Antibody-Based Immunotherapy: Alternative Approaches for the Treatment of Metastatic Melanoma

 ,
,  ,
, 
Abstract
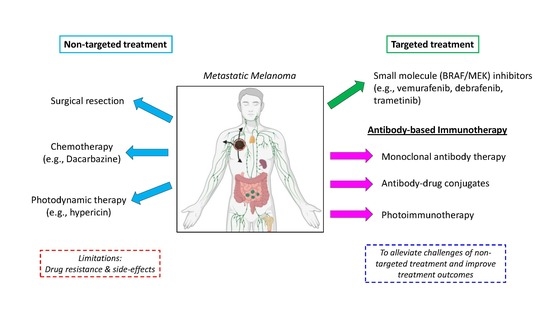
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
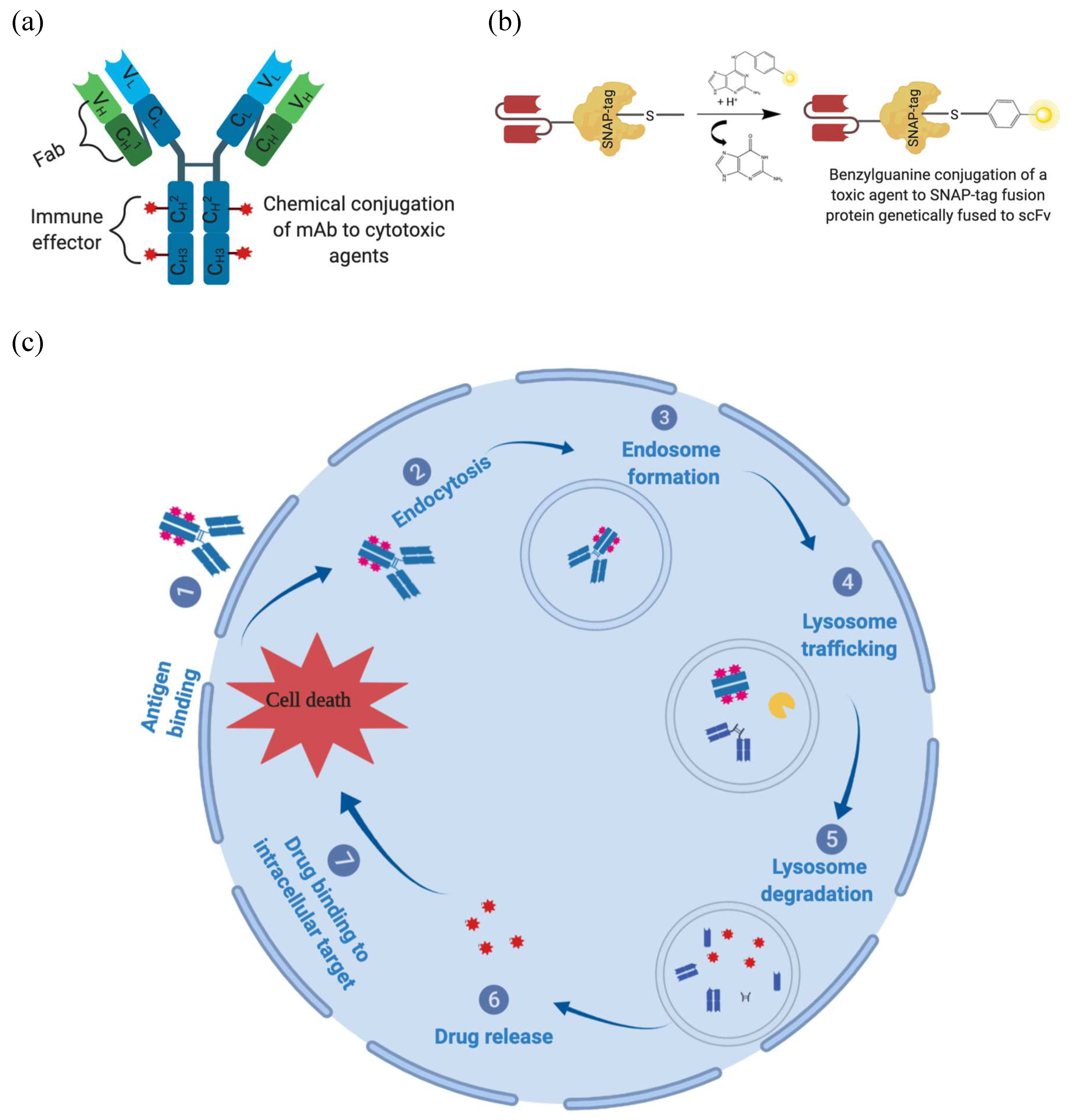{kind=link}
{kind=link}
1. Introduction
1.1. Summary of FDA-Approved Melanoma Chemotherapy and Targeted Therapies
1.2. Photodynamic Therapy Treatment
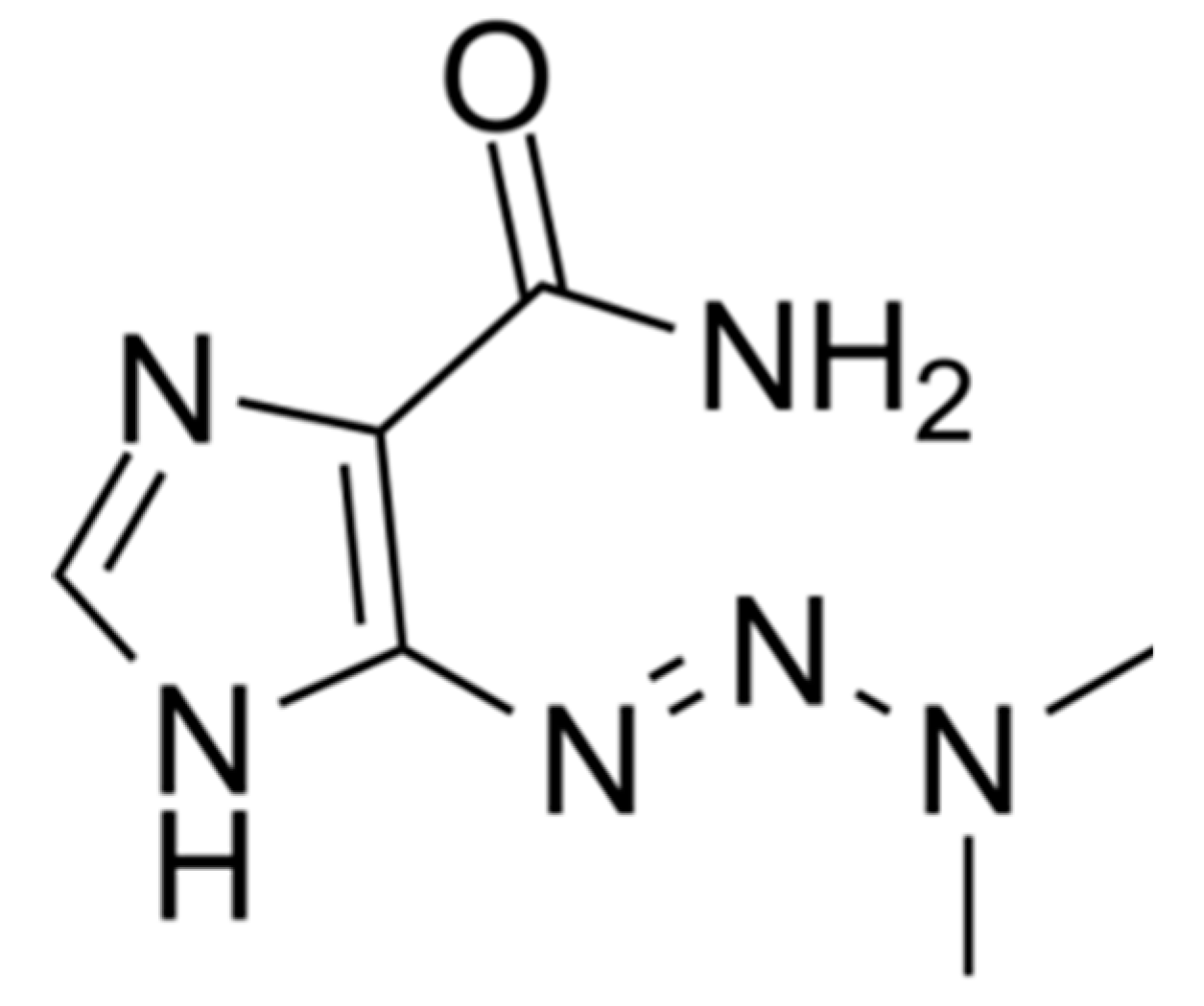
1.2.1. Mechanism of Action
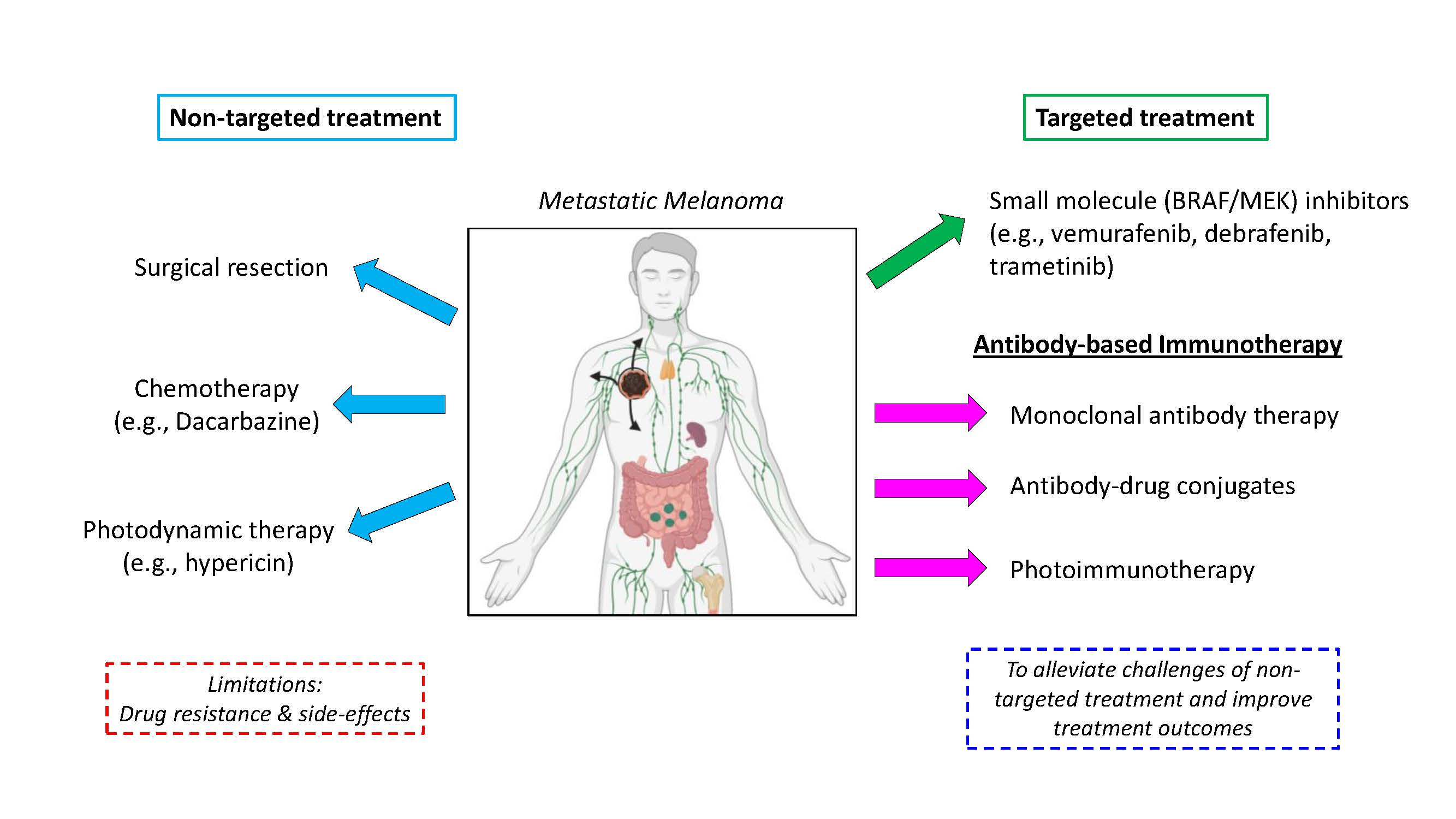
1.2.2. Hypericin
1.2.3. Photodynamic Therapy and Melanoma
1.3. Melanoma Immunotherapy
1.3.1. Introduction to Immunotherapy
1.3.2. Antibody-Based Immunotherapy
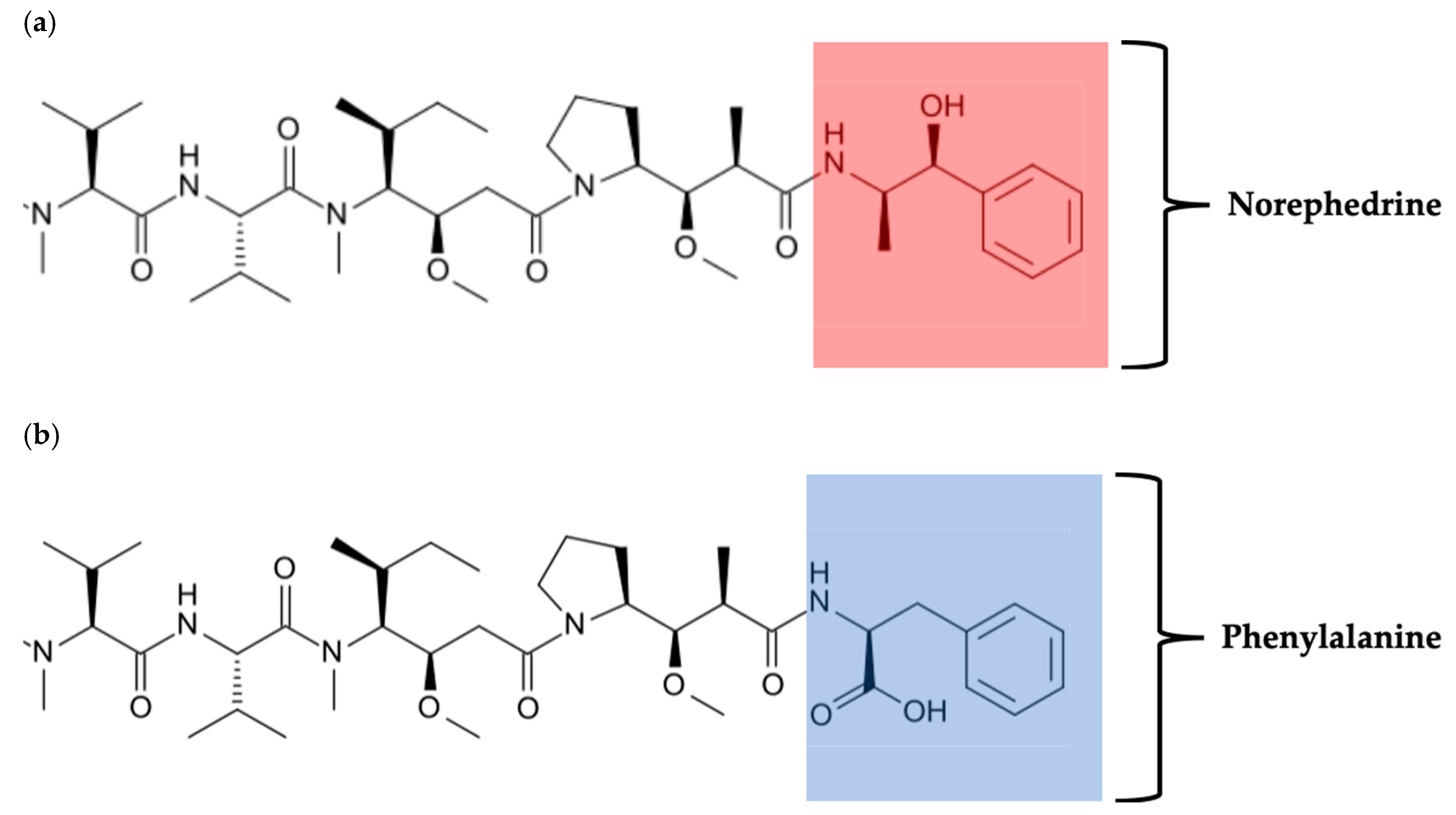
1.3.3. Overcoming Monoclonal Antibody Limitations Using Antibody-Drug Conjugates
1.3.4. Recombinant Antibody-Drug Conjugates for Melanoma Treatment
1.4. Photoimmunotherapy
2. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.H.; Lok, H.C.; Sahni, S.; Lane, D.J.R.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Baldea, I.; Filip, A.G.; Napoca, C. Photodynamic therapy in melanoma an update. J. Physiol. Pharmacol. 2012, 63, 109–118. [Google Scholar] [PubMed]
- Mackie, R.M.; Hauschild, A.; Eggermont, A.M.M. Epidemiology of invasive cutaneous melanoma. Ann. Oncol. 2009, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chartrain, M.; Riond, J.; Stennevin, A.; Vandenberghe, I.; Gomes, B.; Lamant, L.; Meyer, N.; Gairin, J.E.; Guilbaud, N.; Annereau, J.P. Melanoma chemotherapy leads to the selection of ABCB5-expressing cells. PLoS ONE 2012, 7, e36762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of us food and drug administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorenko, I.V.; Paraiso, K.H.T.; Smalley, K.S.M. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem. Pharmacol. 2011, 82, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biteghe, F.N.; Davids, L.M. A combination of photodynamic therapy and chemotherapy displays a differential cytotoxic effect on human metastatic melanoma cells. J. Photochem. Photobiol. B Biol. 2017, 166, 18–27. [Google Scholar] [CrossRef]
