Mitochondrial Transfer by Human Mesenchymal Stromal Cells Ameliorates Hepatocyte Lipid Load in a Mouse Model of NASH


 , , ,
, , ,  ,
,
Abstract
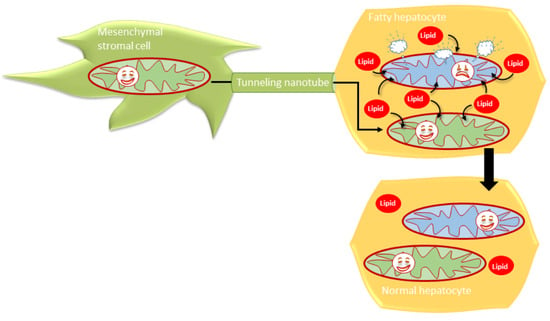
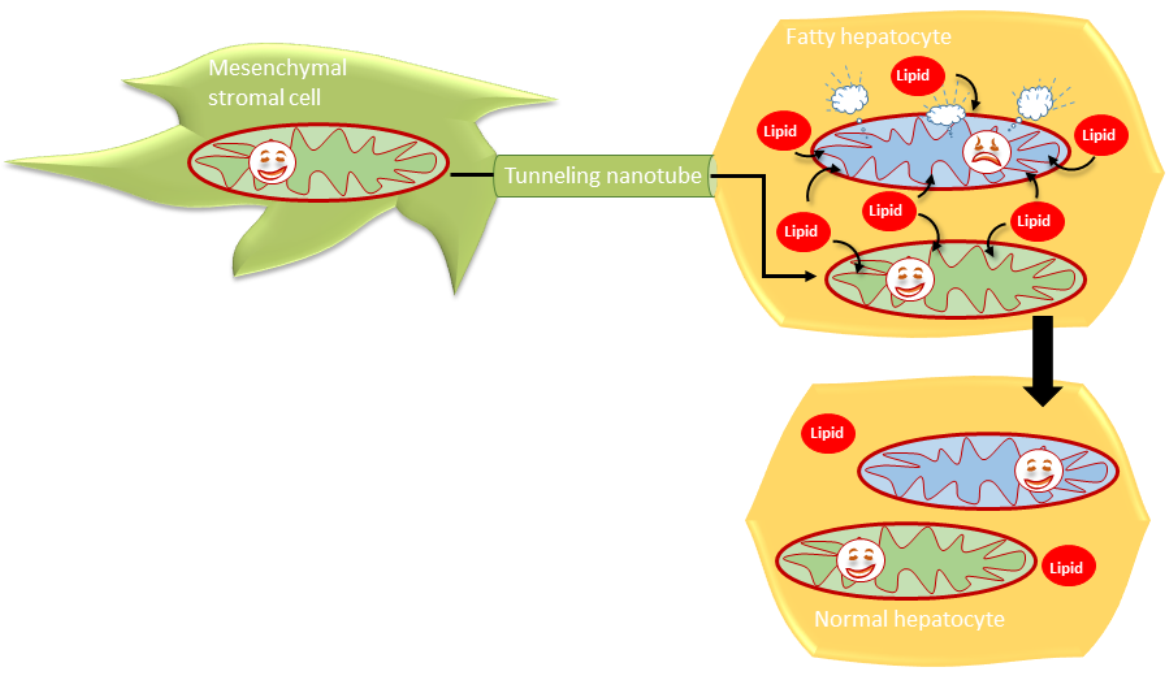
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Experimental Design
2.2. Proteome and Metabolome Analyses
2.2.1. Liver Metabolite and Protein Extraction and Preparation for Protein Quantification by Stable Isotope Labeling of Mammals (SILAM)
2.2.2. In-Gel Tryptic Digestion and Liquid Chromatography-Tandem Mass Spectrometry (GeLC-MS/MS)
2.2.3. Metabolome
2.3. Combined Analysis and Visualization of Proteome and Metabolome Data
2.3.1. Weighted Gene Correlation Network Analysis (WGCNA)
2.3.2. Identification of Key Drivers
2.4. Immunohistochemistry
2.5. Western Blotting
2.6. Quantification of Liver Triglyceride Content
2.7. Isolation of Primary Mouse Hepatocytes and Co-Culture with Hepatocytic Differentiated MSCs
2.8. Cell Labeling with Fluorescent Vital Dyes
2.9. Fluorescence Staining after Cell Fixation
2.10. Image Capture and Analysis
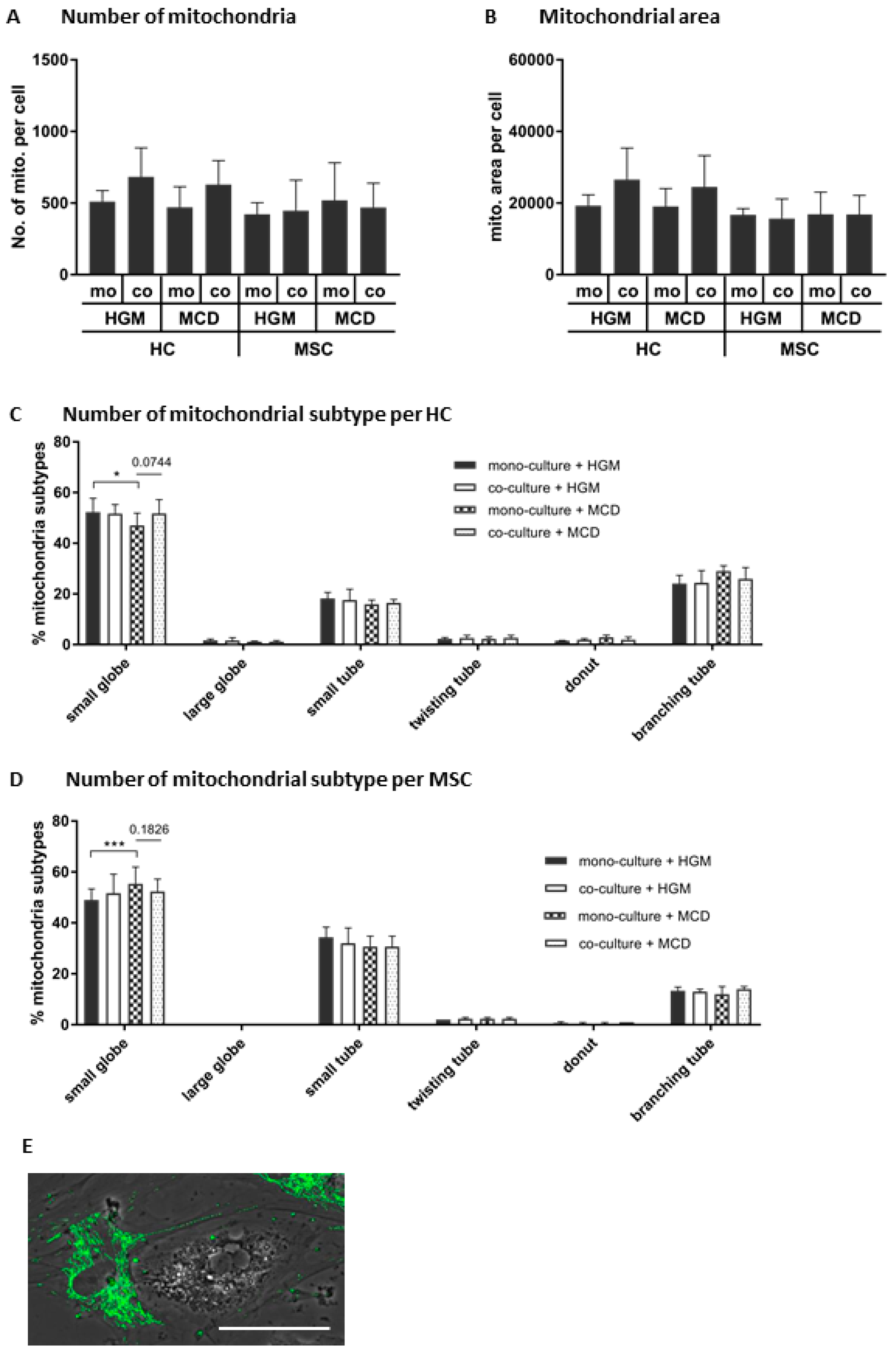
2.11. Morphological Subtyping of Mitochondria
2.12. RNA Isolation, cDNA Synthesis and PCR
2.13. Statistical Analysis
3. Results
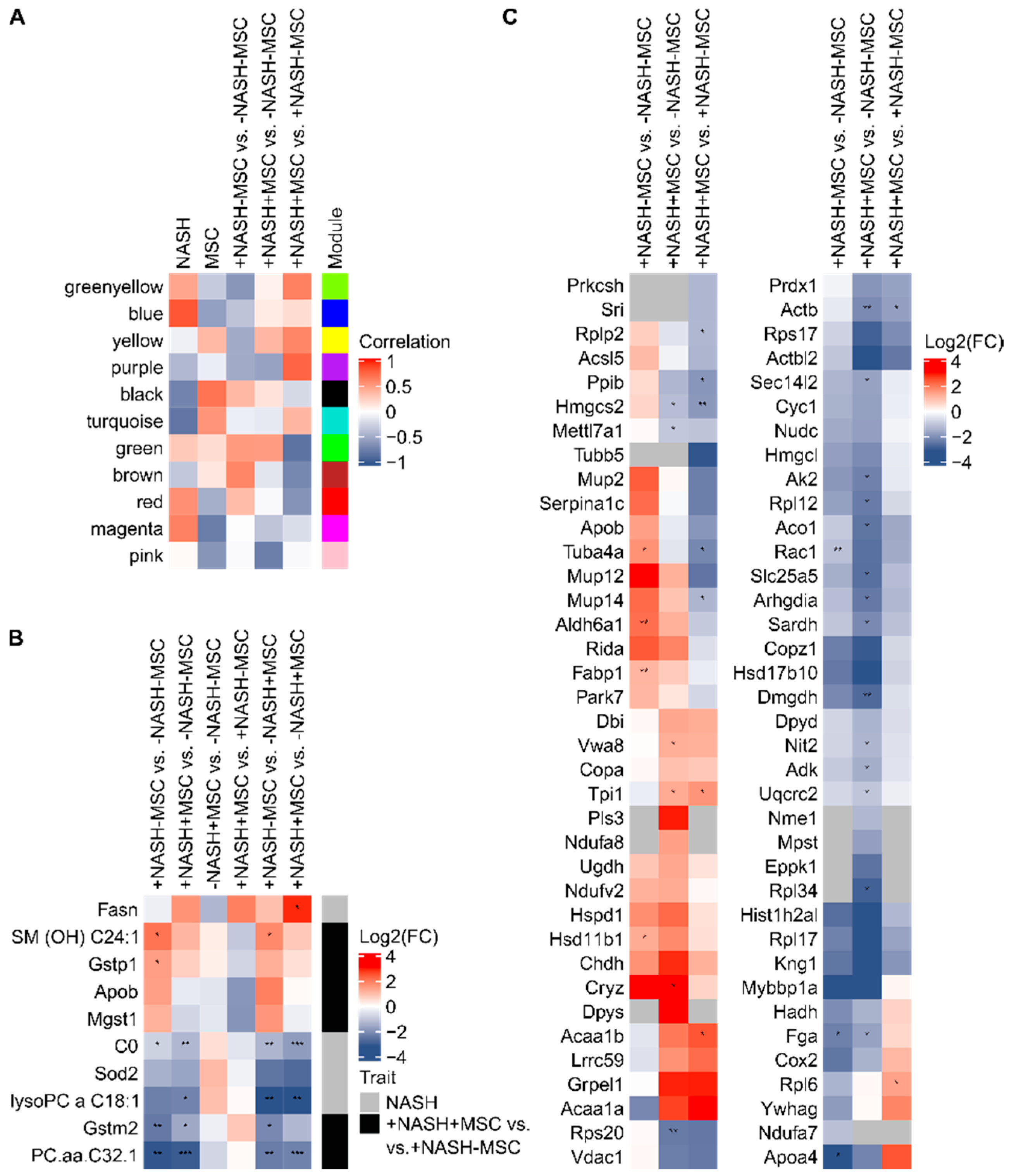
3.1. WGCNA Suggests a Shift from Lipid Storage to Utilization by MSCs
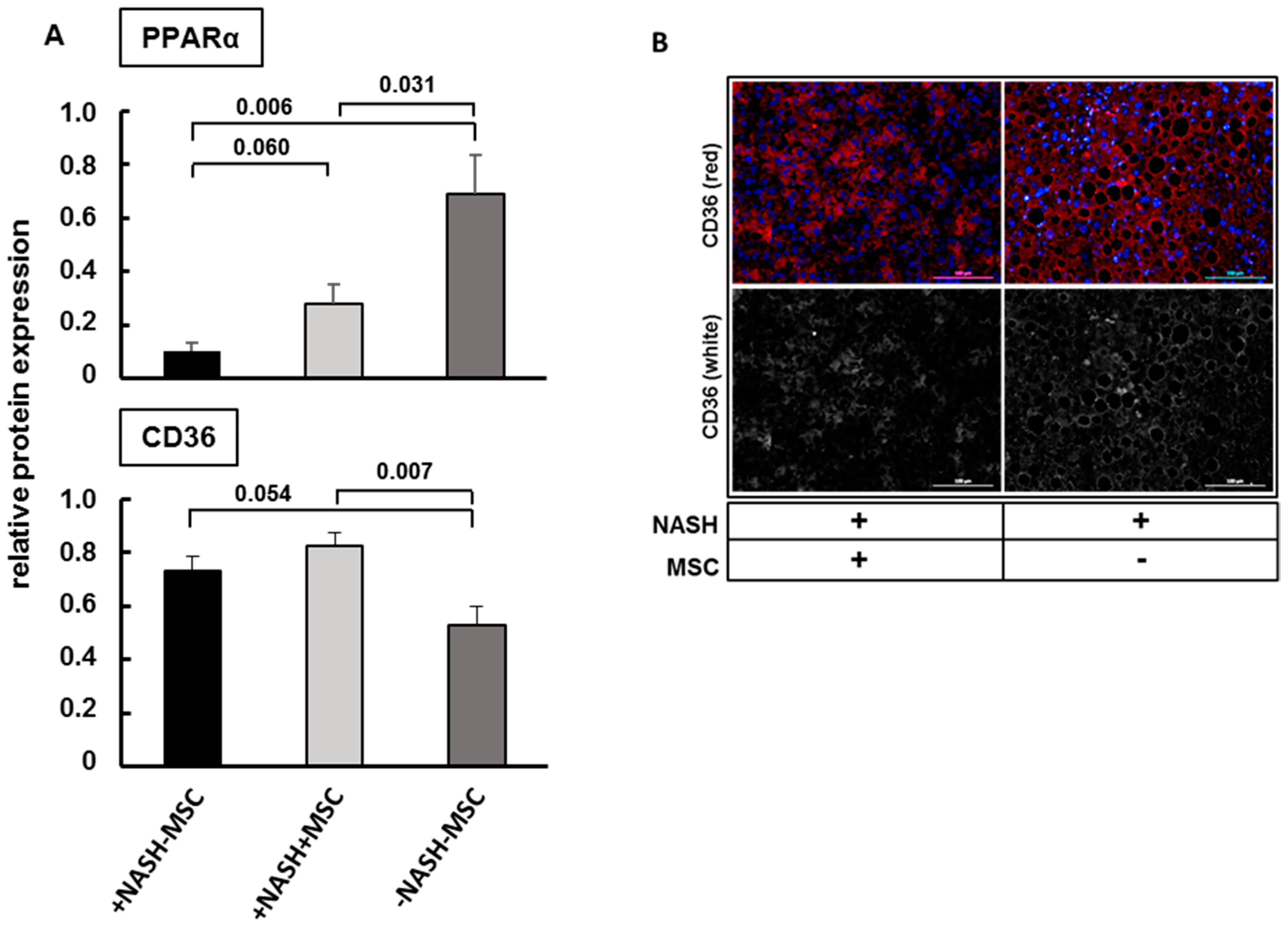
3.2. Improvement of Tissue Homeostasis by MSCs
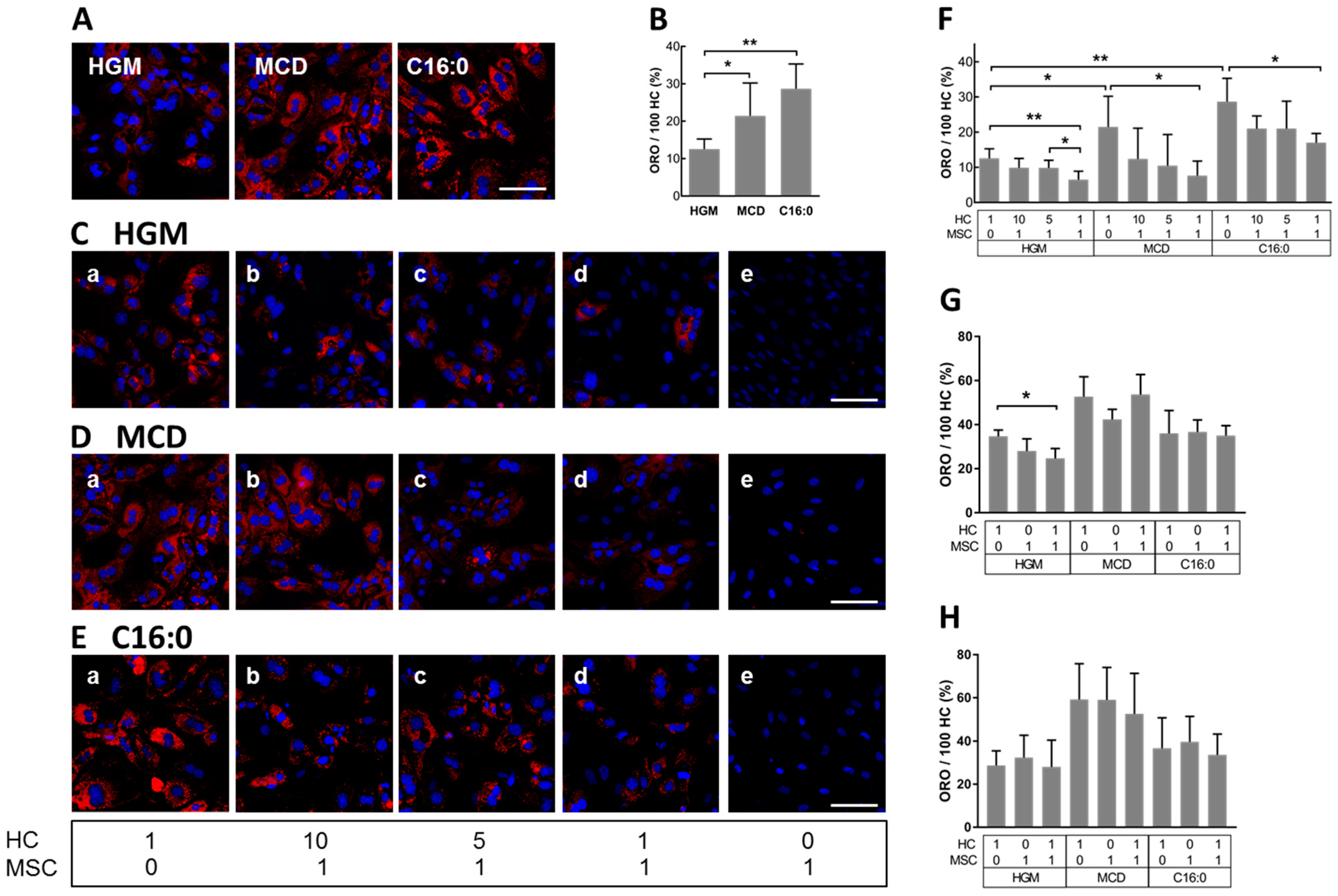
3.3. MSCs Decreased Hepatocyte Lipid Load In Vitro
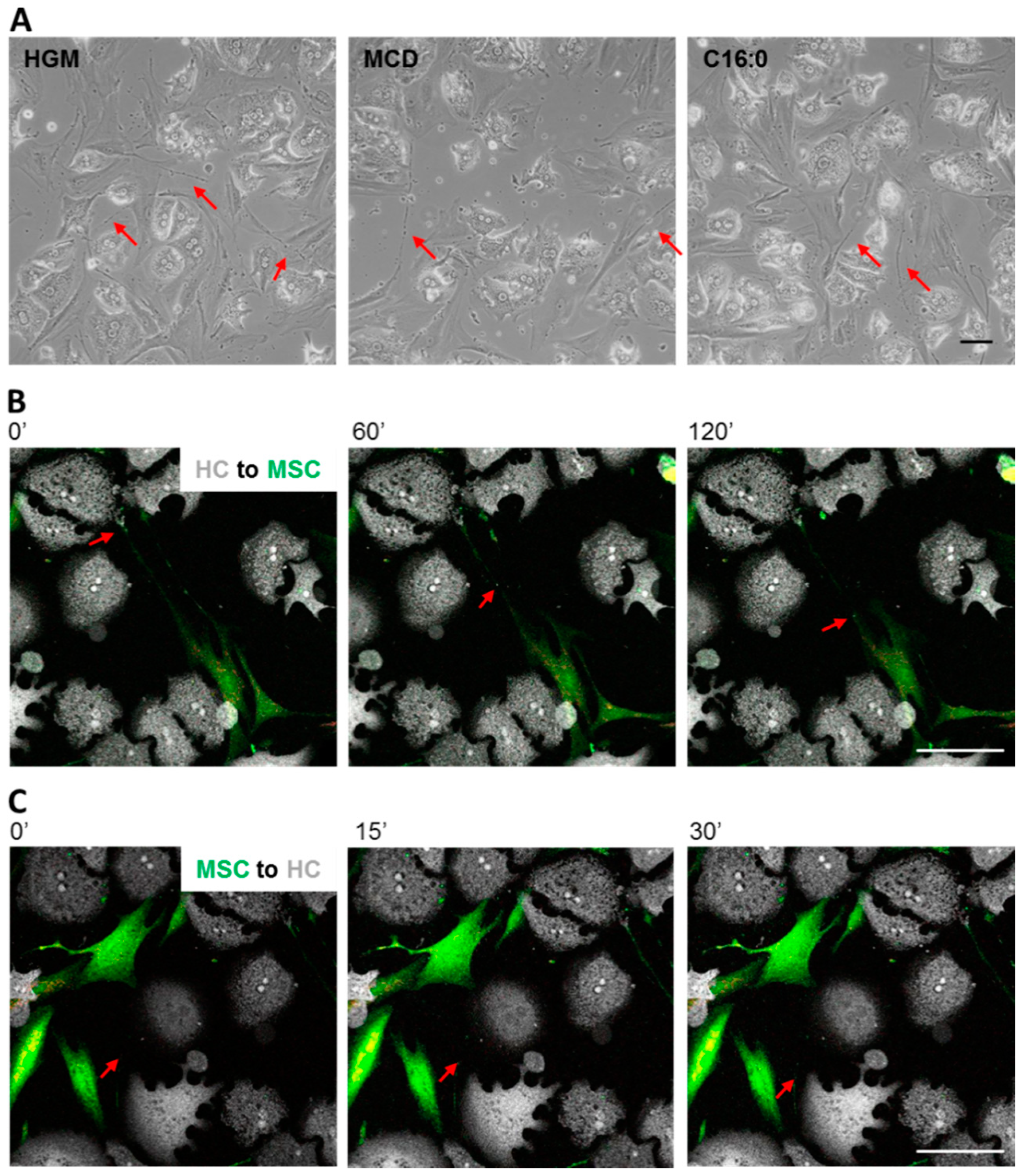
3.4. MSCs Communicated with Hepatocytes via Tunneling Nanotubes in a Microtubule-Dependent Manner
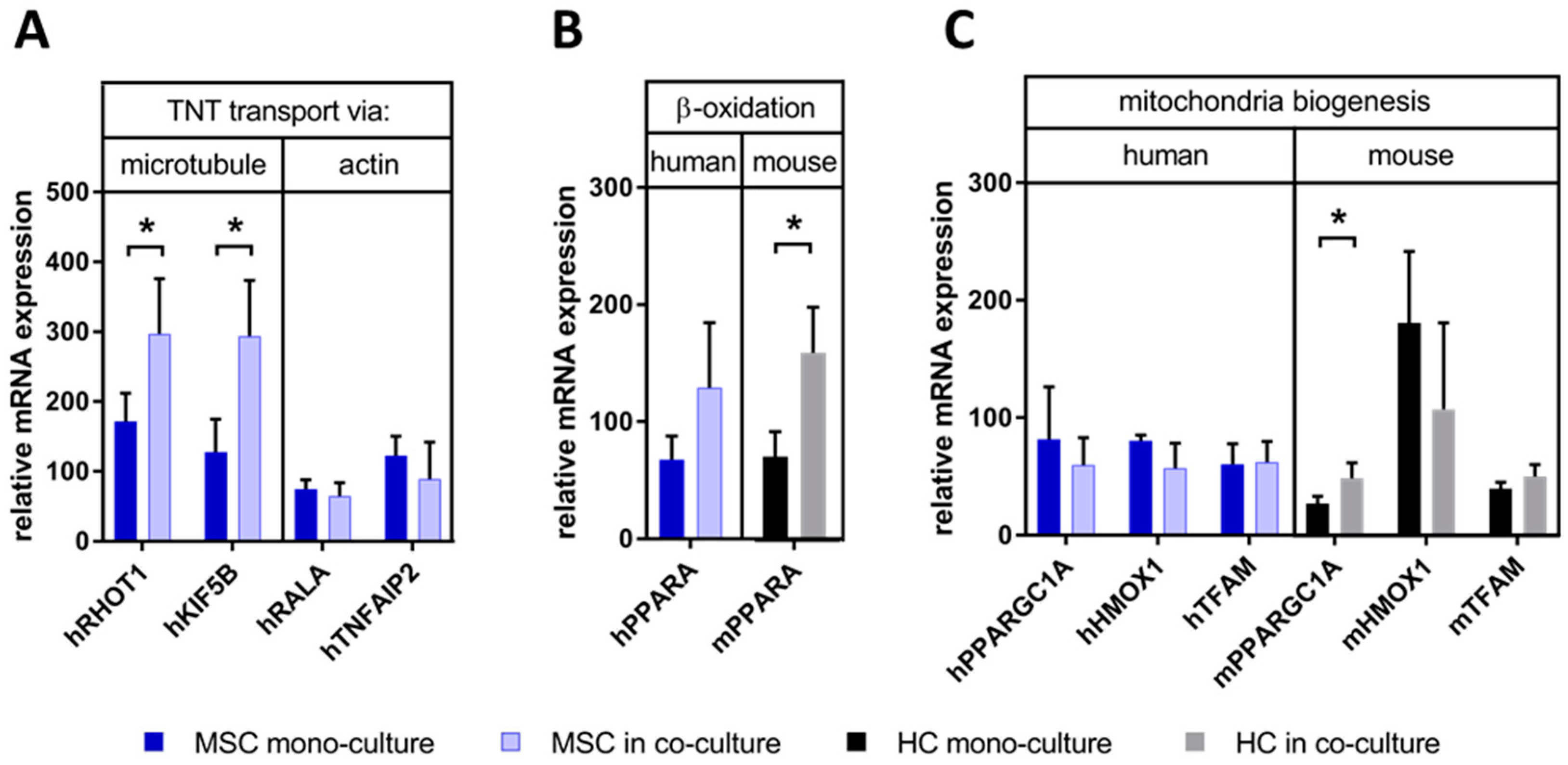
3.5. MSCs May Have Enhanced Lipid Utilization in HCs by Eliciting Oxidative Capacity
