
Inflammatory Bowel Disease Increases the Severity of Myocardial Infarction after Acute Ischemia–Reperfusion Injury in Mice

 ,
,  and
and
Abstract
:1. Introduction
2. Methods
2.1. Animals
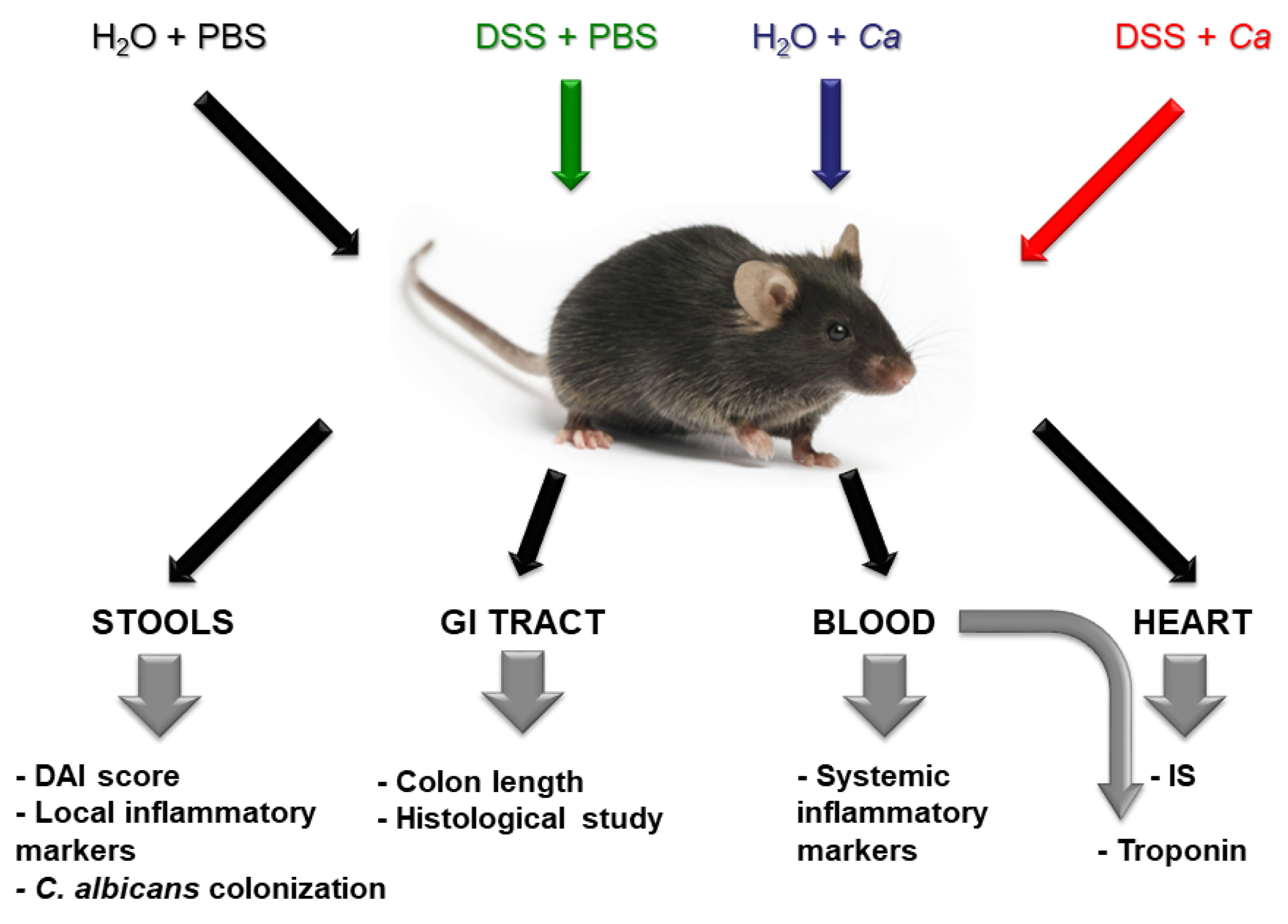
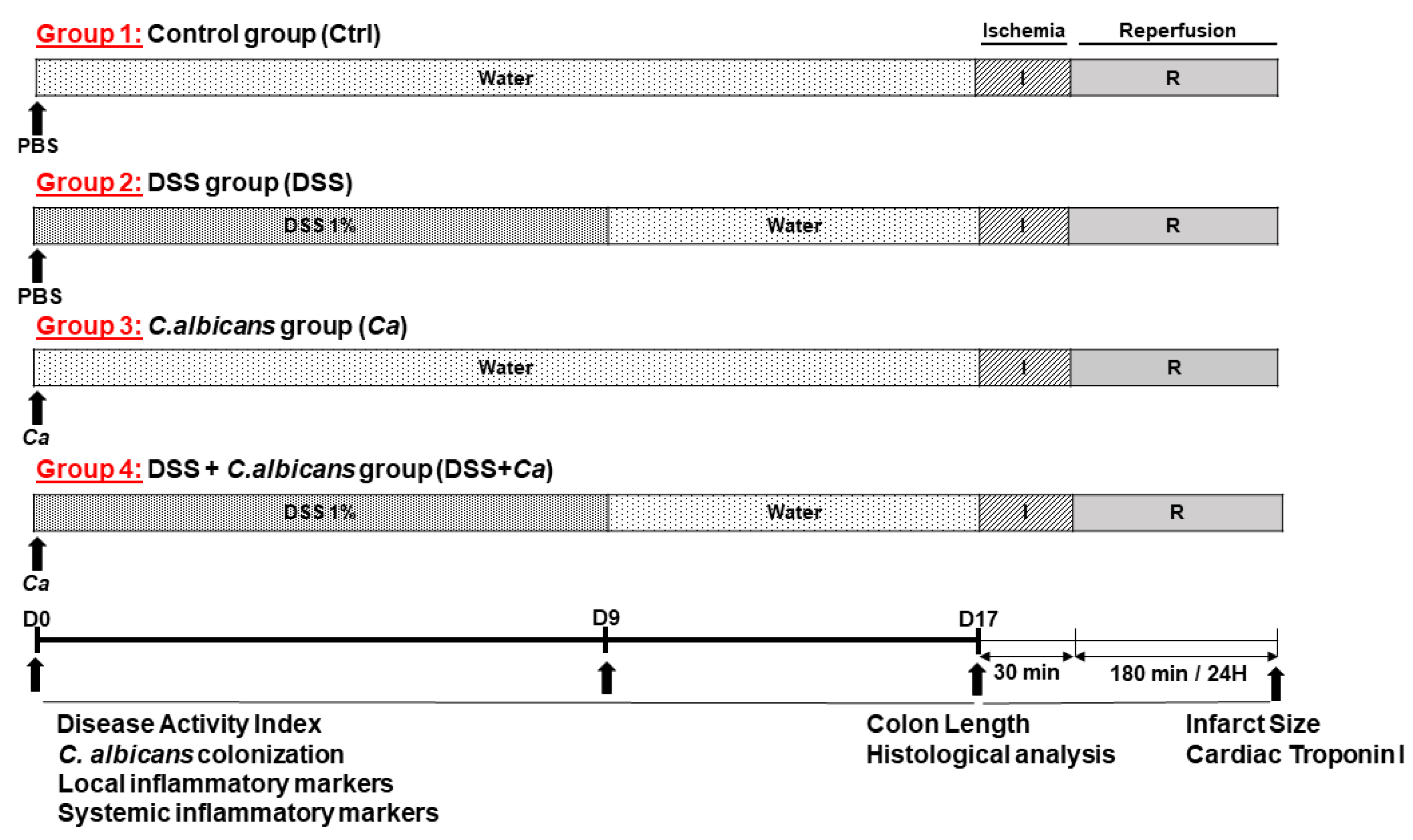
2.2. Experimental Design of the Study