- Ugurel, S.; Paschen, A.; Becker, J.C. Dacarbazine in melanoma: From a chemotherapeutic drug to an immunomodulating agent. J. Investig. Dermatol. 2013, 133, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Koprowska, K.; Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M.E. Parthenolide enhances dacarbazine activity against melanoma cells. Cancer Biol. Ther. 2013, 24, 835–845. [Google Scholar] [CrossRef]
- Marabondo, S.; Kaufman, H.L. High-dose interleukin-2 (IL-2) for the treatment of melanoma: Safety considerations and future directions. Expert Opin. Drug Saf. 2017, 12, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Ridolfi, L.; de Rosa, F.; Ridolfi, R.; Gentili, G.; Valmorri, L.; Scarpi, E.; Parisi, E.; Romeo, A.; Guidoboni, M. Radiotherapy as an immunological booster in patients with metastatic melanoma or renal cell carcinoma treated with high-dose Interleukin-2: Evaluation of biomarkers of immunologic and therapeutic response. J. Transl. Med. 2014, 12, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology (Williston Park) 2009, 23, 488–496. [Google Scholar] [PubMed]
- Palathinkal, D.M.; Sharma, T.R.; Koon, H.B.; Bordeaux, J.S. Current systemic therapies for melanoma. Dermatol. Surg. 2014, 40, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Oh, A.; Tran, D.M.; McDowell, L.C.; Keyvani, D.; Barcelon, J.A.; Merino, O.; Wilson, L. Cost-effectiveness of nivolumab-ipilimumab combination therapy compared with monotherapy for first-line treatment of metastatic melanoma in the United States. J. Manag. Care Spec. Pharm. 2017, 23, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capone, E.; Lamolinara, A.; D’Agostino, D.; Rossi, C.; De Laurenzi, V.; Iezzi, M.; Iacobelli, S.; Sala, G. EV20-mediated delivery of cytotoxic auristatin MMAF exhibits potent therapeutic efficacy in cutaneous melanoma. J. Control. Release 2018, 277, 48–56. [Google Scholar] [CrossRef]
- Von Felbert, V.; Bauerschlag, D.; Maass, N.; Bräutigam, K.; Meinhold-Heerlein, I.; Woitok, M.; Barth, S.; Hussain, A.F. A specific photoimmunotheranostics agent to detect and eliminate skin cancer cells expressing EGFR. J. Cancer Res. Clin. Oncol. 2016, 142, 1003–1011. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Approves Encorafenib and Binimetinib in Combination for Unresectable or Metastatic Melanoma with BRAF Mutations. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-encorafenib-and-binimetinib-combination-unresectable-or-metastatic-melanoma-braf (accessed on 4 August 2020).
