Long Term Pharmacological Perturbation of Autophagy in Mice: Are HCQ Injections a Relevant Choice?

Abstract
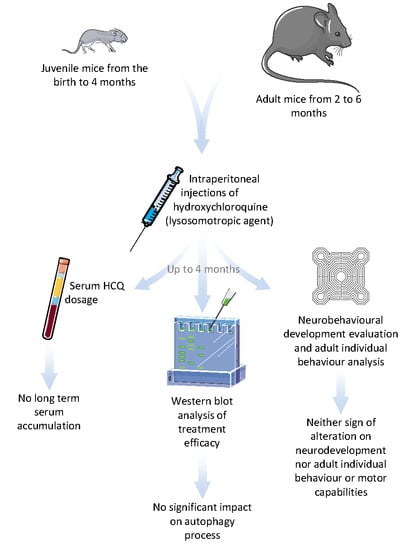
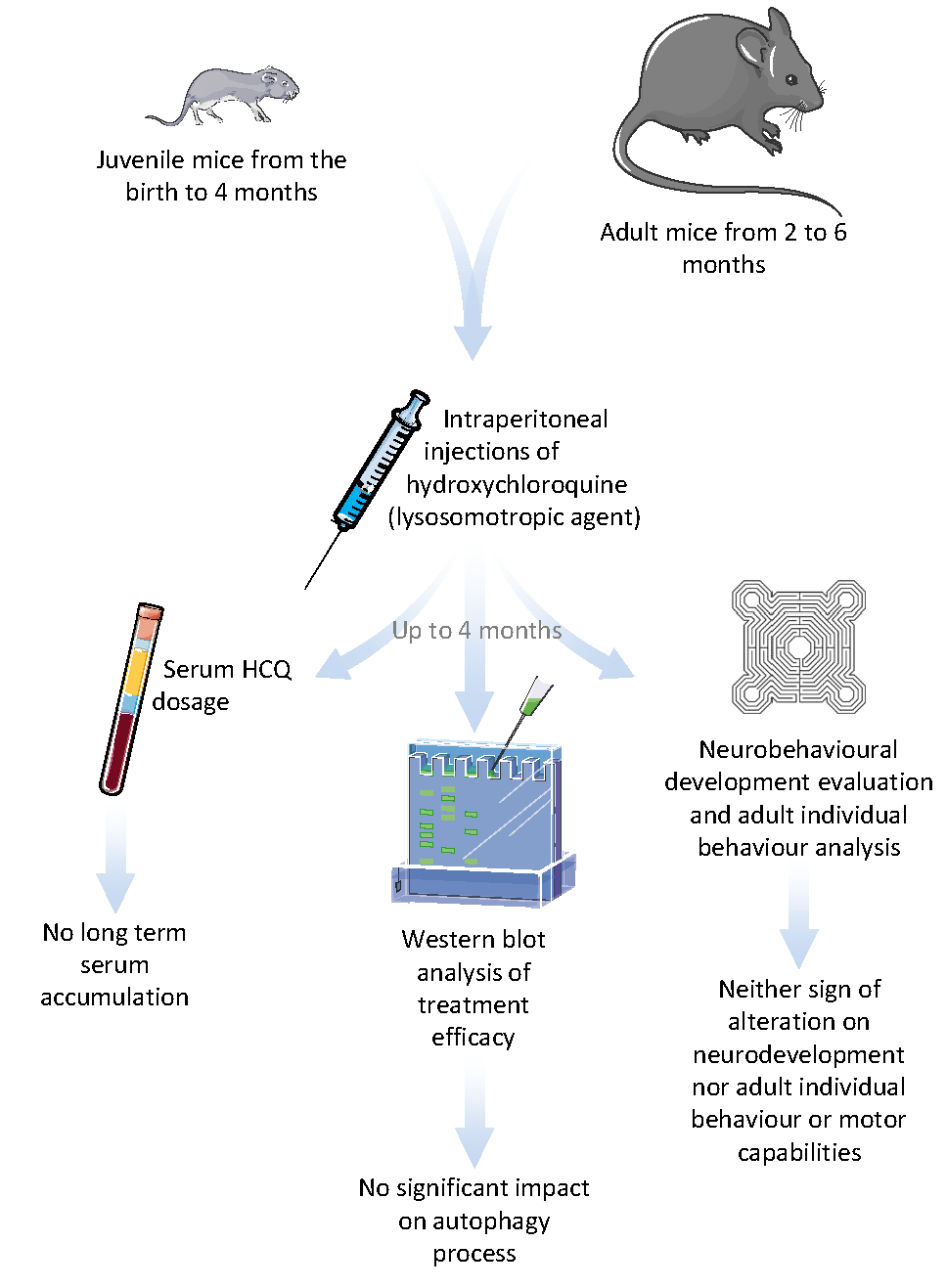
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.1.1. Juvenile Experiment
2.1.2. Adult Experiment
2.2. Doses and Protocol of Exposure
2.2.1. Juvenile Experiment
2.2.2. Adult Experiment
2.3. Removed Sample Analyses
2.3.1. Tissue Preparation
2.3.2. Analytical Dosage of HCQ
2.3.3. Western Blot of Autophagy Proteins
2.4. Behavioural Testing
2.4.1. Neurobehavioural Development in Juvenile Experiment
2.4.2. Maternal Behaviour in the Juvenile Experiment
2.4.3. Mature Behavioural Testing
2.5. Statistical Analysis
- Data from behavioural developmental tests were analysed using a non-parametric Kruskal–Wallis test followed by a Mann–Whitney procedure modified for multiple comparisons when appropriate. Pearson Chi-square procedure was used to analyse the number of successful animals. All males and females were tested together up to PND25 then gender was used as a variable for analysis.
- Pup weight was analysed using a one-way analysis of variance (one-way ANOVA). Post hoc comparisons have been performed using the Dunnett’s test when ANOVA was significant.
- Data from adult behavioural testing in both experiments were analysed using a one-way analysis of variance (one-way ANOVA). Post hoc comparisons have been performed using the Dunnett’s test when ANOVA was significant.
