- Aznar, M.A.; Tinari, N.; Rullán, A.J.; Sánchez-Paulete, A.R.; Rodriguez-Ruiz, M.E.; Melero, I. Intratumoral Delivery of Immunotherapy—Act Locally, Think Globally. J. Immunol. 2017, 198, 31–39. [Google Scholar] [CrossRef]
- Büning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef]
- Eichhoff, A.M.; Börner, K.; Albrecht, B.; Schäfer, W.; Baum, N.; Haag, F.; Körbelin, J.; Trepel, M.; Braren, I.; Grimm, D.; et al. Nanobody-Enhanced Targeting of AAV Gene Therapy Vectors. Mol. Ther. Methods Clin. Dev. 2019, 15, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.; Heukers, R.; Sornkom, J.; Kok, R.J.; Van Bergen En Henegouwen, P.M.P. Targeting tumors with nanobodies for cancer imaging and therapy. J. Control. Release 2013, 172, 607–617. [Google Scholar] [CrossRef]
- Zhao, N.; Qin, Y.; Liu, H.; Cheng, Z. Tumor-Targeting Peptides: Ligands for Molecular Imaging and Therapy. Anticancer Agents Med. Chem. 2017, 18, 74–86. [Google Scholar] [CrossRef]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-Pilipich, N.; Martisova, E.; Ballesteros-Briones, M.C.; Hervas-Stubbs, S.; Casares, N.; González-Sapienza, G.; Smerdou, C.; Vanrell, L. Long-Term Systemic Expression of a Novel PD-1 Blocking Nanobody from an AAV Vector Provides Antitumor Activity without Toxicity. Biomedicines 2020, 8, 562. https://doi.org/10.3390/biomedicines8120562
Silva-Pilipich N, Martisova E, Ballesteros-Briones MC, Hervas-Stubbs S, Casares N, González-Sapienza G, Smerdou C, Vanrell L. Long-Term Systemic Expression of a Novel PD-1 Blocking Nanobody from an AAV Vector Provides Antitumor Activity without Toxicity. Biomedicines. 2020; 8(12):562. https://doi.org/10.3390/biomedicines8120562
Chicago/Turabian StyleSilva-Pilipich, Noelia, Eva Martisova, María Cristina Ballesteros-Briones, Sandra Hervas-Stubbs, Noelia Casares, Gualberto González-Sapienza, Cristian Smerdou, and Lucia Vanrell. 2020. "Long-Term Systemic Expression of a Novel PD-1 Blocking Nanobody from an AAV Vector Provides Antitumor Activity without Toxicity" Biomedicines 8, no. 12: 562. https://doi.org/10.3390/biomedicines8120562