- Michaelis, M.; Wiese, M.; Michaelis, M.; Rothweiler, F.; Nerreter, T.; Van Rikxoort, M.; Zehner, R. Association between acquired resistance to Association between acquired resistance to PLX4032 (vemurafenib) and ATP-binding cassette transporter expression. BMC Res. Notes 2014, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Egger, M.E.; McMasters, K.M.; Hao, H. Differential expression of ABCB5 in BRAF inhibitor-resistant melanoma cell lines. BMC Cancer 2018, 18, 1–10. [Google Scholar] [CrossRef]
- Manzano, J.L.; Layos, L.; Bugés, C.; de Los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martínez-Cardús, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, S.; Seidel, D.; Jahnke, H.G.; Eichler, M.; Simon, J.C.; Robitzki, A.A. Induced cross-resistance of BRAF V600E melanoma cells to standard chemotherapeutic dacarbazine after chronic PLX4032 treatment. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Yue, Q.; Ma, J.; Liu, Y.; Zhao, T.; Guo, W.; Zhu, G.; Guo, S.; Wang, S.; Gao, T.; et al. POU4F1 promotes the resistance of melanoma to BRAF inhibitors through MEK/ERK pathway activation and MITF up-regulation. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Wolchok, J.D. Immune checkpoint blockade. Hematol. Oncol. Clin. N. Am. 2014, 28, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Padayachee, E.R.; Biteghe, F.A.N.; Malindi, Z.; Bauerschlag, D.; Barth, S. Human Antibody Fusion Proteins/Antibody Drug Conjugates in Breast and Ovarian Cancer. Transfus. Med. Hemother. 2017, 44, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.H.; et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Van Wyk, J.C.; Rebecca, E.; Henry, P.; Adeola, A.; Augustine, F.; Biteghe, N.; Chetty, S.; Patience, N.; Barth, S. Applications of SNAP—Tag technology in skin cancer therapy. Health Sci. Rep. 2019, 2, e103. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan- refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Bakhtiar, R. Antibody drug conjugates. Biotechnol. Lett. 2016, 38, 1655–1664. [Google Scholar] [CrossRef]
- Ho, R.M.; Crescioli, S.; Mele, S.; Sachouli, E.; Cheung, A.; Chui, C.K.; Andriollo, P.; Jackson, P.J.M.; Lacy, K.E.; Spicer, J.F.; et al. A Novel Antibody-Drug Conjugate (ADC) Delivering a DNA Mono-Alkylating Payload to Chondroitin Sulfate Proteoglycan (CSPG4)-Expressing Melanoma Ricarda. Cancers 2020, 12, 1–22. [Google Scholar]
- Carter, P.J.; Senter, P.D. Antibody-Drug Conjugates for Cancer Therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef]
- Vezina, H.E.; Cotreau, M.; Han, T.H.; Gupta, M. Antibody-Drug Conjugates as Cancer Therapeutics: Past, Present, and Future. J. Clin. Pharmacol. 2017, 57, S11–S25. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Nesterova, A.; Alley, S.C.; Torgov, M.Y.; Carter, P.J. Potent cytotoxicity of an auristatin-containing antibody-drug conjugate targeting melanoma cells expressing melanotransferrin/p97. Mol. Cancer Ther. 2006, 5, 1474–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chalouni, C.; Tan, C.; Clark, R.; Venook, R.; Ohri, R.; Raab, H.; Firestein, R.; Mallet, W.; Polakis, P. The melanosomal protein PMEL17 as a target for antibody drug conjugate therapy in melanoma. J. Biol. Chem. 2012, 287, 24082–24091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, H.; Griffiths, G.L.; Choyke, P.L. Near-Infrared Photoimmunotherapy: Photoactivatable Antibody–Drug Conjugates (ADCs). Bioconjug. Chem. 2019. [Google Scholar] [CrossRef]
- Amoury, M.; Bauerschlag, D.; Zeppernick, F.; von Felbert, V.; Berges, N.; Di Fiore, S.; Mintert, I.; Bleilevens, A.; Maass, N.; Bräutigam, K.; et al. Photoimmunotheranostic agents for triple-negative breast cancer diagnosis and therapy that can be activated on demand. Oncotarget 2016, 7, 54925–54936. [Google Scholar] [CrossRef] [Green Version]
- Maawy, A.A.; Hiroshima, Y.; Zhang, Y.; Heim, R.; Makings, L.; Garcia-Guzman, M.; Luiken, G.A.; Kobayashi, H.; Hoffman, R.M.; Bouvet, M. Near infra-red photoimmunotherapy with anti-CEA-IR700 results in extensive tumor lysis and a significant decrease in tumor burden in orthotopic mouse models of pancreatic cancer. PLoS ONE 2015, 10, e0121989. [Google Scholar] [CrossRef]
- Bauerschlag, D.; Meinhold-Heerlein, I.; Maass, N.; Bleilevens, A.; Bräutigam, K.; Al Rawashdeh, W.; Di Fiore, S.; Haugg, A.M.; Gremse, F.; Steitz, J.; et al. Detection and Specific Elimination of EGFR+ Ovarian Cancer Cells Using a Near Infrared Photoimmunotheranostic Approach. Pharm. Res. 2017, 34, 696–703. [Google Scholar] [CrossRef]
- Saxena, A.; Khosraviani, S.; Noel, S.; Mohan, D.; Donner, T.; Hamad, A.R.A. Photoimmunotherapy lowers recurrence after pancreatic cancer surgery in orthotopic nude mouse models. J. Surg. Res. 2015, 197, 5–11. [Google Scholar] [CrossRef]
- Nakajima, T.; Sano, K.; Choyke, P.L.; Kobayashi, H. Improving the efficacy of photoimmunotherapy (PIT) using a cocktail of antibody conjugates in a multiple antigen tumor model. Theranostics 2013, 3, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Nagaya, T.; Nakamura, Y.; Okuyama, S.; Ogata, F.; Maruoka, Y.; Choyke, P.L.; Kobayashi, H. Near-Infrared Photoimmunotherapy Targeting Prostate Cancer with Prostate-Specific Membrane Antigen (PSMA) Antibody. Mol. Cancer Res. 2017, 15, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- Ogata, F.; Nagaya, T.; Nakamura, Y.; Sato, K. Near-infrared photoimmunotherapy: A comparison of light dosing schedules. Oncotarget 2017, 8, 35069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foletto, M.C.; Haas, S.E. Cutaneous melanoma: New advances in treatment *. An. Bras. Dermatol. 2014, 89, 301–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, D.P.; Moriyama, L.T.; De Oliveira, E.R.; Inada, N.M.; Bagnato, V.S.; Kurachi, C.; Salvio, A.G. Single visit PDT for basal cell carcinoma—A new therapeutic protocol. Photodiagnosis Photodyn. Ther. 2019, 26, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, C.; Kruger, C.A.; Abrahamse, H. Photodynamic Therapy for Metastatic Melanoma Treatment: A Review. Technol. Cancer Res. Treat. 2018, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Goler-Baron, V.; Assaraf, Y.G. Overcoming multidrug resistance via photodestruction of ABCG2-rich extracellular vesicles sequestering photosensitive chemotherapeutics. PLoS ONE 2012, 7, e35487. [Google Scholar] [CrossRef] [Green Version]
- Madan, V.; Lear, J.T.; Szeimies, R.-M. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.V.; Davids, L.M. Depigmentation in melanomas increases the efficacy of hypericin-mediated photodynamic-induced cell death. Photodiagnosis Photodyn. Ther. 2011, 9, 156–163. [Google Scholar] [CrossRef]
- Kawczyk-Krupka, A.; Bugaj, A.M.; Latos, W.; Zaremba, K.; Sieroń, A. Photodynamic therapy in treatment of cutaneous and choroidal melanoma. Photodiagnosis Photodyn. Ther. 2013, 10, 503–509. [Google Scholar] [CrossRef]
- Wargo, J.A.; Gogdill, A.; Dang, P.; Gupta, R.; Piris, A.; Boni, A.; Garber, H.R.; Ott, H.; Newton, L.P.; Flaherty, K.T.; et al. Treatment with a selective inhibitor of BRAFV600E increases melanocyte antigen expression and CD8 T cell infiltrate in tumors of patients with metastatic melanoma. Cancer Res. 2011, 71, 958. [Google Scholar]
- Nathanson, K.L.; Martin, A.M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.T.; et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryabaya, O.O.; Inshakov, A.N.; Egorova, A.V.; Emelyanova, M.A.; Nasedkina, T.V.; Zasedatelev, A.S.; Khochenkov, D.A.; Stepanova, E.V. Autophagy inhibitors chloroquine and LY294002 enhance temozolomide cytotoxicity on cutaneous melanoma cell lines in vitro. Anticancer Drugs 2017, 3, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, 14504. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, I.; Celli, J.P.; Evans, C.L.; Abu-Yousif, A.O.; Muzikansky, A.; Pogue, B.W.; Finkelstein, D.; Hasan, T. Synergistic enhancement of carboplatin efficacy with photodynamic therapy in a three-dimensional model for micrometastatic ovarian cancer. Cancer Res. 2010, 70, 9319–9328. [Google Scholar] [CrossRef] [Green Version]
- Tudor, D.; Nenu, I.; Filip, G.A.; Olteanu, D.; Cenariu, M.; Tabaran, F.; Ion, R.M.; Gligor, L.; Baldea, I. Combined regimen of photodynamic therapy mediated by Gallium phthalocyanine chloride and Metformin enhances anti-melanoma efficacy. PLoS ONE 2017, 12, e0173241. [Google Scholar] [CrossRef]
- Davids, L.M.; Kleemann, B. Combating melanoma: The use of photodynamic therapy as a novel, adjuvant therapeutic tool. Cancer Treat. Rev. 2011, 37, 465–475. [Google Scholar] [CrossRef]
- Nsole Biteghe, F.A.; Padayachee, E.; Davids, L.M.; Nyangone, C.E.T.; Jean Delacroix, N.; Barth, S. Desensitization of metastatic melanoma cells to therapeutic treatment through repeated exposure to dacarbazine. J. Photochem. Photobiol. B Biol. 2020, 116544. [Google Scholar] [CrossRef]