- Western-blot data and analytical HCQ dosages were analysed using a non-parametric Kruskal–Wallis test followed by a Mann–Whitney procedure modified for multiple comparisons when appropriate, due to the low number of animals in each group. For statistical comparison reasons, each gender was analysed separately because they were generated with two successive Western blots and multiple comparisons were exclusively performed for treated groups compared to controls.
3. Results
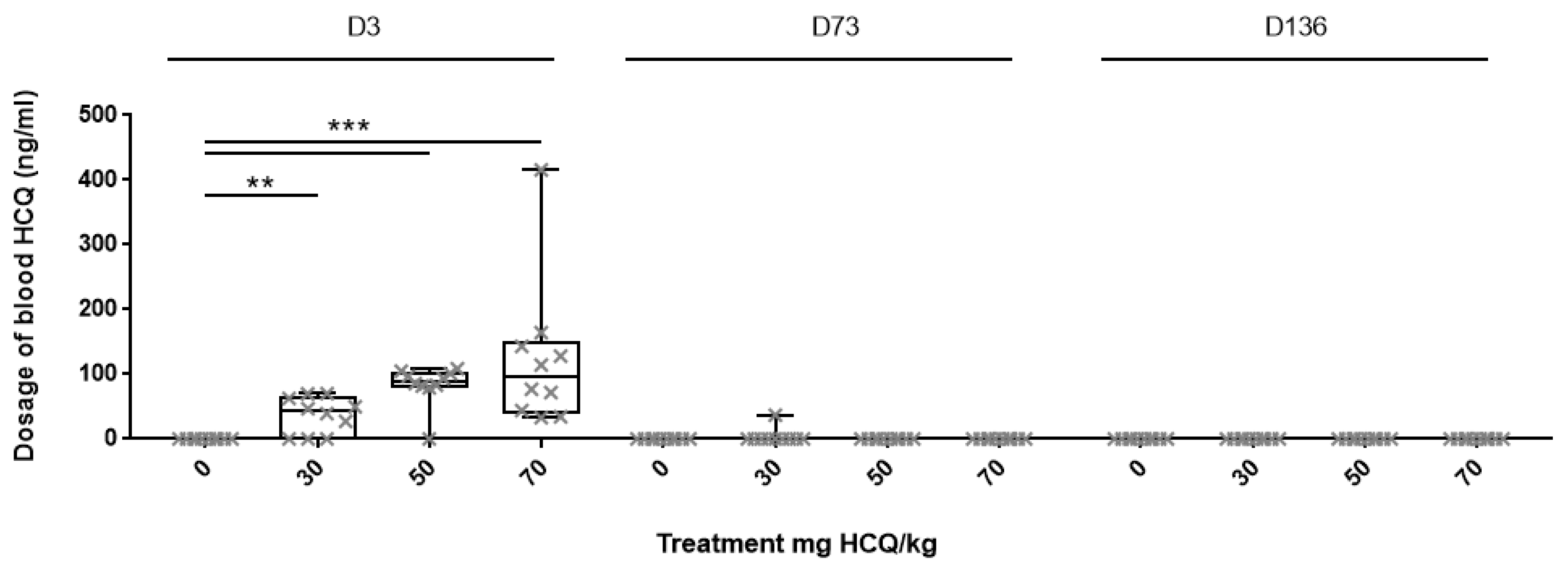
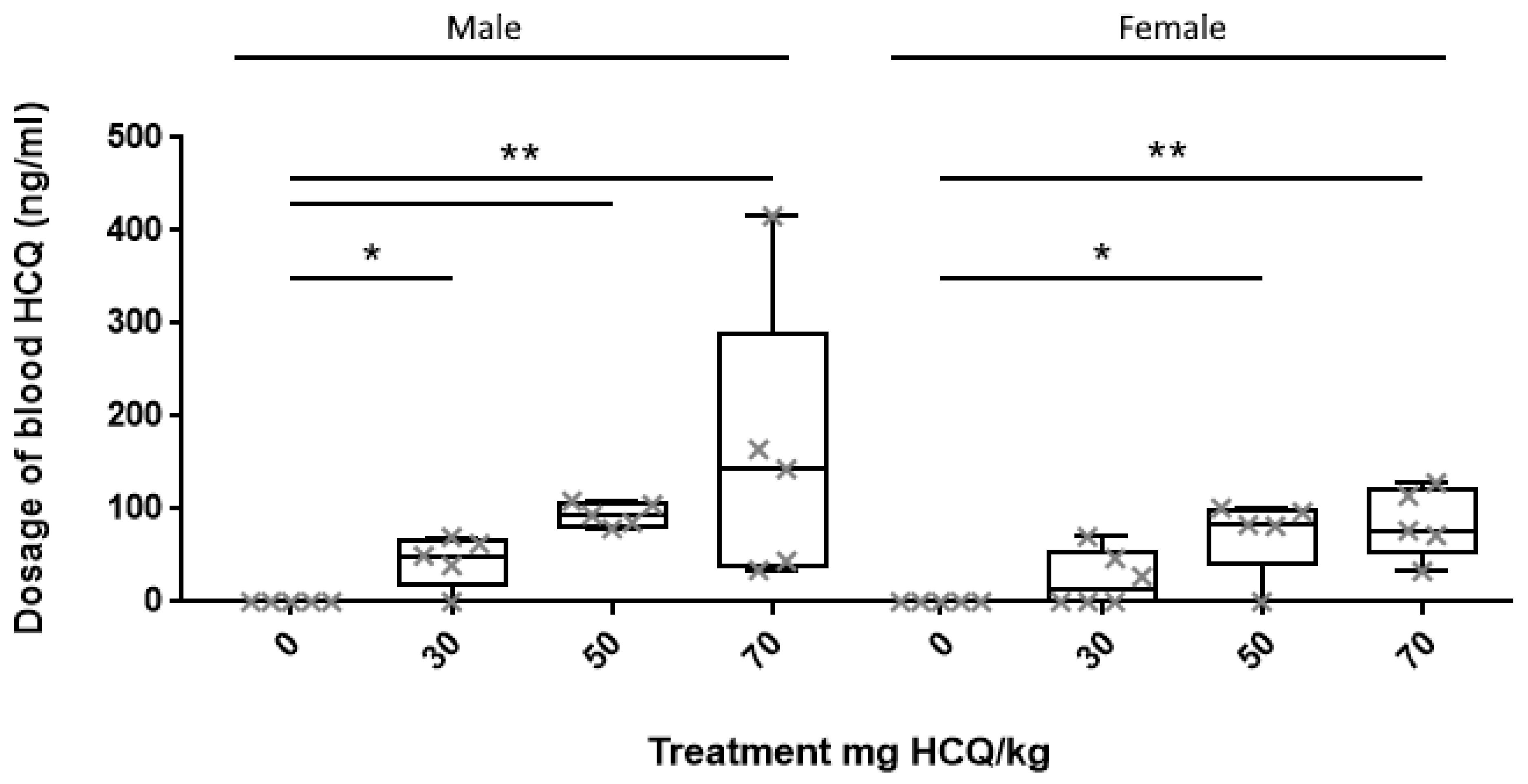
3.1. HCQ Assay in Plasma
3.2. Western Blot of Autophagy Proteins
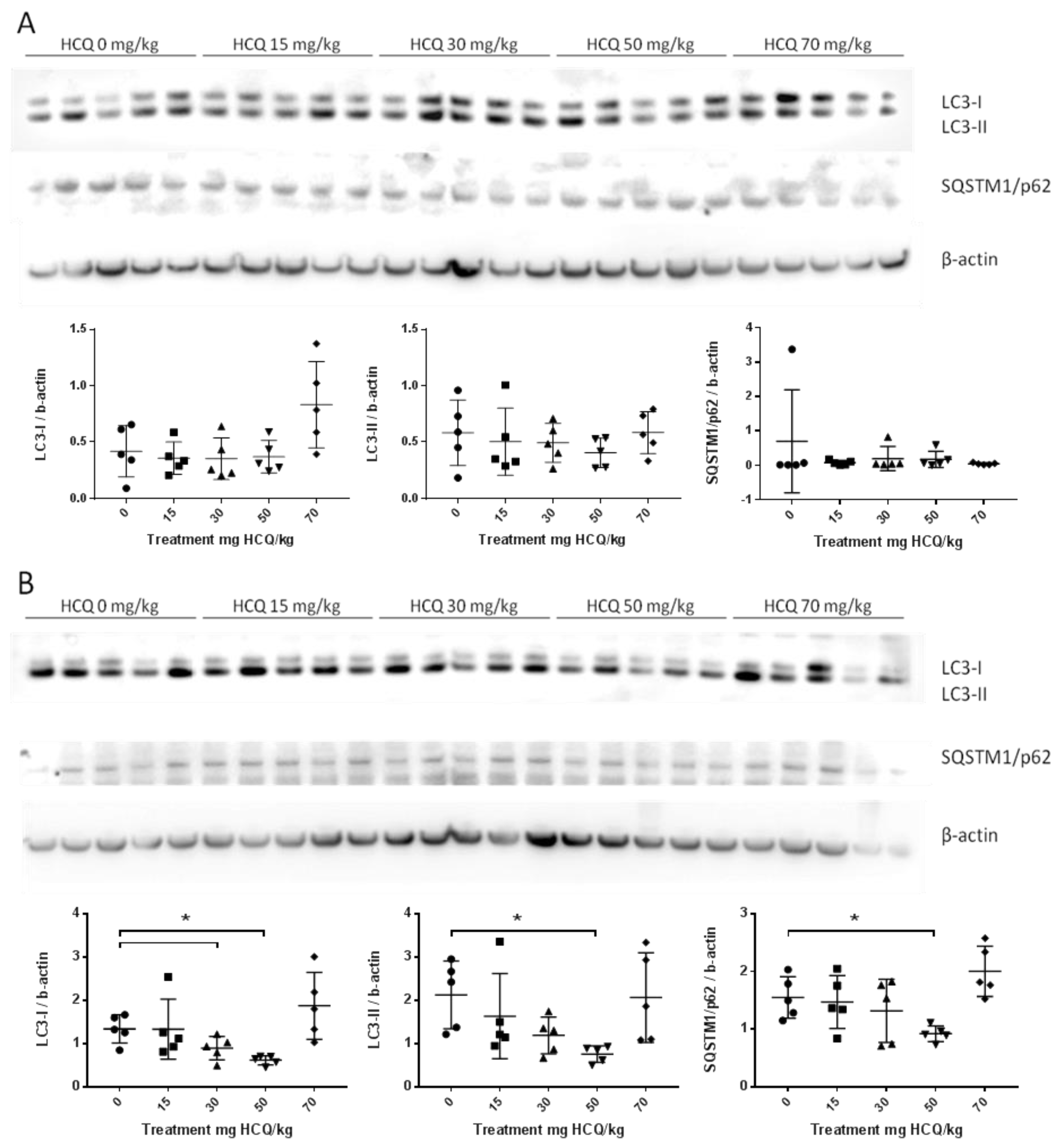
3.2.1. Juvenile Experiments
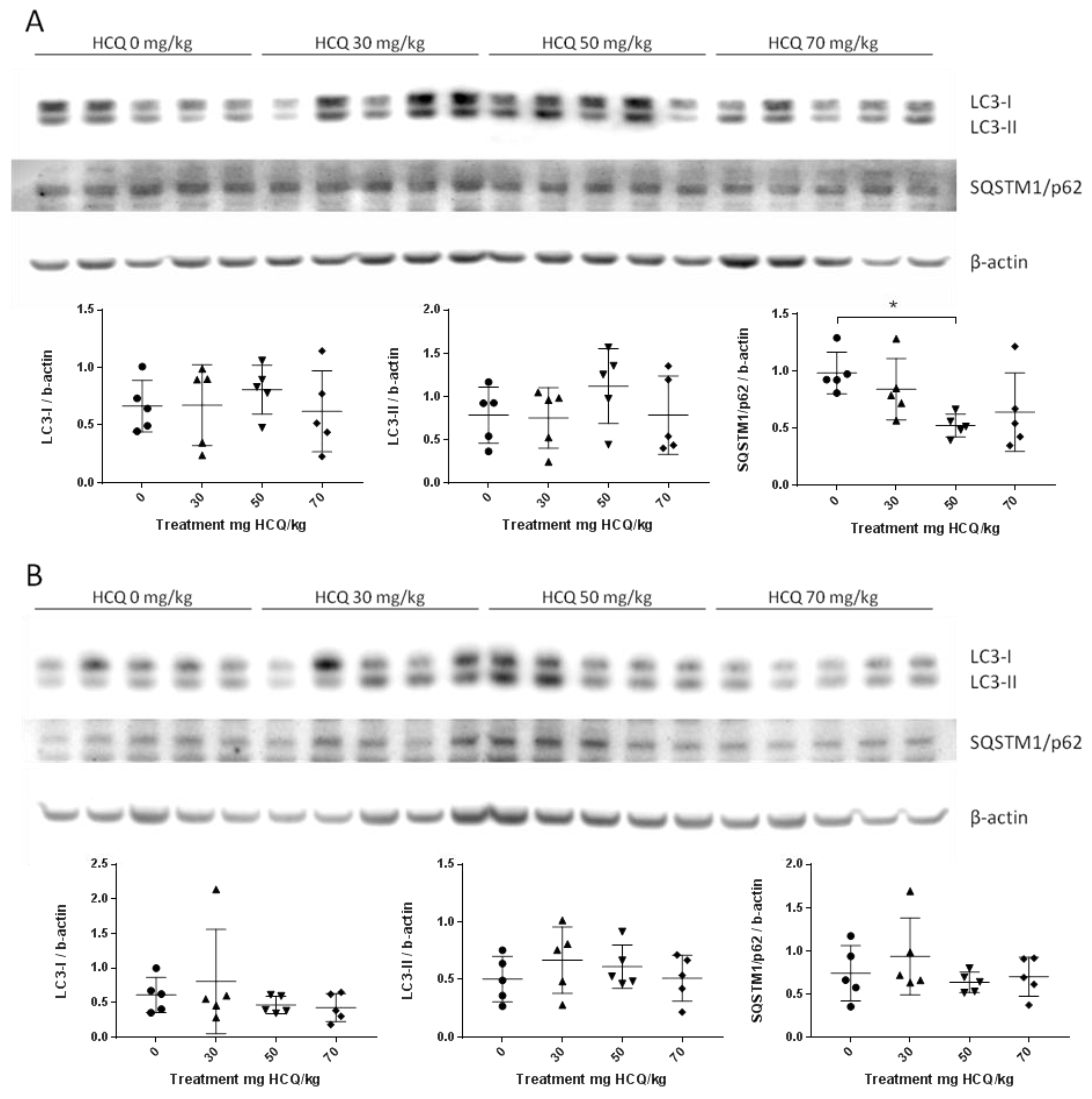
3.2.2. Adult Experiments
3.3. Behavioural Testing
3.3.1. Juvenile Experiments
3.3.2. Adult Experiments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Al | Aluminium |
| ATG | Autophagy related |
| CQ | Chloroquine |
| HCQ | Hydroxychloroquine |
| ip | Intra-peritoneal |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| MMF | Macrophagic myofasciitis |
| PND | Post-natal day |
| PVDF | Polyvinylidene difluoride |
| ROS | Reactive oxygen species |
| RT | Room temperature |
| TA | Tibialis anterior |
References
- Stern, S.T.; Adiseshaiah, P.P.; Crist, R.M. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part. Fibre Toxicol. 2012, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Li, W.; Li, J.; Bao, J. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Berg, T.O.; Fengsrud, M.; Strømhaug, P.E.; Berg, T.; Seglen, P.O. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J. Biol. Chem. 1998, 273, 21883–21892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brest, P.; Corcelle, E.A.; Cesaro, A.; Chargui, A.; Belaïd, A.; Klionsky, D.J.; Vouret-Craviari, V.; Hebuterne, X.; Hofman, P.; Mograbi, B. Autophagy and Crohn’s disease: At the crossroads of infection, inflammation, immunity, and cancer. Curr. Mol. Med. 2010, 10, 486–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesonen, M.; Vähäkangas, K. Autophagy in exposure to environmental chemicals. Toxicol. Lett. 2019, 305, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Eidi, H.; Joubert, O.; Némos, C.; Grandemange, S.; Mograbi, B.; Foliguet, B.; Tournebize, J.; Maincent, P.; Le Faou, A.; Aboukhamis, I.; et al. Drug delivery by polymeric nanoparticles induces autophagy in macrophages. Int. J. Pharm. 2012, 422, 495–503. [Google Scholar] [CrossRef]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, B.; Toborek, M. Autophagy is involved in nanoalumina-induced cerebrovascular toxicity. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 212–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohignac, V.; Landry, M.; Boczkowski, J.; Lanone, S. Autophagy as a Possible Underlying Mechanism of Nanomaterial Toxicity. Nanomaterials 2014, 4, 548–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Lima, H.; Jacobson, L.S.; Goldberg, M.F.; Chandran, K.; Diaz-Griffero, F.; Lisanti, M.P.; Brojatsch, J. Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle Georget. Tex 2013, 12, 1868–1878. [Google Scholar] [CrossRef] [Green Version]
- Sabella, S.; Carney, R.P.; Brunetti, V.; Malvindi, M.A.; Al-Juffali, N.; Vecchio, G.; Janes, S.M.; Bakr, O.M.; Cingolani, R.; Stellacci, F.; et al. A general mechanism for intracellular toxicity of metal-containing nanoparticles. Nanoscale 2014, 6, 7052–7061. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Sarkar, S.; Bhattacharya, S. Toxic metals and autophagy. Chem. Res. Toxicol. 2014, 27, 1887–1900. [Google Scholar] [CrossRef]