2.3. Inflammatory Bowel Disease Studies
2.3.1. Candida albicans Strain SC5314 Preparation
2.3.2. Disease Activity Index Assessment
2.3.3. Histological Analysis
2.3.4. Candida albicans Gastrointestinal Colonization Analysis
2.3.5. Fecal Lipocalin 2 Analysis
2.3.6. Serum Cytokine Analysis
2.4. Ischemia–Reperfusion Studies In Vivo
2.4.1. Surgical Preparation and Induction of Myocardial Infarction
2.4.2. Infarct Size Measurement
2.4.3. Serum Cardiac Troponin I (cTnI) Determination
2.5. Cardiomyocyte Studies In Vitro
2.6. Statistical Analysis
3. Results
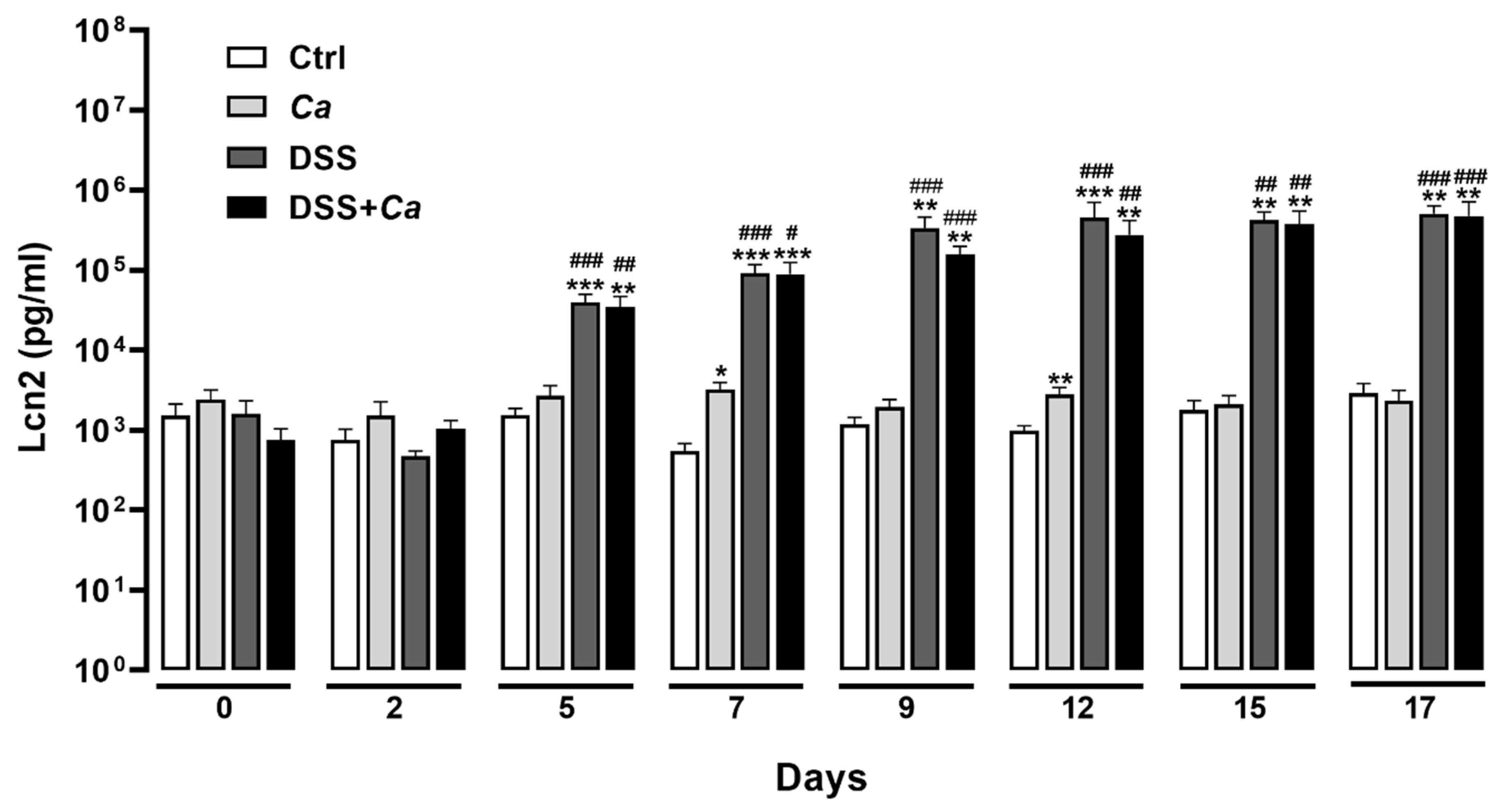