- Sharma, K.V.; Davids, L.M. Hypericin-PDT-induced rapid necrotic death in human squamous cell carcinoma cultures after multiple treatment. Cell Biol. Int. 2012, 36, 1261–1266. [Google Scholar] [CrossRef]
- Jendzelovský, R.; Mikes, J.; Koval’, J.; Soucek, K.; Procházková, J.; Kello, M.; Sacková, V.; Hofmanová, J.; Kozubík, A.; Fedorocko, P. Drug efflux transporters, MRP1 and BCRP, affect the outcome of hypericin-mediated photodynamic therapy in HT-29 adenocarcinoma cells. Photochem. Photobiol. Sci. 2009, 8, 1716–1723. [Google Scholar] [CrossRef]
- Cohen, D.K.; Lee, P.K. Photodynamic therapy for non-melanoma skin cancers. Cancers 2016, 8, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskaran, R.; Lee, J.; Yang, S.-G. Clinical development of photodynamic agents and therapeutic applications. Biomater. Res. 2018, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.L.; Oliveira, J.; Monteiro, E.; Santos, J.; Sarmento, C. Treatment of Head and Neck Cancer with Photodynamic Therapy with Redaporfin: A Clinical Case Report. Case Rep. Oncol. 2018, 11, 769–776. [Google Scholar] [CrossRef]
- Nelson, J.S.; McCullough, J.L.; Berns, M.W. Photodynamic therapy of human malignant melanoma xenografts in athymic nude mice. J. Natl. Cancer Inst. 1988, 80, 56–60. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D. Photodynamic Therapy of Cancer: An Update. CA. Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Wagnières, G.A.; Star, W.M.; Wilson, B.C. In Vivo Fluorescence Spectroscopy and Imaging for Oncological Applications. Photochem. Photobiol. 1998, 68, 603–632. [Google Scholar] [CrossRef] [PubMed]
- Van Straten, D.; Mashayekhi, V.; de Bruijn, H.S.; Oliveira, S.; Robinson, D.J. Oncologic photodynamic therapy: Basic principles, current clinical status and future directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef]
- Moan, J.; Berg, K. Differential effects of glucose deprivation on the cellular sensitivity towards photodynamic treatment-based production of reactive oxygen species and apoptosis-induction. Photochem. Photobiol. 1991, 53, 1991. [Google Scholar]
- Uzdensky, A.B.; Ma, L.W.; Iani, V.; Hjortland, G.O.; Steen, H.B.; Moan, J. Intracellular localisation of hypericin in human glioblastoma and carcinoma cell lines. Lasers Med. Sci. 2001, 16, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Ritz, R.; Roser, F.; Radomski, N.; Strauss, W.S.L.; Tatagiba, M.; Gharabaghi, A. Subcellular colocalization of hypericin with respect to endoplasmic reticulum and Golgi apparatus in glioblastoma cells. Anticancer Res. 2008, 28, 2033–2038. [Google Scholar] [PubMed]
- Robertson, C.; Evans, D.H.; Abrahamse, H. Photodynamic therapy (PDT): A short review on cellular mechanisms and cancer research applications for PDT. J. Photochem. Photobiol. B. 2009, 96, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Tan, L.C.; Dong, L.W.; Zhang, W.Q.; Shen, X.X.; Lu, X.; Zheng, H.; Lu, Y.G. Susceptibility and Resistance Mechanisms During Photodynamic Therapy of Melanoma. Front. Oncol. 2020, 10, 597. [Google Scholar] [CrossRef] [PubMed]
- Mikeš, J.; Jendželovský, R.; Fedoročko, P. Cellular Aspects of Photodynamic Therapy with Hypericin. In Photodynamic Therapy: New Research; Mohamed Lotfy Taha, E., Ed.; Nova Science Publishers: New York, NY, USA, 2013; ISBN 9781624176357. [Google Scholar]
- Theodossiou, T.A.; Hothersall, J.S.; De Witte, P.A.; Pantos, A.; Agostinis, P. The multifaceted photocytotoxic profile of hypericin. Mol. Pharm. 2009, 6, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.; Vepachedu, R.; Lawrence, C.B.; Stermitz, F.R.; Vivanco, J.M.; Bais, H.P. Molecular and Biochemical Characterization of an Enzyme Responsible for the Formation of Hypericin in St. John’s Wort (Hypericum perforatum L.). J. Biol. Chem. 2003, 278, 32413–32422. [Google Scholar] [CrossRef] [Green Version]
- Karioti, A.; Bilia, A.R. Hypericins as Potential Leads for New Therapeutics. Int. J. Mol. Sci. 2010, 11, 562–594. [Google Scholar] [CrossRef] [Green Version]
- Kleemann, B.; Loos, B.; Scriba, T.J.; Lang, D.; Davids, L.M. St John’s Wort (Hypericum perforatum L.) photomedicine: Hypericin-photodynamic therapy induces metastatic melanoma cell death. PLoS ONE 2014, 9, e103762. [Google Scholar] [CrossRef] [Green Version]
- Falk, H. From the Photosensitizer Hypericin to the Photoreceptor Stentorin—The Chemistry of Phenanthroperylene Quinones. Angew. Chem. Int. Ed. Engl. 1999, 38, 3116–3136. [Google Scholar] [CrossRef]
- Ott, M.; Huls, M.; Cornelius, M.G.; Fricker, G. St. John’s Wort constituents modulate P-glycoprotein transport activity at the blood-brain barrier. Pharm. Res. 2010, 27, 811–822. [Google Scholar] [CrossRef]
- Butterweck, V.; Schmidt, M. St. John’s wort: Role of active compounds for its mechanism of action and efficacy. Wien. Med. Wochenschr. 2007, 157, 356–361. [Google Scholar] [CrossRef]
- Ritz, R.; Daniels, R.; Noell, S.; Gc, F.; Schmidt, V.; Bornemann, A.; Ramina, K.; Mayer, D.; Ws, S.; Tatagiba, M. Hypericin for visualization of high grade gliomas: First clinical experience. Eur. J. Surg. Oncol. 2012, 38, 2012. [Google Scholar] [CrossRef]
- Noell, S.; Feigl, G.C.; Serifi, D.; Mayer, D.; Naumann, U.; Göbel, W.; Ehrhardt, A.; Ritz, R. Microendoscopy for hypericin fluorescence tumor diagnosis in a subcutaneous glioma mouse model. Photodiagnosis Photodyn. Ther. 2013, 10, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Penjweini, R.; Loew, H.G.; Breit, P.; Kratky, K.W. Optimizing the antitumor selectivity of PVP-Hypericin re A549 cancer cells and HLF normal cells through pulsed blue light. Photodiagnosis Photodyn. Ther. 2013, 10, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.D.; Liu, J.K.; Sheng, X.; Chin, S.S.; Schmidt, M.H.; Weiss, M.H.C.W. Hypericin-mediated photodynamic therapy of pituitary tumors: Preclinical study in a GH4C1 rat tumor model. J. Neurooncol. 2008, 87, 11060. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, H.B.; Hinton, D.R.; Law, R.E.; Gopalakrishna, R.; Su, Y.Z.; Chen, Z.H.; Weiss, M.H.C.W. Inhibition of cellular growth and induction of apoptosis in pituitary adenoma cell lines by the protein kinase C inhibitor hypericin: Potential therapeutic application. J. Neurosurg. 1996, 85, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, P.S.; Rhee, C.K.; Kim, K.H.; Paek, W.; Chung, J.; Paiva, M.B.; Eshraghi, A.A.; Castro, D.J.S.R. Intratumoral hypericin and KTP laser therapy for transplanted squamous cell carcinoma. Laryngoscope 2000, 110, 1312–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyenge, E.B.; Forny, P.; Lüscher, D.; Laass, A.; Walt, H.; Maake, C. Effects of hypericin and a chlorin based photosensitizer alone or in combination in squamous cell carcinoma cells in the dark. Photodiagnosis Photodyn. Ther. 2012, 9, 321–331. [Google Scholar] [CrossRef]
- Ali, S.M.; Olivo, M. Bio-distribution and subcellular localization of Hypericin and its role in PDT induced apoptosis in cancer cells. Int. J. Oncol. 2002, 21, 531–540. [Google Scholar] [CrossRef]
- Chung, P.-S.; Saxton, R.E.; Paiva, M.B.; Rhee, C.-K.; Soudant, J.; Mathey, A.; Foote, C.; Castro, D.J. Hypericin uptake in rabbits and nude mice transplanted with human squamous cell carcinomas: Study of a new sensitizer for laser phototherapy. Laryngoscope 1994, 104, 1471–1476. [Google Scholar] [CrossRef]
- Huntosova, V.; Alvarez, L.; Bryndzova, L.; Nadova, Z.; Jancura, D.; Buriankova, L.; Bonneau, S.; Brault, D.; Miskovsky, P.; Sureau, F. Interaction dynamics of hypericin with low-density lipoproteins and U87-MG cells. Int. J. Pharm. 2010, 389, 32–40. [Google Scholar] [CrossRef]
- Delaey, E.M.; Obermuëller, R.; Zupkó, I.; De Vos, D.; Falk, H.; Witte, P.A. In Vitro Study of the Photocytotoxicity of Some Hypericin Analogs on Different Cell Lines. Photochem. Photobiol. 2007, 74, 164–171. [Google Scholar] [CrossRef]
- Kascakova, S.; Nadova, Z.; Mateasik, A.; Mikes, J.; Huntosova, V.; Refregiers, M. High level of low-density lipoprotein receptors enhance hypericin uptake by U-87 MG cells in the presence of LDL. Photochem. Photobiol. 2008, 84, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Redmond, R.W.; Gamlin, J.N. A compilation of singlet oxygen yields from biologically relevant molecules. Photochem. Photobiol. 1999, 70, 391–475. [Google Scholar] [CrossRef] [PubMed]
- Solár, P.; Cavarga, I.; Hofmanová, J.; Cekanová-Figurová, M.; Miskovský, P.; Brezáni, P.; Hrcková, G.; Kozubík, A.; Fedorocko, P. Effect of acetazolamide on hypericin photocytotoxicity. Planta Med. 2002, 68, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Kamuhabwa, A.R.; Agostinis, P.M.; D’Hallewin, M.A.; Baert, L.; de Witte, P.A. Cellular photodestruction induced by hypericin in AY-27 rat bladder carcinoma cells. Photochem. Photobiol. 2001, 74, 126–132. [Google Scholar] [CrossRef]
- Agostinis, P.; Buytaert, E.; Breyssens, H.; Hendrickx, N. Regulatory pathways in photodynamic therapy induced apoptosis. Photochem. Photobiol. Sci. 2004, 3, 721–729. [Google Scholar] [CrossRef]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. Sci. 2002, 1, 1–21. [Google Scholar]