- Zhang, Z.; Miah, M.; Culbreth, M.; Aschner, M. Autophagy in Neurodegenerative Diseases and Metal Neurotoxicity. Neurochem. Res. 2016, 41, 409–422. [Google Scholar] [CrossRef]
- Corcelle, E.; Djerbi, N.; Mari, M.; Nebout, M.; Fiorini, C.; Fénichel, P.; Hofman, P.; Poujeol, P.; Mograbi, B. Control of the autophagy maturation step by the MAPK ERK and p38: Lessons from environmental carcinogens. Autophagy 2007, 3, 57–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chargui, A.; Zekri, S.; Jacquillet, G.; Rubera, I.; Ilie, M.; Belaid, A.; Duranton, C.; Tauc, M.; Hofman, P.; Poujeol, P.; et al. Cadmium-induced autophagy in rat kidney: An early biomarker of subtoxic exposure. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 121, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Belaid, A.; Roméo, B.; Filippakis, H.; Meyer, M.; Grosjean, I.; Yazbeck, N.; Domdom, M.A.; Crépeaux, G.; Gherardi, R.K.; Lagadic-Gossmann, D.; et al. Autophagy-Driven Cancer Drug Development. In Autophagy and Cardiometabolic Diseases; Ren, J., Sowers, J.R., Zhang, Y., Eds.; Academic Press: Cambridge, UK, 2018; pp. 255–275. ISBN 978-0-12-805253-2. [Google Scholar]
- Pellacani, C.; Costa, L.G. Role of autophagy in environmental neurotoxicity. Environ. Pollut. 2018, 235, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, B.; Schall, N.; Wilhelm, M.; Muller, S. Assessing Autophagy in Mouse Models and Patients with Systemic Autoimmune Diseases. Cells 2017, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.-F.; Hébuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; DeVoss, J.; Diehl, L.; Graham, R.R.; et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.-C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef] [Green Version]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. AJP Gastrointest. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef] [Green Version]
- Gherardi, R.K.; Crépeaux, G.; Authier, F.-J. Myalgia and chronic fatigue syndrome following immunization: Macrophagic myofasciitis and animal studies support linkage to aluminum adjuvant persistency and diffusion in the immune system. Autoimmun. Rev. 2019, 18, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Flach, T.L.; Ng, G.; Hari, A.; Desrosiers, M.D.; Zhang, P.; Ward, S.M.; Seamone, M.E.; Vilaysane, A.; Mucsi, A.D.; Fong, Y.; et al. Alum interaction with dendritic cell membrane lipids is essential for its adjuvanticity. Nat. Med. 2011, 17, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Combadiere, C.; Authier, F.-J.; Itier, V.; Lux, F.; Exley, C.; Mahrouf-Yorgov, M.; Decrouy, X.; Moretto, P.; Tillement, O.; et al. Slow CCL2-dependent translocation of biopersistent particles from muscle to brain. BMC Med. 2013, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-L.; Shao, B.-Z.; Zhao, S.-B.; Chang, X.; Wang, P.; Miao, C.-Y.; Li, Z.-S.; Bai, Y. Intestinal autophagy links psychosocial stress with gut microbiota to promote inflammatory bowel disease. Cell Death Dis. 2019, 10, 391. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-monitoring and autophagy-deficient mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Cho, M.-H.; Shim, W.H.; Kim, J.K.; Jeon, E.-Y.; Kim, D.-H.; Yoon, S.-Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Fox, R.I. Mechanism of action of hydroxychloroquine as an antirheumatic drug. Semin. Arthritis Rheum. 1993, 23, 82–91. [Google Scholar] [CrossRef]