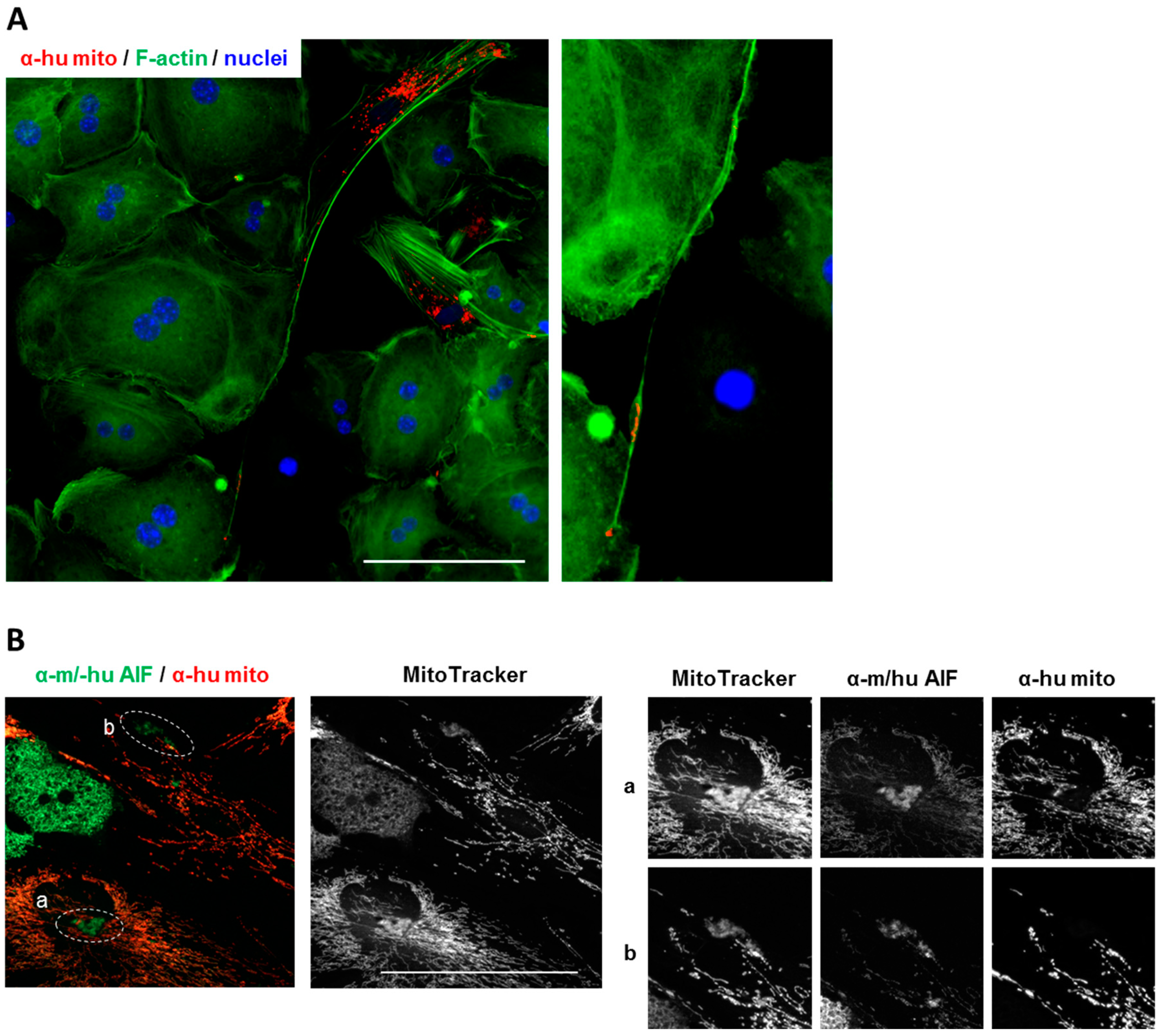
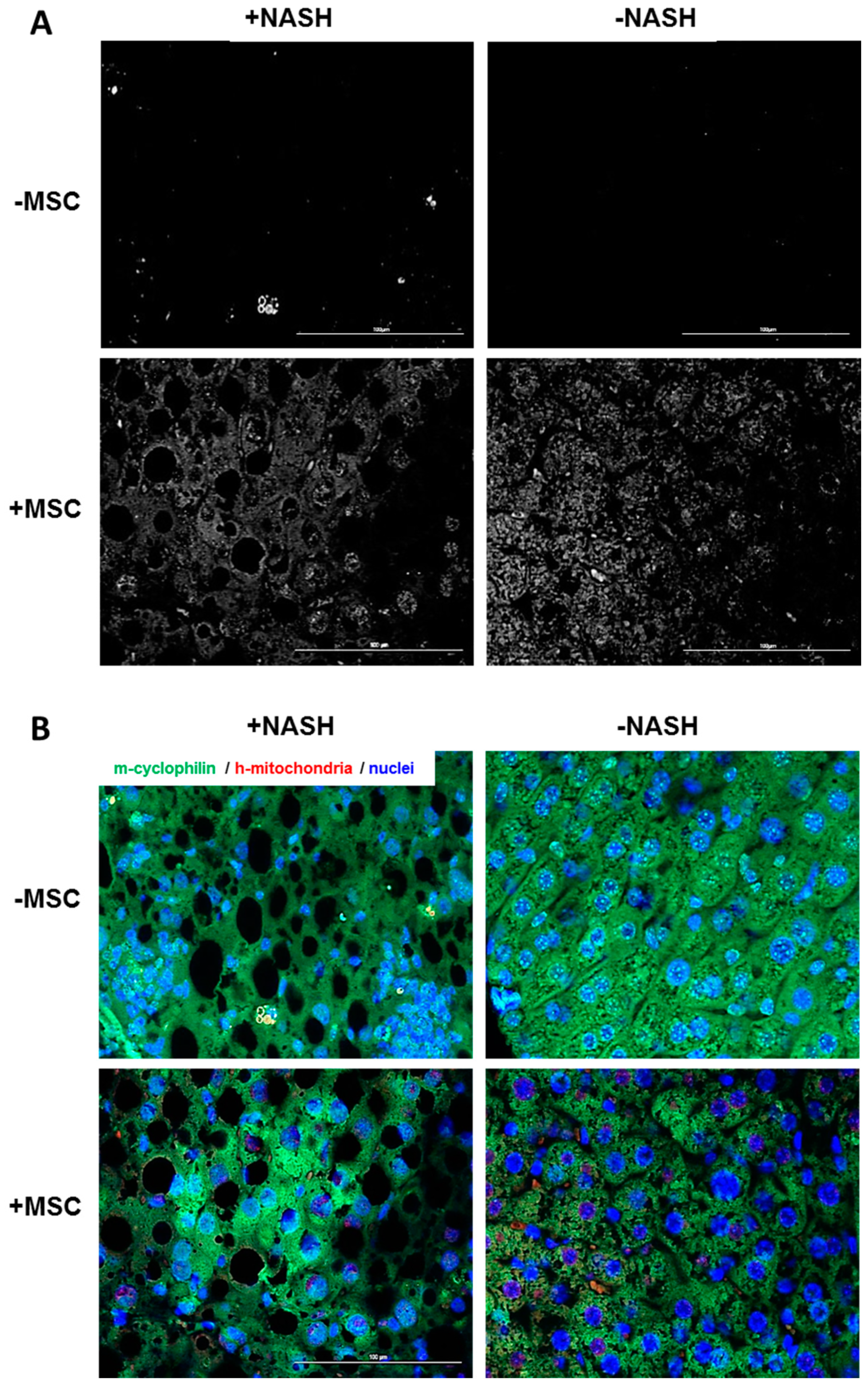
3.6. Human BM-MSCs Delivered Mitochondria to Mouse Hepatocytes In Vivo
4. Discussion
4.1. Pathobiochemical Consequences of Changes in Metabolic Protein Expression and Correction by MSCs
4.2. Histopathological Consequences of Hepatocyte Lipid Load and Improvement by MSCs
4.3. Mitochondrial Transfer by MSCs May Attenuate Hepatocyte Lipid Load
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of nafld and nash: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Mechanisms behind the link between obesity and gastrointestinal cancers. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Park, H.W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.H.; Kim, M.; Cazares, V.A.; Stuenkel, E.L.; Kim, J.J.; Kim, J.S.; et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [Green Version]
- Pessayre, D.; Fromenty, B. Nash: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in nash: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.; Poussin, C.; Velagapudi, V.R.; Empsen, C.; Joffraud, M.; Beckmann, J.S.; Geerts, A.E.; Ravussin, Y.; Ibberson, M.; Oresic, M.; et al. Peroxisomal and microsomal lipid pathways associated with resistance to hepatic steatosis and reduced pro-inflammatory state. J. Biol. Chem. 2010, 285, 31011–31023. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, F.; Jiang, S.; Sun, X.; Xu, J. Induction of liver steatosis in bap31-deficient mice burdened with tunicamycin-induced endoplasmic reticulum stress. Int. J. Mol. Sci. 2018, 19, 2291. [Google Scholar] [CrossRef] [Green Version]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for nafld/nash. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, S.M.; Mehta, N.; Kelley, R.K.; Roberts, J.P.; Yao, F.Y.; Brandman, D. Liver transplantation recipients with nonalcoholic steatohepatitis have lower risk hepatocellular carcinoma. Liver Transplant. Off. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transplant. Soc. 2017, 23, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Cholankeril, G.; Wong, R.J.; Hu, M.; Perumpail, R.B.; Yoo, E.R.; Puri, P.; Younossi, Z.M.; Harrison, S.A.; Ahmed, A. Liver transplantation for nonalcoholic steatohepatitis in the us: Temporal trends and outcomes. Dig. Dis. Sci. 2017, 62, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Vipani, A.; Bresee, C.; Todo, T.; Kim, I.K.; Alkhouri, N.; Setiawan, V.W.; Tran, T.; Ayoub, W.S.; Lu, S.C.; et al. Nash leading cause of liver transplant in women: Updated analysis of indications for liver transplant and ethnic and gender variances. Am. J. Gastroenterol. 2018, 113, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Alfaifi, M.; Eom, Y.W.; Newsome, P.N.; Baik, S.K. Mesenchymal stromal cell therapy for liver diseases. J. Hepatol. 2018, 68, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Christ, B.; Bruckner, S.; Winkler, S. The therapeutic promise of mesenchymal stem cells for liver restoration. Trends Mol. Med. 2015, 21, 673–686. [Google Scholar] [CrossRef]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076. [Google Scholar] [CrossRef] [Green Version]
- Pelz, S.; Stock, P.; Bruckner, S.; Christ, B. A methionine-choline-deficient diet elicits nash in the immunodeficient mouse featuring a model for hepatic cell transplantation. Exp. Cell Res. 2012, 318, 276–287. [Google Scholar] [CrossRef]