- Ndhundhuma, I.M.; Abrahamse, H. Susceptibility of In Vitro Melanoma Skin Cancer to Photoactivated Hypericin versus Aluminium(III) Phthalocyanine Chloride Tetrasulphonate. Biomed. Res. Int. 2017, 2017, 5407012. [Google Scholar] [CrossRef] [Green Version]
- Davids, L.M.; Kleemann, B.; Kacerovská, D.; Pizinger, K.; Kidson, S.H. Hypericin phototoxicity induces different modes of cell death in melanoma and human skin cells. J. Photochem. Photobiol. B Biol. 2008, 91, 67–76. [Google Scholar] [CrossRef]
- Maduray, K.; Odhav, B.; Nyokong, T. In vitro photodynamic effect of aluminum tetrasulfophthalocyanines on melanoma skin cancer and healthy normal skin cells. Photodiagnosis Photodyn. Ther. 2012, 9, 32–39. [Google Scholar] [CrossRef]
- Maduray, K.; Karsten, A.; Odhav, B.; Nyokong, T. In vitro toxicity testing of zinc tetrasulfophthalocyanines in fibroblast and keratinocyte cells for the treatment of melanoma cancer by photodynamic therapy. J. Photochem. Photobiol. B. 2011, 103, 98–104. [Google Scholar] [CrossRef]
- Huang, Y.; Vecchio, D.; Avci, P.; Yin, R.; Garcia-diaz, M.; Hamblin, M.R. Melanoma resistance to photodynamic therapy: New insights. Biol. Chem. 2013, 394, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Sheleg, S.V.; Zhavrid, E.A.; Khodina, T.V.; Kochubeev, G.A.; Istomin, Y.P.; Chalov, V.N.; Zhuravkin, I.N. Photodynamic therapy with chlorin e(6) for skin metastases of melanoma. Photodermatol. Photoimmunol. Photomed. 2004, 20, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Saczko, J.; Kulbacka, J.; Chwiłkowska, A.; Drag-Zalesińiska, M.; Wysocka, T.; Lugowski, M.; Banaś, T. The influence of photodynamic therapy on apoptosis in human melanoma cell line. Folia Histochem. Cytobiol. 2005, 43, 129–132. [Google Scholar] [PubMed]
- Robertson, C.A.; Abrahamse, H.; Evans, D. The in vitro PDT efficacy of a novel metallophthalocyanine (MPc) derivative and established 5-ALA photosensitizing dyes against human metastatic melanoma cells. Lasers Surg. Med. 2010, 42, 766–776. [Google Scholar] [CrossRef]
- Choromańska, A.; Saczko, J.; Kulbacka, J.; Skolucka, N.; Majkowski, M. The potential role of photodynamic therapy in the treatment of malignant melanoma—An in vitro study. Adv. Clin. Exp. Med. 2012, 21, 179–185. [Google Scholar] [PubMed]
- Garg, A.D.; Agostinis, P. ER stress, autophagy and immunogenic cell death in photodynamic therapy-induced anti-cancer immune responses. Photochem. Photobiol. Sci. 2014, 13, 474–487. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.V.P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Zheng, Y.; Yin, G.; Le, V.; Zhang, A.; Chen, S.Y.; Liang, X.; Liu, J.W. Photodynamic-therapy activates immune response by disrupting immunity homeostasis of tumor cells, which generates vaccine for cancer therapy. Int. J. Biol. Sci. 2016, 12, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.G.; Valencia, J.C.; Gillet, J.-P.; Hearing, V.J.; Gottesman, M.M. Involvement of ABC transporters in melanogenesis and the development of multidrug resistance of melanoma. Pigment. Cell Melanoma Res. 2009, 22, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Su, Y.S.; Wang, W.; Kardosh, A.; Liebes, L.F.; Hofman, F.M.; Schönthal, A.H.; Chen, T.C. Enhancement of glioblastoma cell killing by combination treatment with temozolomide and tamoxifen or hypericin. Neurosurg. Focus 2006, 20, E20. [Google Scholar] [CrossRef] [PubMed]
- Theodossiou, T.A.; Ali, M.; Grigalavicius, M.; Grallert, B.; Dillard, P.; Schink, K.O.; Olsen, C.E.; Wälchli, S.; Inderberg, E.M.; Kubin, A.; et al. Simultaneous defeat of MCF7 and MDA-MB-231 resistances by a hypericin PDT—Tamoxifen hybrid therapy. NPJ Breast Cancer 2019, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Yang, L.; Shi, H.; Du, W.; Qi, Y.; Qiu, C.; Liang, X. Endoplasmic reticulum-targeting photosensitizer Hypericin confers chemo- sensitization towards oxaliplatin through inducing pro-death autophagy. Int. J. Biochem. Cell Biol. 2017, 87, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, B.; Feng, X.; Qu, F.; Wang, S.; Wu, L.; Wang, X. Photodiagnosis and Photodynamic Therapy Apoptosis and autophagy induced by DVDMs-PDT on human esophageal cancer Eca-109 cells. Photodiagnosis Photodyn. Ther. 2018, 24, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Kaizhen, Y.; Ting, N.; Mengyu, L.; Lihua, T.; Ling, K. Enhanced cytotoxicity and apoptosis through inhibiting autophagy in metastatic potential colon cancer SW620 cells treated with Chlorin e6 photodynamic therapy. Photodiagnosis Photodyn. Ther. 2018, 24, 332–341. [Google Scholar] [CrossRef]
- Sun, Q.; You, Q.; Pang, X.; Tan, X.; Wang, J.; Liu, L.; Guo, F.; Tan, F.; Li, N. A photoresponsive and rod-shape nanocarrier: Single wavelength of light triggered photothermal and photodynamic therapy based on AuNRs-capped & Ce6-doped mesoporous silica nanorods. Biomaterials 2017, 122, 188–200. [Google Scholar] [CrossRef]
- Wu, C.; Li, D.; Wang, L.; Guan, X.; Tian, Y.; Yang, H.; Li, S.; Liu, Y. Single wavelength light-mediated, synergistic bimodal cancer photoablation and amplified photothermal performance by graphene/gold nanostar/photosensitizer theranostics. Acta Biomater. 2017, 53, 631–642. [Google Scholar] [CrossRef]
- Yan, J.; Sun, H.; Li, J.; Qi, W.; Wang, H. A theranostic plaster combining photothermal therapy and photodynamic therapy based on chlorin e6/gold nanorods (Ce6/Au nrs) composite. Colloids Surf. A Physicochem. Eng. Asp. 2018, 537, 460–466. [Google Scholar] [CrossRef]
- Xu, W.; Qian, J.; Hou, G.; Wang, Y.; Wang, J.; Sun, T.; Ji, L.; Suo, A.; Yao, Y. A dual-targeted hyaluronic acid-gold nanorod platform with triple-stimuli responsiveness for photodynamic/photothermal therapy of breast cancer. Acta Biomater. 2019, 83, 400–413. [Google Scholar] [CrossRef]
- Kareliotis, G.; Tremi, I.; Kaitatzi, M.; Drakaki, E.; Serafetinides, A.A.; Makropoulou, M.; Georgakilas, A.G. Combined radiation strategies for novel and enhanced cancer treatment. Int. J. Radiat. Biol. 2020, 96, 1087–1103. [Google Scholar] [CrossRef]
- Nowis, D.; Makowski, M.; Stokłosa, T.; Legat, M.; Issat, T.; Gołąb, J. Direct tumor damage mech anisms of photodynamic therapy. Acta Biochim. Pol. 2005, 52, 339–352. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Casares, N.; Péquignot, M.O.; Chaput, N.; Albert, M.L.; Kroemer, G. Immune response against dying tumor cells. Adv. Immunol. 2004, 84, 131–179. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.D.; Manadas, B.J.; Carvalho, A.P.; Duarte, C.B. Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta 2004, 1704, 59–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammela, T.; Saaristo, A.; Holopainen, T.; Ylä-Herttuala, S.; Andersson, L.C.; Virolainen, S.; Immonen, I.; Alitalo, K. Photodynamic ablation of lymphatic vessels and intralymphatic cancer cells prevents metastasis. Sci. Transl. Med. 2011, 3, 69ra11. [Google Scholar] [CrossRef] [PubMed]
- Turubanova, V.D.; Balalaeva, I.V.; Mishchenko, T.A.; Catanzaro, E.; Alzeibak, R.; Peskova, N.N.; Efimova, I.; Bachert, C.; Mitroshina, E.V.; Krysko, O.; et al. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J. Immunother. Cancer 2019, 7, 350. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Vandecasteele, K.; Bachert, C.; Krysko, O.; Krysko, D.V. Immunogenic Apoptotic Cell Death and Anticancer Immunity. Adv. Exp. Med. Biol. 2016, 930, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, S.O.; Evans, S.S.; Baumann, H.; Owczarczak, B.; Maier, P.; Vaughan, L.; Wang, W.C.; Unger, E.; Henderson, B.W. Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br. J. Cancer 2003, 88, 1772–1779. [Google Scholar] [CrossRef] [Green Version]
- Firczuk, M.; Nowis, D.; Golab, J. PDT-induced inflammatory and host responses. Photochem. Photobiol. Sci. 2011, 10, 653–663. [Google Scholar] [CrossRef]
- Henderson, B.W.; Gollnick, S.O.; Snyder, J.W.; Busch, T.M.; Kousis, P.C.; Cheney, R.T.; Morgan, J. Choice of Oxygen-Conserving Treatment Regimen Determines the Inflammatory Response and Outcome of Photodynamic Therapy of Tumors. Cancer Res. 2004, 64, 2120–2126. [Google Scholar] [CrossRef] [Green Version]