- McChesney, E.W. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am. J. Med. 1983, 75, 11–18. [Google Scholar] [CrossRef]
- Wolf, R.; Wolf, D.; Ruocco, V. Antimalarials: Unapproved uses or indications. Clin. Dermatol. 2000, 18, 17–35. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, A.M.; Krise, J.P. Lysosomal sequestration of amine-containing drugs: Analysis and therapeutic implications. J. Pharm. Sci. 2007, 96, 729–746. [Google Scholar] [CrossRef]
- Ochsendorf, F.R. Use of antimalarials in dermatology. J. Dtsch. Dermatol. Ges. J. Ger. Soc. Dermatol. JDDG 2010, 8, 829–844, quiz 845. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; Merlini, L.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Haspel, J.; Shaik, R.S.; Ifedigbo, E.; Nakahira, K.; Dolinay, T.; Englert, J.A.; Choi, A.M.K. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy 2011, 7, 629–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, I.; Lee, Y.; Cosio-Lima, L.M.; Cho, J.-Y.; Yeom, D.-C. Effects of long-term resistance exercise training on autophagy in rat skeletal muscle of chloroquine-induced sporadic inclusion body myositis. J. Exerc. Nutr. Biochem. 2015, 19, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodicka, P.; Lim, J.; Williams, D.T.; Kegel, K.B.; Chase, K.; Park, H.; Marchionini, D.; Wilkinson, S.; Mead, T.; Birch, H.; et al. Assessment of chloroquine treatment for modulating autophagy flux in brain of WT and HD mice. J. Huntingt. Dis. 2014, 3, 159–174. [Google Scholar] [CrossRef]
- An, N.; Chen, Y.; Wang, C.; Yang, C.; Wu, Z.-H.; Xue, J.; Ye, L.; Wang, S.; Liu, H.-F.; Pan, Q. Chloroquine Autophagic Inhibition Rebalances Th17/Treg-Mediated Immunity and Ameliorates Systemic Lupus Erythematosus. Cell. Physiol. Biochem. 2017, 44, 412–422. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Iwai-Kanai, E.; Yuan, H.; Huang, C.; Sayen, M.R.; Perry-Garza, C.N.; Kim, L.; Gottlieb, R.A. A method to measure cardiac autophagic flux in vivo. Autophagy 2008, 4, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Collins, K.P.; Jackson, K.M.; Gustafson, D.L. Hydroxychloroquine: A Physiologically-Based Pharmacokinetic Model in the Context of Cancer-Related Autophagy Modulation. J. Pharmacol. Exp. Ther. 2018, 365, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.A.; Huang, C.; Ramil, J.M.; Ravindran, N.; Andres, A.M.; Sin, J.; Linton, P.-J.; Gottlieb, R.A. Measuring cardiac autophagic flux in vitro and in vivo. Methods Mol. Biol. Clifton NJ 2015, 1219, 187–197. [Google Scholar]
- Campos, J.C.; Queliconi, B.B.; Bozi, L.H.M.; Bechara, L.R.G.; Dourado, P.M.M.; Andres, A.M.; Jannig, P.R.; Gomes, K.M.S.; Zambelli, V.O.; Rocha-Resende, C.; et al. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy 2017, 13, 1304–1317. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.D.; Pfeil, J.; Mueller, A.-K. Continuous oral chloroquine as a novel route for Plasmodium prophylaxis and cure in experimental murine models. BMC Res. Notes 2011, 4, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulis, M.; Vindis, C. Methods for Measuring Autophagy in Mice. Cells 2017, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Chen, K.; Lu, X.; Gao, H.; Qin, Z.; Lin, F. Exercise ameliorates the detrimental effect of chloroquine on skeletal muscles in mice via restoring autophagy flux. Acta Pharmacol. Sin. 2014, 35, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalbandian, A.; Llewellyn, K.J.; Nguyen, C.; Yazdi, P.G.; Kimonis, V.E. Rapamycin and Chloroquine: The In Vitro and In Vivo Effects of Autophagy-Modifying Drugs Show Promising Results in Valosin Containing Protein Multisystem Proteinopathy. PLOS ONE 2015, 10, e0122888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabin, J.C.; Ramirez, K. Hydroxychloroquine Ocular Toxicity: Lessons Learned. J. Rheumatol. 