- Wang, H.; Wang, D.; Yang, L.; Wang, Y.; Jia, J.; Na, D.; Chen, H.; Luo, Y.; Liu, C. Compact bone-derived mesenchymal stem cells attenuate nonalcoholic steatohepatitis in a mouse model by modulation of cd4 cells differentiation. Int. Immunopharmacol. 2017, 42, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Winkler, S.; Christ, B. Treatment of nash with human mesenchymal stem cells in the immunodeficient mouse. Methods Mol. Biol. 2014, 1213, 51–56. [Google Scholar]
- Tanimoto, H.; Terai, S.; Taro, T.; Murata, Y.; Fujisawa, K.; Yamamoto, N.; Sakaida, I. Improvement of liver fibrosis by infusion of cultured cells derived from human bone marrow. Cell Tissue Res. 2013, 354, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, S.; Zipprich, A.; Hempel, M.; Thonig, A.; Schwill, F.; Roderfeld, M.; Roeb, E.; Christ, B. Improvement of portal venous pressure in cirrhotic rat livers by systemic treatment with adipose tissue-derived mesenchymal stromal cells. Cytotherapy 2017, 19, 1462–1473. [Google Scholar] [CrossRef] [PubMed]
- Banas, A.; Teratani, T.; Yamamoto, Y.; Tokuhara, M.; Takeshita, F.; Osaki, M.; Kato, T.; Okochi, H.; Ochiya, T. Rapid hepatic fate specification of adipose-derived stem cells and their therapeutic potential for liver failure. J. Gastroenterol. Hepatol. 2009, 24, 70–77. [Google Scholar] [CrossRef]
- Stock, P.; Bruckner, S.; Winkler, S.; Dollinger, M.M.; Christ, B. Human bone marrow mesenchymal stem cell-derived hepatocytes improve the mouse liver after acute acetaminophen intoxication by preventing progress of injury. Int. J. Mol. Sci. 2014, 15, 7004–7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, A.; Prigent, J.; Lombard, C.; Rosseels, V.; Daujat-Chavanieu, M.; Breckpot, K.; Najimi, M.; Deblandre, G.; Sokal, E.M. Adult-derived human liver stem/progenitor cells infused 3 days postsurgery improve liver regeneration in a mouse model of extended hepatectomy. Cell Transplant. 2017, 26, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Tautenhahn, H.M.; Bruckner, S.; Baumann, S.; Winkler, S.; Otto, W.; von Bergen, M.; Bartels, M.; Christ, B. Attenuation of postoperative acute liver failure by mesenchymal stem cell treatment due to metabolic implications. Ann. Surg. 2016, 263, 546–556. [Google Scholar] [CrossRef]