- Shams, M.; Owczarczak, B.; Manderscheid-Kern, P.; Bellnier, D.A.; Gollnick, S.O. Development of photodynamic therapy regimens that control primary tumor growth and inhibit secondary disease. Cancer Immunol. Immunother. 2015, 64, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Lamberti, M.J.; Mentucci, F.M.; Roselli, E.; Araya, P.; Rivarola, V.A.; Rumie Vittar, N.B.; Maccioni, M. Photodynamic Modulation of Type 1 Interferon Pathway on Melanoma Cells Promotes Dendritic Cell Activation. Front. Immunol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Guan, Z.; Wang, X.; Wang, Z.; Zeng, R.; Xu, L. ALA-PDT promotes HPV-positive cervical cancer cells apoptosis and DCs maturation via miR-34a regulated HMGB1 exosomes secretion. Photodiagnosis Photodyn. Ther. 2018, 24, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Theodoraki, M.N.; Lorenz, K.; Lotfi, R.; Fürst, D.; Tsamadou, C.; Jaekle, S.; Mytilineos, J.; Brunner, C.; Theodorakis, J.; Hoffmann, T.K.; et al. Influence of photodynamic therapy on peripheral immune cell populations and cytokine concentrations in head and neck cancer. Photodiagnosis Photodyn. Ther. 2017, 19, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Naylor, M.F.; Chen, W.R.; Teague, T.K.; Perry, L.A.; Nordquist, R.E. In situ photoimmunotherapy: A tumour-directed treatment for melanoma. Br. J. Dermatol. 2006, 155, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Naylor, M.F.; Le, H.; Nordquist, R.E.; Teague, T.K.; Howard, C.A.; Murray, C.; Chen, W.R. Clinical effects of in situ photoimmunotherapy on late-stage melanoma patients: A preliminary study. Cancer Biol. Ther. 2010, 10, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, T.; Liu, J.; Yu, H.; Jiao, S.; Feng, B.; Zhou, F.; Fu, Y.; Yin, Q.; Zhang, P.; et al. Acid-Activatable Versatile Micelleplexes for PD-L1 Blockade-Enhanced Cancer Photodynamic Immunotherapy. Nano Lett. 2016, 16, 5503–5513. [Google Scholar] [CrossRef] [PubMed]
- Saji, H.; Song, W.; Furumoto, K.; Kato, H.; Engleman, E.G. Systemic antitumor effect of intratumoral injection of dendritic cells in combination with local photodynamic therapy. Clin. Cancer Res. 2006, 12, 2568–2574. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Duan, X.; Guo, N.; Chan, C.; Poon, C.; Weichselbaum, R.R.; Lin, W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, P.; Nomula, R.; Zhao, J. Activatable triplet photosensitizers: Magic bullets for targeted photodynamic therapy. J. Mater. Chem. C 2014, 2, 5982–5997. [Google Scholar] [CrossRef]
- Huang, Z. A review of progress in clinical photodynamic therapy. Technol. Cancer Res. Treat. 2005, 4, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Popovic, A.; Wiggins, T.; Davids, L.M. Differential susceptibility of primary cultured human skin cells to hypericin PDT in an in vitro model. J. Photochem. Photobiol. B Biol. 2015, 149, 249–256. [Google Scholar] [CrossRef]
- Idris, N.M.; Jayakumar, M.K.G.; Bansal, A.; Zhang, Y. Upconversion nanoparticles as versatile light nanotransducers for photoactivation applications. Chem. Soc. Rev. 2015, 44, 1449–1478. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Mancini, M.C.; Nie, S. Second window for in vivo imaging. Nat. Nantechnol. 2009, 4, 710–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Yu, H.; Feng, B.; Gao, W.; Yan, M.; Zhang, Z.; Li, Y.; Liu, S. Highly efficient ablation of metastatic breast cancer using ammonium-tungsten-bronze nanocube as a novel 1064nm-laser-driven photothermal agent. Biomaterials 2015, 52, 407–416. [Google Scholar] [CrossRef]
- Jerjes, W.; Upile, T.; Mosse, C.A.; Hamdoon, Z.; Morcos, M.; Morley, S.; Hopper, C. Prospective evaluation of 110 patients following ultrasound-guided photodynamic therapy for deep seated pathologies. Photodiagnosis Photodyn. Ther. 2011, 8, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Mallidi, S.; Anbil, S.; Bulin, A.L.; Obaid, G.; Ichikawa, M.; Hasan, T. Beyond the barriers of light penetration: Strategies, perspectives and possibilities for photodynamic therapy. Theranostics 2016, 6, 2458–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining immune checkpoint inhibitors: Established and emerging targets and strategies to improve outcomes in melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef] [Green Version]
- Bender, C.; Hassel, J.C.; Enk, A. Immunotherapy of Melanoma. Oncol. Res. Treat. 2016, 39, 369–376. [Google Scholar] [CrossRef]
- Zhang, H.; Ye, Z.L.; Yuan, Z.G.; Luo, Z.Q.; Jin, H.J.; Qian, Q.J. New strategies for the treatment of solid tumors with CAR-T cells. Int. J. Biol. Sci. 2016, 12, 718–729. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Shangguan, J.; Eresen, A.; Li, Y.; Wang, J.; Zhang, Z. Dendritic cells in pancreatic cancer immunotherapy: Vaccines and combination immunotherapies. Pathol. Res. Pract. 2019, 215, 152691. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Geskin, L.J.; Damiano, J.J.; Patrone, C.C.; Butterfield, L.H.; Kirkwood, J.M.; Falo, L.D. Three antigen-loading methods in dendritic cell vaccines for metastatic melanoma. Melanoma Res. 2018, 28, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Eagles, M.E.; Nassiri, F.; Badhiwala, J.H.; Suppiah, S.; Almenawer, S.A.; Zadeh, G.; Aldape, K.D. Dendritic cell vaccines for high-grade gliomas. Ther. Clin. Risk Manag. 2018, 14, 1299–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Bae, Y.-S. Dendritic cell-based therapeutic cancer vaccines: Past, present and future. Clin. Exp. Vaccine Res. 2014, 3, 113–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguille, S.; Smits, E.L.; Lion, E.; Van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 8, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current progress in car-t cell therapy for solid tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Wang, M.; Yin, B.; Wang, H.Y.; Wang, R.F. Current advances in T-cell-based cancer immunotherapy. Immunotherapy 2014, 6, 1265–1278. [Google Scholar] [CrossRef] [Green Version]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome Release and Low pH Belong to a Framework of Resistance of Human Melanoma Cells to Cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef] [Green Version]
- Stock, S.; Schmitt, M.; Sellner, L. Optimizing Manufacturing Protocols of Chimeric Antigen Receptor T Cells for Improved Anticancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, J.S.; Hawkins, R.E.; Hombach, A.A.; Abken, H.; Gilham, D.E. Building Better Chimeric Antigen Receptors for Adoptive T Cell Therapy. Curr. Gene Ther. 2010, 10, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, B.; Uslu, U. CAR-T cell therapy in melanoma: A future success story? Exp. Dermatol. 2018, 27, 1315–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, S.; Kerr, K.M.; Stahel, R. PD-1 blockade in advanced NSCLC: A focus on pembrolizumab. Cancer Treat. Rev. 2018, 62, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Gray, K.; Gray, G.S.; Worland, P.J.; Rolfe, M. Anticancer antibodies. Am. J. Clin. Pathol. 2003, 119, 472–485. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Tai, Y.T.; Anderson, K.C. Antibody-based therapies in multiple myeloma. Bone Marrow Res. 2011, 2011, 924058. [Google Scholar] [CrossRef] [Green Version]
- Smith, F.O.; Downey, S.G.; Klapper, J.A.; Yang, J.C.; Richard, M.; Royal, R.E.; Kammula, U.S.; Hughes, M.S.; Restifo, N.P.; Levy, C.L.; et al. Treatment of Metastatic Melanoma Using Interleukin-2 Alone or in Conjunction with Vaccines. Clin. Cancer Res. 2008, 14, 5610–5618. [Google Scholar] [CrossRef] [Green Version]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods—A review and update. Biotechnol. Genet. Eng. Rev. 2013, 29, 175–186. [Google Scholar] [CrossRef]
- Sanz, L.; Cuesta, Á.M.; Compte, M.; Álvarez-Vallina, L. Antibody engineering: Facing new challenges in cancer therapy. Acta Pharmacol. Sin. 2005, 26, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Morisada, M.; Clavijo, P.E.; Moore, E.; Sun, L.; Chamberlin, M.; Van Waes, C.; Hodge, J.W.; Mitchell, J.B.; Friedman, J.; Allen, C.T. PD-1 blockade reverses adaptive immune resistance induced by high-dose hypofractionated but not low-dose daily fractionated radiation. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Hudson, P.J. Recombinant antibody constructs in cancer therapy. Curr. Opin. Immunol. 1999, 11, 548–557. [Google Scholar] [CrossRef]
- Michaelis, M.; Rothweiler, F.; Nerreter, T.; Van Rikxoort, M.; Sharifi, M.; Wiese, M.; Ghafourian, T.; Cinatl, J. Differential effects of the oncogenic BRAF inhibitor PLX4032 (vemurafenib) and its progenitor PLX4720 on ABCB1 function. J. Pharm. Pharm. Sci. 2014, 17, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Boudewijns, S.; Koornstra, R.H.T.; Westdorp, H.; Schreibelt, G.; Eertwegh, A.J.M.V.D.; Foppen, M.H.G.; Haanen, J.B.; De Vries, I.J.M.; Figdor, C.G.; Bol, K.F.; et al. Ipilimumab administered to metastatic melanoma patients who progressed after dendritic cell vaccination. OncoImmunology 2016, 5, e1201625. [Google Scholar] [CrossRef]
- Huang, M.-N.; Nicholson, L.T.; Batich, K.A.; Swartz, A.M.; Kopin, D.; Wellford, S.; Prabhakar, V.K.; Woroniecka, K.; Nair, S.K.; E Fecci, P.; et al. Antigen-loaded monocyte administration induces potent therapeutic antitumor T cell responses. J. Clin. Investig. 2020, 130, 774–788. [Google Scholar] [CrossRef]