2019, 46, 1640–1641. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; McNeill, J.H. To scale or not to scale: The principles of dose extrapolation. Br. J. Pharmacol. 2009, 157, 907–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Noe, G.; Breaud, A.R.; Vidal, M.; Clarke, W.A.; Zahr, N.; Dervieux, T.; Costedoat-Chalumeau, N.; Blanchet, B. Development and validation of a clinical HPLC method for the quantification of hydroxychloroquine and its metabolites in whole blood. Future Sci. OA 2015, 1, FSO26. [Google Scholar] [CrossRef]
- Fox, W.M. Reflex-ontogeny and behavioural development of the mouse. Anim. Behav. 1965, 13, 234–241. [Google Scholar] [CrossRef]
- Fox, M.W. Natural environment: Theoretical and practical aspects for breeding and rearing laboratory animals. Lab. Anim. Care 1966, 16, 316–321. [Google Scholar] [PubMed]
- Altman, J.; Sudarshan, K. Postnatal development of locomotion in the laboratory rat. Anim. Behav. 1975, 23, 896–920. [Google Scholar] [CrossRef]
- Elnar, A.A.; Diesel, B.; Desor, F.; Feidt, C.; Bouayed, J.; Kiemer, A.K.; Soulimani, R. Neurodevelopmental and behavioral toxicity via lactational exposure to the sum of six indicator non-dioxin-like-polychlorinated biphenyls (∑6 NDL-PCBs) in mice. Toxicology 2012, 299, 44–54. [Google Scholar] [CrossRef]
- Crépeaux, G.; Grova, N.; Bouillaud-Kremarik, P.; Sikhayeva, N.; Salquèbre, G.; Rychen, G.; Soulimani, R.; Appenzeller, B.; Schroeder, H. Short-term effects of a perinatal exposure to a 16 polycyclic aromatic hydrocarbon mixture in rats: Assessment of early motor and sensorial development and cerebral cytochrome oxidase activity in pups. Neurotoxicology 2014, 43, 90–101. [Google Scholar] [CrossRef]
- Meyer, O.A.; Tilson, H.A.; Byrd, W.C.; Riley, M.T. A method for the routine assessment of fore- and hindlimb grip strength of rats and mice. Neurobehav. Toxicol. 1979, 1, 233–236. [Google Scholar]
- Bouayed, J.; Desor, F.; Rammal, H.; Kiemer, A.K.; Tybl, E.; Schroeder, H.; Rychen, G.; Soulimani, R. Effects of lactational exposure to benzo[alpha]pyrene (B[alpha]P) on postnatal neurodevelopment, neuronal receptor gene expression and behaviour in mice. Toxicology 2009, 259, 97–106. [Google Scholar] [CrossRef]
- Walsh, R.N.; Cummins, R.A. The Open-Field Test: A critical review. Psychol. Bull. 1976, 83, 482–504. [Google Scholar] [CrossRef]
- Kondziella, W. A new method for the measurement of muscle relaxation in white mice. Arch. Int. Pharmacodyn. Ther. 1964, 152, 277–284. [Google Scholar]
- Maurissen, J.P.J.; Marable, B.R.; Andrus, A.K.; Stebbins, K.E. Factors affecting grip strength testing. Neurotoxicol. Teratol. 2003, 25, 543–553. [Google Scholar] [CrossRef]
- Pratte, M.; Panayotis, N.; Ghata, A.; Villard, L.; Roux, J.-C. Progressive motor and respiratory metabolism deficits in post-weaning Mecp2-null male mice. Behav. Brain Res. 2011, 216, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Chhonker, Y.S.; Sleightholm, R.L.; Li, J.; Oupický, D.; Murry, D.J. Simultaneous quantitation of hydroxychloroquine and its metabolites in mouse blood and tissues using LC–ESI–MS/MS: An application for pharmacokinetic studies. J. Chromatogr. B 2018, 1072, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods San Diego Calif 2015, 75, 13–18. [Google Scholar] [CrossRef]