- Sokal, E.M.; Stephenne, X.; Ottolenghi, C.; Jazouli, N.; Clapuyt, P.; Lacaille, F.; Najimi, M.; de Lonlay, P.; Smets, F. Liver engraftment and repopulation by in vitro expanded adult derived human liver stem cells in a child with ornithine carbamoyltransferase deficiency. Jimd Rep. 2014, 13, 65–72. [Google Scholar]
- Sokal, E.M.; Lombard, C.A.; Roelants, V.; Najimi, M.; Varma, S.; Sargiacomo, C.; Ravau, J.; Mazza, G.; Jamar, F.; Versavau, J.; et al. Biodistribution of liver-derived mesenchymal stem cells after peripheral injection in a hemophilia a patient. Transplantation 2017, 101, 1845–1851. [Google Scholar] [CrossRef]
- Kharaziha, P.; Hellstrom, P.M.; Noorinayer, B.; Farzaneh, F.; Aghajani, K.; Jafari, F.; Telkabadi, M.; Atashi, A.; Honardoost, M.; Zali, M.R.; et al. Improvement of liver function in liver cirrhosis patients after autologous mesenchymal stem cell injection: A phase i-ii clinical trial. Eur. J. Gastroenterol. Hepatol. 2009, 21, 1199–1205. [Google Scholar] [CrossRef]
- Jang, Y.O.; Kim, Y.J.; Baik, S.K.; Kim, M.Y.; Eom, Y.W.; Cho, M.Y.; Park, H.J.; Park, S.Y.; Kim, B.R.; Kim, J.W.; et al. Histological improvement following administration of autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: A pilot study. Liver Int. Off. J. Int. Assoc. Study Liver 2014, 34, 33–41. [Google Scholar] [CrossRef]