- Letendre, P.; Monga, V.V.; Milhem, M.M.; Zakharia, Y. Ipilimumab: From preclinical development to future clinical perspectives in melanoma. Future Oncol. 2017, 13, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. Eur. J. Surg. Oncol. (EJSO) 2017, 43, 604–611. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhang, M.; Li, J.; Young, K.H. PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Long, G.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Schachter, J.; Long, G.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Sanlorenzo, M.; Vujic, I.; Floris, A.; Novelli, M.; Gammaitoni, L.; Giraudo, L.; Macagno, M.; Leuci, V.; Rotolo, R.; Donini, C.; et al. BRAF and MEK Inhibitors Increase PD-1-Positive Melanoma Cells Leading to a Potential Lymphocyte-Independent Synergism with Anti–PD-1 Antibody. Clin. Cancer Res. 2018, 24, 3377–3385. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, H. Human monoclonal antibodies: The benefits of humanization. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2019; Volume 1904, pp. 1–10. [Google Scholar]
- Brennan, F.R.; Morton, L.D.; Spindeldreher, S.; Kiessling, A.; Allenspach, R.; Hey, A.; Muller, P.Y.; Frings, W.; Sims, J. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs 2010, 2, 233–255. [Google Scholar] [CrossRef] [Green Version]
- Weiner, L.M.; Dhodapkar, M.V.; Ferrone, S. Monoclonal Antibodies for Cancer Immunotherapy. Lancet 2009, 373, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Green, L.L. Transgenic Mouse Strains as Platforms for the Successful Discovery and Development of Human Therapeutic Monoclonal Antibodies. Curr. Drug Discov. Technol. 2014, 11, 74–84. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Goyal, A.; Kaur, P.; Singh, R.; Kalra, S. Anticancer Drug-induced Thyroid Dysfunction. Eur. Endocrinol. 2020, 16, 32–39. [Google Scholar] [CrossRef]
- Igg, M.; Shawler, D.L.; Bartholomew, R.M.; Linda, M.; Dillman, R. Human immune response to multiple injections of murine monoclonal IgG. J. Immunol. 1985, 135, 1530–1535. [Google Scholar]
- Schroff, R.W.; Foon, K.A.; Beatty, S.M.; Oldham, R.K.; Morgan, A.C. Human Anti-Murine Immunoglobulin Responses in Patients Receiving Monoclonal Antibody Therapy. Cancer Res. 1985, 45, 879–885. [Google Scholar] [PubMed]
- Maloney, D.G. Preclinical and phase I and II trials of rituximab. Semin. Oncol. 1999, 26, 74–78. [Google Scholar]
- Press, O.W. Radiolabeled Antibody Therapy of B-Cell Lymphomas. Semin. Oncol. 1999, 26, 58–65. [Google Scholar] [PubMed]
- Winkler, J.K.; Schiller, M.; Bender, C.; Enk, A.H.; Hassel, J.C. Rituximab as a therapeutic option for patients with advanced melanoma. Cancer Immunol. Immunother. 2018, 67, 917–924. [Google Scholar] [CrossRef]
- Velter, C.; Pagès, C.; Schneider, P.; Osio, A.; Brice, P.; Lebbé, C. Four cases of rituximab-associated melanoma. Melanoma Res. 2014, 24, 401–403. [Google Scholar] [CrossRef]
- Padlan, E.A. Anatomy of the antibody molecule. Mol. Immunol. 1994, 31, 169–217. [Google Scholar] [CrossRef] [Green Version]
- Welschof, M.; Krauss, J. Recombinant Antibodies for Cancer Therapy; Humana: Totowa, NJ, USA, 2003; ISBN 0896039188. [Google Scholar]
- Queen, C.; Schneider, W.P.; Selick, H.E.; Payne, P.W.; Landolfi, N.F.; Duncan, J.F.; Avdalovic, N.M.; Levitt, M.; Junghans, R.P.; Waldmann, T.A. A humanized antibody that binds to the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 1989, 86, 10029–10033. [Google Scholar] [CrossRef] [Green Version]
- Co, M.S.; Queen, C. Humanized antibodies for therapy. Nature 1991, 351, 501–502. [Google Scholar] [CrossRef]
- Padlan, E.A. A possible procedure for reducing the immunogenicity of antibody variable domains while preserving their ligand-binding properties. Mol. Immunol. 1991, 28, 489–498. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Henry, A.H.; Searle, S.J.; Guild, B.C.; Roguska, M.; Rees, A.R. Comparison of surface accessible residues in human and murine immunoglobulin Fv domains. Implication for humanization of murine antibodies. J. Mol. Biol. 1994, 235, 959–973. [Google Scholar] [CrossRef]
- Roguska, M.A.; Pedersen, J.T.; Henry, A.H.; Searle, S.M.; Roja, M.; Avery, B.; Hoffee, M.; Cook, S.; Lambert, J.M.; Blattler, W.A.; et al. A comparison of two murine monoclonal antibodies humanized by CDR-grafting and variable domain resurfacing. Protein Eng. 1996, 9, 895–904. [Google Scholar] [CrossRef] [Green Version]
- DM, F.; Sl, O.D.; Bengoechea, T.; Dv, H.; Harding, F.; Sl, B.; Rm, K.; Km, H.; Sr, S.; Lonberg, N. High-avidity human IgG kappa monoclonal antibodies from a novel strain of minilocus transgenic mice. Nat. Biotechnol. 1996, 14, 845–851. [Google Scholar]
- Mendez, M.J.; Green, L.L.; Corvalan, J.R.; Jia, X.-C.; Maynard-Currie, C.E.; Yang, X.-D.; Gallo, M.L.; Louie, D.M.; Lee, D.V.; Erickson, K.L.; et al. Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat. Genet. 1997, 15, 146–156. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. MAbs 2019, 11, 219–238. [Google Scholar] [CrossRef]
- Malas, S.; Harrasser, M.; Lacy, K.E.; Karagiannis, S.N. Antibody therapies for melanoma: New and emerging, opportunities to activate immunity (Review). Oncol. Rep. 2014, 32, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Albertini, M.R. The age of enlightenment in melanoma immunotherapy. J. Immunother. Cancer 2018, 6, 80. [Google Scholar] [CrossRef]
- Callahan, M.K.; Kluger, H.; Postow, M.A.; Segal, N.H.; Lesokhin, A.; Atkins, M.B.; Kirkwood, J.M.; Krishnan, S.; Bhore, R.; Horak, C.; et al. Nivolumab plus ipilimumab in patients with advanced melanoma: Updated survival, response, and safety data in a phase i dose-escalation study. J. Clin. Oncol. 2018, 36, 391–398. [Google Scholar] [CrossRef]
- Beatty, G.L.; Moon, E.K. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology 2014, 3, e970027. [Google Scholar] [CrossRef] [Green Version]
- Amoury, M.; Mladenov, R.; Nachreiner, T.; Pham, A.T.; Hristodorov, D.; Di Fiore, S.; Helfrich, W.; Pardo, A.; Fey, G.; Schwenkert, M.; et al. A novel approach for targeted elimination of CSPG4-positive triple-negative breast cancer cells using a MAP tau-based fusion protein. Int. J. Cancer 2016, 139, 916–927. [Google Scholar] [CrossRef] [Green Version]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef] [Green Version]
- Shefet-Carasso, L.; Benhar, I. Antibody-targeted drugs and drug resistance—Challenges and solutions. Drug Resist. Updat. 2015, 18, 36–46. [Google Scholar] [CrossRef]
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase enzyme-mediated generation of site-specifically conjugated antibody drug conjugates with high In Vitro and In Vivo potency. PLoS ONE 2015, 10, e0131177. [Google Scholar] [CrossRef] [Green Version]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef]
- Siemers, N.O.; Kerr, D.E.; Yarnold, S.; Stebbins, M.R.; Vrudhula, V.M.; Hellström, I.; Hellström, K.E.; Senter, P.D. Construction, expression, and activities of L49-sFv-lactamase, a single-chain antibody fusion protein for anticancer prodrug activation. Bioconjug. Chem. 1997, 8, 510–519. [Google Scholar] [CrossRef]
- Adem, Y.T.; Schwarz, K.A.; Duenas, E.; Patapoff, T.W.; Galush, W.J.; Esue, O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjug. Chem. 2014, 25, 656–664. [Google Scholar] [CrossRef]
- Beckley, N.S.; Lazzareschi, K.P.; Chih, H.W.; Sharma, V.K.; Flores, H.L. Investigation into temperature-induced aggregation of an antibody drug conjugate. Bioconjug. Chem. 2013, 24, 1674–1683. [Google Scholar] [CrossRef]
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E.M. Immunogenicity of Therapeutic Protein Aggregates. J. Pharm. Sci. 2016, 105, 417–430. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, C.F.; Turcott, E.; Westendorf, L.; Webster, J.B.; Alley, S.C.; Kim, K.; Andreyka, J.; Stone, I.; Hamblett, K.J.; Francisco, J.A.; et al. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng. Des. Sel. 2006, 19, 299–307. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef]
- Chudasama, V.; Maruani, A.; Caddick, S. Corrigendum: Recent advances in the construction of antibody – drug conjugates. Nat. Chem. 2016, 8, 281. [Google Scholar] [CrossRef]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Blackstock, D.; Chen, W. Halo-tag mediated self-labeling of fluorescent proteins to molecular beacons for nucleic acid detection. Chem. Commun. 2014, 50, 13735–13738. [Google Scholar] [CrossRef]
- Hoehnel, S.; Lutolf, M.P. Capturing Cell-Cell Interactions via SNAP-tag and CLIP-tag Technology. Bioconjug. Chem. 2015, 26, 1678–1686. [Google Scholar] [CrossRef]
- Kolberg, K.; Puettmann, C.; Pardo, A.; Fitting, J.; Barth, S. SNAP-Tag Technology: A General Introduction. Curr. Pharm. Des. 2013, 5406–5413. [Google Scholar] [CrossRef]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa, I.R.; Kindermann, M.; Beaufils, F.; Johnsson, K. An Engineered Protein Tag for Multiprotein Labeling in Living Cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Amoury, M.; Blume, T.; Brehm, H.; Niesen, J.; Tenhaef, N.; Barth, S.; Gattenlohner, S.; Helfrich, W.; Fitting, J.; Nachreiner, T.; et al. SNAP-tag based Agents for Preclinical In Vitro Imaging in Malignant Diseases. Curr. Pharm. Des. 2013, 19, 5429–5436. [Google Scholar] [CrossRef]