- Crépeaux, G. Exposition périnatale à un mélange d’Hydrocarbures Aromatiques Polycycliques chez le rat: Evaluation des effets neurotoxiques à court et à long terme; Sciences Agronomiques, Université de Lorraine: Nancy, France, 2012. [Google Scholar]
- Carreira, M.B.; Cossio, R.; Britton, G.B. Individual and sex differences in high and low responder phenotypes. Behav. Processes 2017, 136, 20–27. [Google Scholar] [CrossRef]
- Ndolo, R.A.; Forrest, M.L.; Krise, J.P. The Role of Lysosomes in Limiting Drug Toxicity in Mice. J. Pharmacol. Exp. Ther. 2010, 333, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Wolfram, J.; Nizzero, S.; Liu, H.; Li, F.; Zhang, G.; Li, Z.; Shen, H.; Blanco, E.; Ferrari, M. A chloroquine-induced macrophage-preconditioning strategy for improved nanodelivery. Sci. Rep. 2017, 7, 13738. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Hou, H.; Zou, Y.; Guo, L. Defective autophagy is associated with neuronal injury in a mouse model of multiple sclerosis. Bosn. J. Basic Med. Sci. 2017, 17, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Jin, X.; Li, F.; Liu, X.; Liu, Y.; Ye, F.; Li, P.; Zhao, T.; Li, Q. Inhibiting autophagy with chloroquine enhances the anti-tumor effect of high-LET carbon ions via ER stress-related apoptosis. Med. Oncol. Northwood Lond. Engl. 2017, 34, 25. [Google Scholar] [CrossRef]
- Yi, H.; Ye, T.; Ge, M.; Yang, M.; Zhang, L.; Jin, S.; Ye, X.; Long, B.; Li, L. Inhibition of autophagy enhances the targeted therapeutic effect of sorafenib in thyroid cancer. Oncol. Rep. 2018, 39, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.-S.; Im, J.-S.; Cho, J.-Y.; Bae, K.-S.; Klein, T.A.; Yeom, J.-S.; Kim, T.-S.; Choi, J.-S.; Jang, I.-J.; Park, J.-W. Pharmacokinetics of Hydroxychloroquine and Its Clinical Implications in Chemoprophylaxis against Malaria Caused by Plasmodium vivax. Antimicrob. Agents Chemother. 2009, 53, 1468–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, M.K.; Boberg, J.R.; Walsh, M.T.; Wolf, V.; Trujillo, A.; Duke, M.S.; Palme, R.; Felton, L.A. A less stressful alternative to oral gavage for pharmacological and toxicological studies in mice. Toxicol. Appl. Pharm. 2012, 260, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, H.C.; Hugill, A.; Dear, N.T.; Ashcroft, F.M.; Cox, R.D. Deletion of nicotinamide nucleotide transhydrogenase: A new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes 2006, 55, 2153–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, K.; Galvanovskis, J.; Goldsworthy, M.; Hugill, A.; Kaizak, S.; Lee, A.; Meadows, N.; Quwailid, M.M.; Rydström, J.; Teboul, L.; et al. Insulin secretion from beta-cells is affected by deletion of nicotinamide nucleotide transhydrogenase. Methods Enzymol. 2009, 457, 451–480. [Google Scholar]
- Ripoll, V.M.; Meadows, N.A.; Bangert, M.; Lee, A.W.; Kadioglu, A.; Cox, R.D. Nicotinamide nucleotide transhydrogenase (NNT) acts as a novel modulator of macrophage inflammatory responses. FASEB J. 2012, 26, 3550–3562. [Google Scholar] [CrossRef]
- Arnoult, D.; Grodet, A.; Lee, Y.-J.; Estaquier, J.; Blackstone, C. Release of OPA1 during Apoptosis Participates in the Rapid and Complete Release of Cytochrome c and Subsequent Mitochondrial Fragmentation. J. Biol. Chem. 2005, 280, 35742–35750. [Google Scholar] [CrossRef] [Green Version]
- Taveira-DaSilva, A.M.; Moss, J. Optimizing treatments for lymphangioleiomyomatosis. Expert Rev. Respir. Med. 2012, 6, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.G.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.K.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef]