- Suk, K.T.; Yoon, J.H.; Kim, M.Y.; Kim, C.W.; Kim, J.K.; Park, H.; Hwang, S.G.; Kim, D.J.; Lee, B.S.; Lee, S.H.; et al. Transplantation with autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: Phase 2 trial. Hepatology 2016, 64, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, Y.; Verma, I.M. Little evidence of bone marrow-derived hepatocytes in the replacement of injured liver. Proc. Natl. Acad. Sci. USA 2003, 100 (Suppl. 1), 11850–11853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashiyama, R.; Moro, T.; Nakao, S.; Mikami, K.; Fukumitsu, H.; Ueda, Y.; Ikeda, K.; Adachi, E.; Bou-Gharios, G.; Okazaki, I.; et al. Negligible contribution of bone marrow-derived cells to collagen production during hepatic fibrogenesis in mice. Gastroenterology 2009, 137, 1459–1466.e1451. [Google Scholar] [CrossRef] [PubMed]
- Winkler, S.; Borkham-Kamphorst, E.; Stock, P.; Bruckner, S.; Dollinger, M.; Weiskirchen, R.; Christ, B. Human mesenchymal stem cells towards non-alcoholic steatohepatitis in an immunodeficient mouse model. Exp. Cell Res. 2014, 326, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Stock, P.; Bruckner, S.; Ebensing, S.; Hempel, M.; Dollinger, M.M.; Christ, B. The generation of hepatocytes from mesenchymal stem cells and engraftment into murine liver. Nat. Protoc. 2010, 5, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.A.; Kohajda, T.; Findeiss, S.; Stadler, P.F.; Washietl, S.; Kellis, M.; von Bergen, M.; Kalkhof, S. Optimization of parameters for coverage of low molecular weight proteins. Anal. Bioanal. Chem. 2010, 398, 2867–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Römisch-Margl, W.; Prehn, C.; Bogumil, R.; Röhring, C.; Suhre, K.; Adamski, J. Procedure for tissue sample preparation and metabolite extraction for high-throughput targeted metabolomics. Metabolomics 2012, 8, 133–142. [Google Scholar] [CrossRef]
- Wickham, H.; Bryan, J. Readxl: Read Excel Files. Available online: https://CRAN.R-project.org/package=readxl (accessed on 10 September 2020).