- Stagge, F.; Mitronova, G.Y.; Belov, V.N.; Wurm, C.A.; Jakobs, S. Snap-, CLIP- and Halo-Tag Labelling of Budding Yeast Cells. PLoS ONE 2013, 8, e78745. [Google Scholar] [CrossRef] [Green Version]
- Los, G.V.; Encell, L.P.; Mcdougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. Halo Tag: Protein labeling Technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef]
- Algar, W.R.; Prasuhn, D.E.; Stewart, M.H.; Jennings, T.L.; Blanco-Canosa, J.B.; Dawson, P.E.; Medintz, I.L. The controlled display of biomolecules on nanoparticles: A challenge suited to bioorthogonal chemistry. Bioconjug. Chem. 2011, 22, 825–858. [Google Scholar] [CrossRef]
- Gronemeyer, T.; Chidley, C.; Juillerat, A.; Heinis, C.; Johnsson, K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for applications in protein labeling. Protein Eng. Des. Sel. 2006, 19, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanugula, S.; Pegg, A.E. Alkylation damage repair protein O6-alkylguanine-DNA alkyltransferase from the hyperthermophiles Aquifex aeolicus and Archaeoglobus fulgidus. Biochem. J. 2003, 375, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Anguille, S.; Smits, E.L.; Bryant, C.; Van Acker, H.H.; Goossens, H.; Lion, E.; Fromm, P.D.; Hart, D.N.; Van Tendeloo, V.F.; Berneman, Z.N. Dendritic cells as pharmacological tools for cancer immunotherapys. Pharmacol. Rev. 2015, 67, 731–753. [Google Scholar] [CrossRef]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef]
- Gong, H.; Kovar, J.L.; Baker, B.; Zhang, A.; Cheung, L.; Draney, D.R.; Corrêa, I.R.; Xu, M.Q.; Olive, D.M. Near-infrared fluorescence imaging of mammalian cells and xenograft tumors with SNAP-tag. PLoS ONE 2012, 7, e34003. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A.F.; Kampmeier, F.; Von Felbert, V.; Merk, H.F.; Tur, M.K.; Barth, S. SNAP-tag technology mediates site specific conjugation of antibody fragments with a photosensitizer and improves target specific phototoxicity in tumor cells. Bioconjug. Chem. 2011, 22, 2487–2495. [Google Scholar] [CrossRef]
- Kampmeier, F.; Niesen, J.; Koers, A.; Ribbert, M.; Brecht, A.; Fischer, R.; Kiessling, F.; Barth, S.; Thepen, T. Rapid optical imaging of EGF receptor expression with a single-chain antibody SNAP-tag fusion protein. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1926–1934. [Google Scholar] [CrossRef]
- Woitok, M.; Klose, D.; Di Fiore, S.; Richter, W.; Stein, C.; Gresch, G.; Grieger, E.; Barth, S.; Fischer, R.; Kolberg, K.; et al. Comparison of a mouse and a novel human scFv-SNAP-auristatin F drug conjugate with potent activity against EGFR-overexpressing human solid tumor cells. Onco Targets Ther. 2017, 10, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Woitok, M.; Klose, D.; Niesen, J.; Richter, W.; Abbas, M.; Stein, C.; Fendel, R.; Bialon, M.; Püttmann, C.; Fischer, R.; et al. The efficient elimination of solid tumor cells by EGFR-specific and HER2-specific scFv-SNAP fusion proteins conjugated to benzylguanine-modified auristatin F. Cancer Lett. 2016, 381, 323–330. [Google Scholar] [CrossRef]
- Bouchard, H.; Viskov, C.; Garcia-Echeverria, C. Antibody-drug conjugates—A new wave of cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.G.; Kim, K.M. Strategies and advancement in antibody-drug conjugate optimization for targeted cancer therapeutics. Biomol. Ther. 2015, 23, 493–509. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yu, C.; Jiang, J.; Huang, C.; Yao, X.; Xu, Q.; Yu, F.; Lou, L.; Fang, J. An anti-HER2 antibody conjugated with monomethyl auristatin E is highly effective in HER2-positive human gastric cancer. Cancer Biol. Ther. 2016, 17, 346–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, A.; Kopitz, C.; Schatz, C.A.; Nising, C.F.; Mahlert, C.; Lerchen, H.G.; Stelte-Ludwig, B.; Hammer, S.; Greven, S.; Schuhmacher, J.; et al. Preclinical efficacy of the auristatin-based antibody-drug conjugate BAY 1187982 for the treatment of FGFR2-positive solid tumors. Cancer Res. 2016, 76, 6331–6339. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.; Pettit, G.R.; Hamel, E. Structure-activity studies with chiral isomers and with segments of the antimitotic marine peptide dolastatin 10. Biochem. Pharmacol. 1990, 40, 1859–1864. [Google Scholar] [CrossRef]
- Polson, A.G.; Calemine-fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; De Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-Drug Conjugates for the Treatment of Non-Hodgkin’s Lymphoma: Target and Linker-Drug Selection. Cancer Res. 2009, 65, 2358–2364. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M. Brentuximab vedotin. Blood 2014, 124, 3197–3201. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Yao, X.; Jiang, J.; Wang, X.; Huang, C.; Li, D.; Xie, K.; Xu, Q.; Li, H.; Li, Z.; Lou, L.; et al. A novel humanized anti-HER2 antibody conjugated with MMAE exerts potent anti-tumor activity. Breast Cancer Res. Treat. 2015, 153, 123–133. [Google Scholar] [CrossRef]
- Kerr, D.E.; Vrudhula, V.M.; Svensson, H.P.; Siemers, N.O.; Senter, P.D. Comparison of recombinant and synthetically formed monoclonal antibody-??-lactamase conjugates for anticancer prodrug activation. Bioconjug. Chem. 1999, 10, 1084–1089. [Google Scholar] [CrossRef]
- Nagaya, T.; Friedman, J.; Maruoka, Y.; Ogata, F.; Okuyama, S.; Clavijo, P.E.; Choyke, P.L.; Allen, C.; Kobayashi, H. Host immunity following near-infrared photoimmunotherapy is enhanced with PD-1 checkpoint blockade to eradicate established antigenic tumors. Cancer Immunol. Res. 2019, 7, 401–413. [Google Scholar] [CrossRef]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.; Choyke, P.; Kobayashi, H. Cancer Cell-Selective In Vivo Near Infrared Photoimmunotherapy Targeting Specific Membrane Molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic Therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, W.; Celli, J.P.; Rizvi, I.; Mai, Z.; Spring, B.Q.; Yun, S.H.; Hasan, T. In vivo high-resolution fluorescence microendoscopy for ovarian cancer detection and treatment monitoring. Br. J. Cancer 2009, 101, 2015–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaya, T.; Sato, K.; Harada, T.; Nakamura, Y.; Choyke, P.L.; Kobayashi, H. Near infrared photoimmunotherapy targeting EGFR positive triple negative breast cancer: Optimizing the conjugate-light regimen. PLoS ONE 2015, 10, e0136829. [Google Scholar] [CrossRef]
- Ito, K.; Mitsunaga, M.; Nishimura, T.; Kobayashi, H.; Tajiri, H. Combination photoimmunotherapy with monoclonal antibodies recognizing different epitopes of human epidermal growth factor receptor 2: An assessment of phototherapeutic effect based on fluorescence molecular imaging. Oncotarget 2016, 7, 14143–14152. [Google Scholar] [CrossRef] [Green Version]
- Railkar, R.; Krane, L.S.; Li, Q.Q.; Sanford, T.; Siddiqui, M.R.; Haines, D.; Vourganti, S.; Brancato, S.J.; Choyke, P.L.; Kobayashi, H.; et al. Epidermal Growth Factor Receptor (EGFR) Targeted Photoimmunotherapy (PIT) for the Treatment of EGFR expressing Bladder Cancer. Mol. Cancer Ther. 2017, 2201–2214. [Google Scholar] [CrossRef] [Green Version]
- Burley, T.A.; Mączyńska, J.; Shah, A.; Szopa, W.; Harrington, K.J.; Boult, J.K.R.; Mrozek-Wilczkiewicz, A.; Vinci, M.; Bamber, J.C.; Kaspera, W.; et al. Near-infrared photoimmunotherapy targeting EGFR—Shedding new light on glioblastoma treatment. Int. J. Cancer 2018, 142, 2363–2374. [Google Scholar] [CrossRef] [Green Version]
- Nagaya, T.; Nakamura, Y.; Sato, K.; Zhang, Y.; Ni, M.; Choyke, P.L.; Ho, M.; Kobayashi, H. Near infrared photoimmunotherapy with an anti-mesothelin antibody. Oncotarget 2016, 7, 23361–23369. [Google Scholar] [CrossRef] [Green Version]
- Penet, M.; Krishnamachary, B.; Chen, Z.; Jin, J.; Bhujwalla, Z.M. Molecular Imaging of the Tumor Microenvironment for Precision Medicine and Theranostics. Adv. Cancer Res. 2014, 124, 235–256. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biteghe, F.A.N.; Chalomie, N.E.T.; Mungra, N.; Vignaux, G.; Gao, N.; Vergeade, A.; Okem, A.; Naran, K.; Ndong, J.D.L.C.; Barth, S. Antibody-Based Immunotherapy: Alternative Approaches for the Treatment of Metastatic Melanoma. Biomedicines 2020, 8, 327. https://doi.org/10.3390/biomedicines8090327
Biteghe FAN, Chalomie NET, Mungra N, Vignaux G, Gao N, Vergeade A, Okem A, Naran K, Ndong JDLC, Barth S. Antibody-Based Immunotherapy: Alternative Approaches for the Treatment of Metastatic Melanoma. Biomedicines. 2020; 8(9):327. https://doi.org/10.3390/biomedicines8090327
Chicago/Turabian StyleBiteghe, Fleury Augustin Nsole, Nyangone Ekome Toung Chalomie, Neelakshi Mungra, Guillaume Vignaux, Nan Gao, Aurelia Vergeade, Ambrose Okem, Krupa Naran, Jean De La Croix Ndong, and Stefan Barth. 2020. "Antibody-Based Immunotherapy: Alternative Approaches for the Treatment of Metastatic Melanoma" Biomedicines 8, no. 9: 327. https://doi.org/10.3390/biomedicines8090327