- Kundu, M.; Lindsten, T.; Yang, C.-Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Wang, C.; Dong, H.; Xia, F.; Zhou, H.; Jiang, X.; Pei, C.; Ren, H.; Li, H.; Li, R.; et al. Immune-related GTPase M (IRGM1) regulates neuronal autophagy in a mouse model of stroke. Autophagy 2012, 8, 1621–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | Juvenile Experiment | Adult Experiment | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group (mg HCQ/kg) | Group (mg HCQ/kg) | |||||||||||||||||
| 0 | 15 | 30 | 50 | 70 | 0 | 30 | 50 | 70 | ||||||||||
| Males | Females | Males | Females | Males | Females | Males | Females | Males | Females | Males | Females | Males | Females | Males | Females | Males | Females | |
| Number of animals | 28 | 28 | 30 | 26 | 28 | 28 | 28 | 28 | 27 | 29 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 |
| Numbers of litters | 27 | 28 | 29 | 26 | 28 | 28 | 28 | 28 | 26 | 28 | ||||||||
| Number of pup/litters | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | ||||||||
| Number of animals tested up to PND25 | 21 | 21 | 23 | 19 | 21 | 21 | 21 | 21 | 20 | 22 | ||||||||
| Numbers of litters tested up to PND25 | 21 | 21 | 22 | 19 | 21 | 21 | 21 | 21 | 19 | 21 | ||||||||
| Number of animals tested at PN73 | 7 | 7 | 9 | 6 | 7 | 7 | 7 | 7 | 6 | 8 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Numbers of litters tested at PND73 | 7 | 7 | 9 | 6 | 7 | 7 | 7 | 7 | 6 | 7 | ||||||||
| Number of animals tested at PND136 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | ||||||||
| Numbers of litters tested at PND136 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Time of developmental testing | PND5-PND25 | |||||||||||||||||
| Time of behavioral testing | week before endpoint for PND73 and PND136 | week before endpoint for D73 and D136 | ||||||||||||||||
| Endpoint | PND11; PND26; PND73; PND136 | D3; D73; D136 | ||||||||||||||||
| Organ | Tibial Anterior Muscle | Draining Lymph Nodes | Spleen | Liver | Brain | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group (mg HCQ/kg) | Group (mg HCQ/kg) | Group (mg HCQ/kg) | Group (mg HCQ/kg) | Group (mg HCQ/kg) | ||||||||||||||||||
| Endpoint | Gender | Protein | 15 | 30 | 50 | 70 | 15 | 30 | 50 | 70 | 15 | 30 | 50 | 70 | 15 | 30 | 50 | 70 | 15 | 30 | 50 | 70 |
| PND11 | ♂ | LC3-I | - | - | - | ↓ | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ↓ | ↓ |
| LC3-II | - | ↓ | ↓ | ↓ | - | - | - | - | - | - | - | - | - | - | - | - | ↓ | ↓ | ↓ | ↓ | ||
| SQSTM1/p62 | - | - | ↓ | - | - | - | - | - | - | ↑ | - | - | - | - | - | - | - | - | - | - | ||
| ♀ | LC3-I | - | - | - | - | ↑ | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| LC3-II | - | - | - | - | - | ↑ | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| SQSTM1/p62 | - | - | - | - | ↑ | ↑ | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| PND26 | ♂ | LC3-I | - | - | - | - | - | - | - | - | ↑ | - | - | - | - | - | - | - | ↑ | - | ↑ | - |
| LC3-II | - | ↑ | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ↑ | ↑ | ↑ | - | ||
| SQSTM1/p62 | - | - | - | - | - | - | - | - | ↑ | - | - | ↓ | - | - | - | - | ↑ | - | ↑ | ↑ | ||
| ♀ | LC3-I | - | - | - | - | ↑ | - | - | ↓ | - | - | - | - | - | - | - | - | - | - | ↑ | - | |
| LC3-II | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ↑ | - | ||
| SQSTM1/p62 | ↑ | ↑ | - | - | - | - | - | - | - | ↓ | ↓ | ↓ | - | - | - | - | - | - | - | - | ||