- Spiess, A.N. Qpcr: Modelling and Analysis of Real-Time pcr Data. R Package v. 1.4-1. Available online: https://CRAN.R-project.org/package=qpcR (accessed on 10 September 2020).
- Wickham, H.; Henry, L. Tidyr: EASILY tidy Data with “Spread ()” and “Gather ()” Functions. R Package Version 0.8. 0. Available online: https://CRAN.R-project.org/package=tidyr (accessed on 10 September 2020).
- Wickham, H. The split-apply-combine strategy for data analysis. J. Stat. Softw. 2011, 40, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Mahto, A. Splitstackshape: Stack and Reshape Datasets after Splitting Concatenated Values. Available online: https://CRAN.R-project.org/package=splitstackshape (accessed on 10 September 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Sakai, R. Dendsort: Modular Leaf Ordering Methods for Dendrogram Nodes. R Package Version 0.3. 3. F1000 Res. 2015, 3, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, N. Ggsci: Scientific Journal and Sci-fi Themed Color Palettes For’ggplot2′. R Package Version 2. 2018. Available online: https://CRAN.R-project.org/package=ggsci (accessed on 10 September 2020).
- Langfelder, P.; Horvath, S. Wgcna: An r package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkossa, I.; Bannuscher, A.; Hellack, B.; Bahl, A.; Buhs, S.; Nollau, P.; Luch, A.; Schubert, K.; von Bergen, M.; Haase, A. An in-depth multi-omics analysis in rle-6tn rat alveolar epithelial cells allows for nanomaterial categorization. Part Fibre Toxicol 2019, 16, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An r package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the r/bioconductor package biomart. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. Biomart and bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Carlson, M. Org.Mm.Eg.Db: Genome Wide Annotation for Mouse. 2018. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html (accessed on 10 September 2020). [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Winkler, S.; Hempel, M.; Bruckner, S.; Mallek, F.; Weise, A.; Liehr, T.; Tautenhahn, H.M.; Bartels, M.; Christ, B. Mouse white adipose tissue-derived mesenchymal stem cells gain pericentral and periportal hepatocyte features after differentiation in vitro, which are preserved in vivo after hepatic transplantation. Acta Physiol. 2015, 215, 89–104. [Google Scholar] [CrossRef]
- Block, G.D.; Locker, J.; Bowen, W.C.; Petersen, B.E.; Katyal, S.; Strom, S.C.; Riley, T.; Howard, T.A.; Michalopoulos, G.K. Population expansion, clonal growth, and specific differentiation patterns in primary cultures of hepatocytes induced by hgf/sf, egf and tgf alpha in a chemically defined (hgm) medium. J. Cell Biol. 1996, 132, 1133–1149. [Google Scholar] [CrossRef] [Green Version]
- Quah, B.J.; Warren, H.S.; Parish, C.R. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2007, 2, 2049–2056. [Google Scholar] [CrossRef]
- Winkler, S.; Hempel, M.; Hsu, M.J.; Gericke, M.; Kuhne, H.; Bruckner, S.; Erler, S.; Burkhardt, R.; Christ, B. Immune-deficient pfp/rag2(-/-) mice featured higher adipose tissue mass and liver lipid accumulation with growing age than wildtype c57bl/6n mice. Cells 2019, 8, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.Y.; Lin, C.C.; Chen, Y.J.; Kao, L.S.; Liu, Y.C.; Chou, C.C.; Huang, Y.H.; Chang, F.R.; Wu, Y.C.; Tsai, Y.S.; et al. Automatic morphological subtyping reveals new roles of caspases in mitochondrial dynamics. PLoS Comput. Biol. 2011, 7, e1002212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.W.; Hsiao, W.T.; Lee, O.K. Mesenchymal stromal cell-based therapies reduce obesity and metabolic syndromes induced by a high-fat diet. Transl. Res. J. Lab. Clin. Med. 2017, 182, 61–74.e68. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Sakai, Y.; Komura, T.; Nasti, A.; Yoshida, K.; Higashimoto, M.; Honda, M.; Usui, S.; Takamura, M.; Takamura, T.; et al. Adipose tissue-derived stem cells as a regenerative therapy for a mouse steatohepatitis-induced cirrhosis model. Hepatology 2013, 58, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Ramadori, G.; Christ, B. Cytokines and the hepatic acute-phase response. Semin. Liver Dis. 1999, 19, 141–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Park, J.S.; Roh, Y.S. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch. Pharmacal Res. 2019, 42, 935–946. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (nafld). Prog. Lipid Res. 2009, 48, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of pparalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Bonen, A.; Campbell, S.E.; Benton, C.R.; Chabowski, A.; Coort, S.L.; Han, X.X.; Koonen, D.P.; Glatz, J.F.; Luiken, J.J. Regulation of fatty acid transport by fatty acid translocase/cd36. Proc. Nutr. Soc. 2004, 63, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.A.; Fromenty, B. Increased expression of cytochrome p450 2e1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef]
- Leung, T.M.; Nieto, N. Cyp2e1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Diehl, A.M. Evidence for and against epithelial-to-mesenchymal transition in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G881–G890. [Google Scholar] [CrossRef] [Green Version]
- Hempel, M.; Schmitz, A.; Winkler, S.; Kucukoglu, O.; Bruckner, S.; Niessen, C.; Christ, B. Pathological implications of cadherin zonation in mouse liver. Cell. Mol. Life Sci. Cmls 2015, 72, 2599–2612. [Google Scholar] [CrossRef] [PubMed]
- Aurich, H.; Koenig, S.; Schneider, C.; Walldorf, J.; Krause, P.; Fleig, W.E.; Christ, B. Functional characterization of serum-free cultured rat hepatocytes for downstream transplantation applications. Cell Transplant. 2005, 14, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.; Aurich, H.; Wenkel, R.; Christ, B. Propagation and functional characterization of serum-free cultured porcine hepatocytes for downstream applications. Cell Tissue Res. 2006, 323, 433–442. [Google Scholar] [CrossRef]
- Winkler, S.; Hempel, M.; Bruckner, S.; Tautenhahn, H.M.; Kaufmann, R.; Christ, B. Identification of pathways in liver repair potentially targeted by secretory proteins from human mesenchymal stem cells. Int. J. Mol. Sci. 2016, 17, 1099. [Google Scholar] [CrossRef] [Green Version]
- van Poll, D.; Parekkadan, B.; Cho, C.H.; Berthiaume, F.; Nahmias, Y.; Tilles, A.W.; Yarmush, M.L. Mesenchymal stem cell-derived molecules directly modulate hepatocellular death and regeneration in vitro and in vivo. Hepatology 2008, 47, 1634–1643. [Google Scholar] [CrossRef]
- Varderidou-Minasian, S.; Lorenowicz, M.J. Mesenchymal stromal/stem cell-derived extracellular vesicles in tissue repair: Challenges and opportunities. Theranostics 2020, 10, 5979–5997. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, J.H.; Xiao, H.; Wu, J.M.; He, K.M.; Lv, Z.Z.; Li, Z.J.; Xu, M.; Zhang, Y.Y. Mitochondria are transported along microtubules in membrane nanotubes to rescue distressed cardiomyocytes from apoptosis. Cell Death Dis. 2018, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Vignais, M.L.; Caicedo, A.; Brondello, J.M.; Jorgensen, C. Cell connections by tunneling nanotubes: Effects of mitochondrial trafficking on target cell metabolism, homeostasis, and response to therapy. Stem Cells Int. 2017, 2017, 6917941. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Wang, Z.; Xu, H.; Yang, W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravagnan, L.; Roumier, T.; Kroemer, G. Mitochondria, the killer organelles and their weapons. J. Cell. Physiol. 2002, 192, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.D.; Sparks, C.E. Overindulgence and metabolic syndrome: Is foxo1 a missing link? J. Clin. Investig. 2008, 118, 2012–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover-Plow, J.; Huang, M. Lipoprotein(a) metabolism: Potential sites for therapeutic targets. Metab. Clin. Exp. 2013, 62, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazat, J.P.; Ransac, S. The fate of glutamine in human metabolism. The interplay with glucose in proliferating cells. Metabolites 2019, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Cazanave, S.; Podtelezhnikov, A.; Jensen, K.; Seneshaw, M.; Kumar, D.P.; Min, H.K.; Santhekadur, P.K.; Banini, B.; Mauro, A.G.; Oseini, A.M.; et al. The transcriptomic signature of disease development and progression of nonalcoholic fatty liver disease. Sci. Rep. 2017, 7, 17193. [Google Scholar] [CrossRef] [Green Version]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schonfels, W.; Herrmann, A.; Bruckner, S.; Stickel, F.; et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology 2016, 151, 513–525.e510. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. Cmls 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Hyotylainen, T.; Jerby, L.; Petaja, E.M.; Mattila, I.; Jantti, S.; Auvinen, P.; Gastaldelli, A.; Yki-Jarvinen, H.; Ruppin, E.; Oresic, M. Genome-scale study reveals reduced metabolic adaptability in patients with non-alcoholic fatty liver disease. Nat. Commun. 2016, 7, 8994. [Google Scholar] [CrossRef]
- Hardwick, R.N.; Fisher, C.D.; Canet, M.J.; Lake, A.D.; Cherrington, N.J. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 2293–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.H.; Swiderska-Syn, M.; Jewell, M.L.; Premont, R.T.; Diehl, A.M. Liver regeneration requires yap1-tgfbeta-dependent epithelial-mesenchymal transition in hepatocytes. J. Hepatol. 2018, 69, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Ezquer, M.; Ezquer, F.; Ricca, M.; Allers, C.; Conget, P. Intravenous administration of multipotent stromal cells prevents the onset of non-alcoholic steatohepatitis in obese mice with metabolic syndrome. J. Hepatol. 2011, 55, 1112–1120. [Google Scholar] [CrossRef]
- Watanabe, T.; Tsuchiya, A.; Takeuchi, S.; Nojiri, S.; Yoshida, T.; Ogawa, M.; Itoh, M.; Takamura, M.; Suganami, T.; Ogawa, Y.; et al. Development of a non-alcoholic steatohepatitis model with rapid accumulation of fibrosis, and its treatment using mesenchymal stem cells and their small extracellular vesicles. Regen. Ther. 2020, 14, 252–261. [Google Scholar] [CrossRef]
- Chella Krishnan, K.; Kurt, Z.; Barrere-Cain, R.; Sabir, S.; Das, A.; Floyd, R.; Vergnes, L.; Zhao, Y.; Che, N.; Charugundla, S.; et al. Integration of multi-omics data from mouse diversity panel highlights mitochondrial dysfunction in non-alcoholic fatty liver disease. Cell Syst. 2018, 6, 103–115.e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobaniec-Lotowska, M.E.; Lebensztejn, D.M. Ultrastructure of hepatocyte mitochondria in nonalcoholic steatohepatitis in pediatric patients: Usefulness of electron microscopy in the diagnosis of the disease. Am. J. Gastroenterol. 2003, 98, 1664–1665. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Hase, K.; Ohno, H. The molecular basis of induction and formation of tunneling nanotubes. Cell Tissue Res. 2013, 352, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Jash, E.; Prasad, P.; Kumar, N.; Sharma, T.; Goldman, A.; Sehrawat, S. Perspective on nanochannels as cellular mediators in different disease conditions. Cell Commun. Signal. Ccs 2018, 16, 76. [Google Scholar] [CrossRef] [Green Version]
- Moravcova, A.; Cervinkova, Z.; Kucera, O.; Mezera, V.; Rychtrmoc, D.; Lotkova, H. The effect of oleic and palmitic acid on induction of steatosis and cytotoxicity on rat hepatocytes in primary culture. Physiol. Res. 2015, 64, S627–S636. [Google Scholar] [CrossRef] [PubMed]
- Caballero, F.; Fernandez, A.; Matias, N.; Martinez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial s-adenosyl-l-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alijani, N.; Johari, B.; Moradi, M.; Kadivar, M. A review on transcriptional regulation responses to hypoxia in mesenchymal stem cells. Cell Biol. Int. 2020, 44, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Newell, C.; Sabouny, R.; Hittel, D.S.; Shutt, T.E.; Khan, A.; Klein, M.S.; Shearer, J. Mesenchymal stem cells shift mitochondrial dynamics and enhance oxidative phosphorylation in recipient cells. Front. Physiol. 2018, 9, 1572. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. Cd38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ards. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.M.; Chai, Y.H.; et al. Ipsc-mscs with high intrinsic miro1 and sensitivity to tnf-alpha yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar]
- Han, D.; Zheng, X.; Wang, X.; Jin, T.; Cui, L.; Chen, Z. Mesenchymal stem/stromal cell-mediated mitochondrial transfer and the therapeutic potential in treatment of neurological diseases. Stem Cells Int. 2020, 2020, 8838046. [Google Scholar] [CrossRef]
- Christ, B.; Dollinger, M.M. The generation of hepatocytes from mesenchymal stem cells and engraftment into the liver. Curr. Opin. Organ Transplant. 2011, 16, 69–75. [Google Scholar] [CrossRef]
- Neumann, E. Kraftwerke unserer Zellen. Top Life Aktuell 2010, 1005. Available online: http://www.toplife.at/gesundheit/artikel172.html (accessed on 30 June 2020).
- Kraft, E.; Stickl, H. Comparative measurement of the growth of rat liver by means of square grid and leitz’ integration table on histological slices. Virchows Archiv Fur Pathol. Anat. und Physiol. und fur Klin. Med. 1954, 324, 650–661. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, M.-J.; Karkossa, I.; Schäfer, I.; Christ, M.; Kühne, H.; Schubert, K.; Rolle-Kampczyk, U.E.; Kalkhof, S.; Nickel, S.; Seibel, P.; et al. Mitochondrial Transfer by Human Mesenchymal Stromal Cells Ameliorates Hepatocyte Lipid Load in a Mouse Model of NASH. Biomedicines 2020, 8, 350. https://doi.org/10.3390/biomedicines8090350
Hsu M-J, Karkossa I, Schäfer I, Christ M, Kühne H, Schubert K, Rolle-Kampczyk UE, Kalkhof S, Nickel S, Seibel P, et al. Mitochondrial Transfer by Human Mesenchymal Stromal Cells Ameliorates Hepatocyte Lipid Load in a Mouse Model of NASH. Biomedicines. 2020; 8(9):350. https://doi.org/10.3390/biomedicines8090350
Chicago/Turabian StyleHsu, Mei-Ju, Isabel Karkossa, Ingo Schäfer, Madlen Christ, Hagen Kühne, Kristin Schubert, Ulrike E. Rolle-Kampczyk, Stefan Kalkhof, Sandra Nickel, Peter Seibel, and et al. 2020. "Mitochondrial Transfer by Human Mesenchymal Stromal Cells Ameliorates Hepatocyte Lipid Load in a Mouse Model of NASH" Biomedicines 8, no. 9: 350. https://doi.org/10.3390/biomedicines8090350