| PND73 | ♂ | LC3-I | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| LC3-II | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| SQSTM1/p62 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| ♀ | LC3-I | - | - | - | - | - | - | - | - | - | - | - | - | ↓ | - | - | ↓ | - | - | - | - | |
| LC3-II | - | - | - | - | - | ↑ | - | - | ↓ | ↓ | - | - | - | - | - | - | - | - | - | - | ||
| SQSTM1/p62 | - | - | - | - | - | ↑ | - | ↓ | ↓ | ↓ | - | - | - | - | - | - | - | - | - | - | ||
| PND136 | ♂ | LC3-I | - | - | - | - | ↑ | ↑ | ↑ | ↑ | - | - | - | - | - | - | - | - | - | - | - | - |
| LC3-II | - | - | - | - | - | ↑ | - | ↑ | - | - | - | - | - | - | - | - | - | - | - | - | ||
| SQSTM1/p62 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| ♀ | LC3-I | - | ↓ | ↓ | - | - | - | - | - | - | - | - | - | ↑ | ↑ | - | ↑ | ↑ | - | - | ↓ | |
| LC3-II | - | - | ↓ | - | - | - | - | - | - | - | - | - | ↑ | ↑ | ↑ | ↑ | - | - | ↓ | ↓ | ||
| SQSTM1/p62 | - | - | ↓ | - | - | - | - | - | - | ↑ | - | - | - | - | - | - | - | - | - | - | ||
| Organ | Tibial Anterior Muscle | Draining Lymph Nodes | Spleen | Liver | Brain | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group (mg HCQ/kg) | Group (mg HCQ/kg) | Group (mg HCQ/kg) | Group (mg HCQ/kg) | Group (mg HCQ/kg) | |||||||||||||
| Endpoint | Gender | Protein | 30 | 50 | 70 | 30 | 50 | 70 | 30 | 50 | 70 | 30 | 50 | 70 | 30 | 50 | 70 |
| D3 | ♂ | LC3-I | - | - | - | - | ↑ | ↑ | - | - | - | - | ↓ | ↓ | - | - | - |
| LC3-II | - | - | - | - | - | - | - | - | - | - | - | ↓ | - | - | - | ||
| SQSTM1/p62 | - | - | - | - | - | - | - | - | - | - | ↓ | ↓ | - | - | - | ||
| ♀ | LC3-I | - | - | - | - | - | ↑ | - | - | - | - | - | - | - | - | - | |
| LC3-II | - | - | - | - | ↑ | ↑ | - | - | - | - | - | - | - | - | - | ||
| SQSTM1/p62 | - | - | - | - | - | ↑ | - | - | - | - | - | - | ↓ | - | - | ||
| D73 | ♂ | LC3-I | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| LC3-II | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| SQSTM1/p62 | - | - | ↓ | - | - | - | - | ↓ | - | - | ↓ | ↓ | - | - | - | ||
| ♀ | LC3-I | - | - | - | - | - | - | - | - | - | - | - | - | ↓ | - | - | |
| LC3-II | - | - | - | - | - | - | - | - | - | - | - | - | - | ↓ | - | ||
| SQSTM1/p62 | - | - | - | - | - | - | - | - | ↓ | - | - | - | - | - | - | ||
| D136 | ♂ | LC3-I | - | - | - | - | ↑ | ↑ | - | - | - | - | - | ↓ | ↓ | - | ↓ |
| LC3-II | - | - | - | ↑ | ↑ | - | - | ↓ | - | - | - | ↓ | ↓ | - | ↓ | ||
| SQSTM1/p62 | - | ↓ | - | ↓ | - | - | ↓ | ↓ | ↓ | - | - | ↓ | - | - | - | ||
| ♀ | LC3-I | - | - | - | - | ↑ | - | - | - | - | - | ↑ | ↑ | - | - | - | |
| LC3-II | - | - | - | - | - | - | - | - | - | ↑ | ↑ | ↑ | - | - | - | ||
| SQSTM1/p62 | - | - | - | - | - | - | - | - | ↓ | ↑ | ↑ | ↑ | - | - | - | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masson, J.-D.; Blanchet, B.; Periou, B.; Authier, F.-J.; Mograbi, B.; Gherardi, R.K.; Crépeaux, G. Long Term Pharmacological Perturbation of Autophagy in Mice: Are HCQ Injections a Relevant Choice? Biomedicines 2020, 8, 47. https://doi.org/10.3390/biomedicines8030047
Masson J-D, Blanchet B, Periou B, Authier F-J, Mograbi B, Gherardi RK, Crépeaux G. Long Term Pharmacological Perturbation of Autophagy in Mice: Are HCQ Injections a Relevant Choice? Biomedicines. 2020; 8(3):47. https://doi.org/10.3390/biomedicines8030047
Chicago/Turabian StyleMasson, Jean-Daniel, Benoit Blanchet, Baptiste Periou, François-Jérôme Authier, Baharia Mograbi, Romain K. Gherardi, and Guillemette Crépeaux. 2020. "Long Term Pharmacological Perturbation of Autophagy in Mice: Are HCQ Injections a Relevant Choice?" Biomedicines 8, no. 3: 47. https://doi.org/10.3390/biomedicines8030047