3.1. Disease Activity Index Evolution in IBD Mice
3.2. Histological Analysis
3.3. Candida albicans Colonization Assessment
3.4. Local Inflammatory Marker Analysis
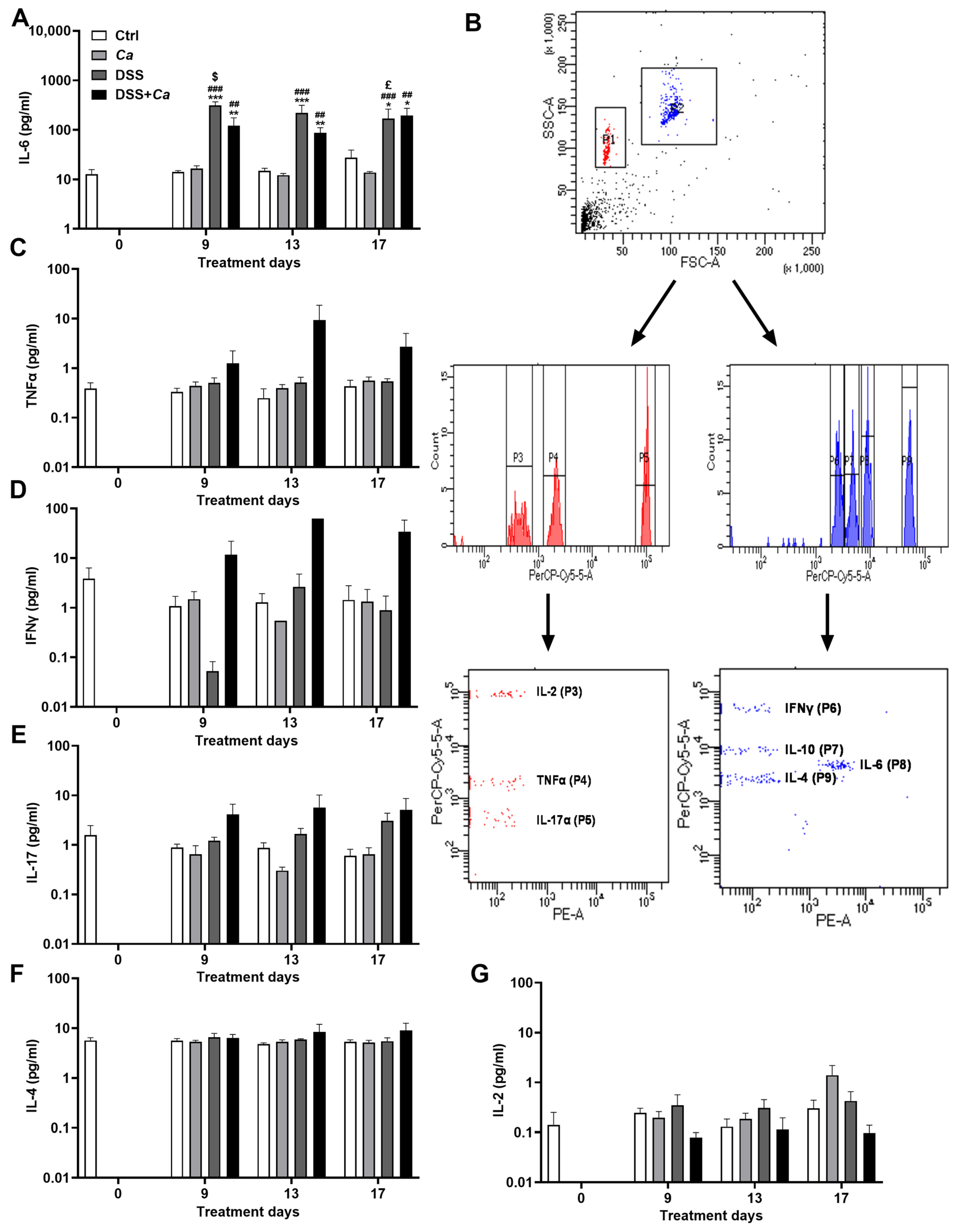
3.5. Circulatory Cytokine Analysis
3.6. Infarct Size Quantification
3.7. Serum Cardiac Troponin I Quantification
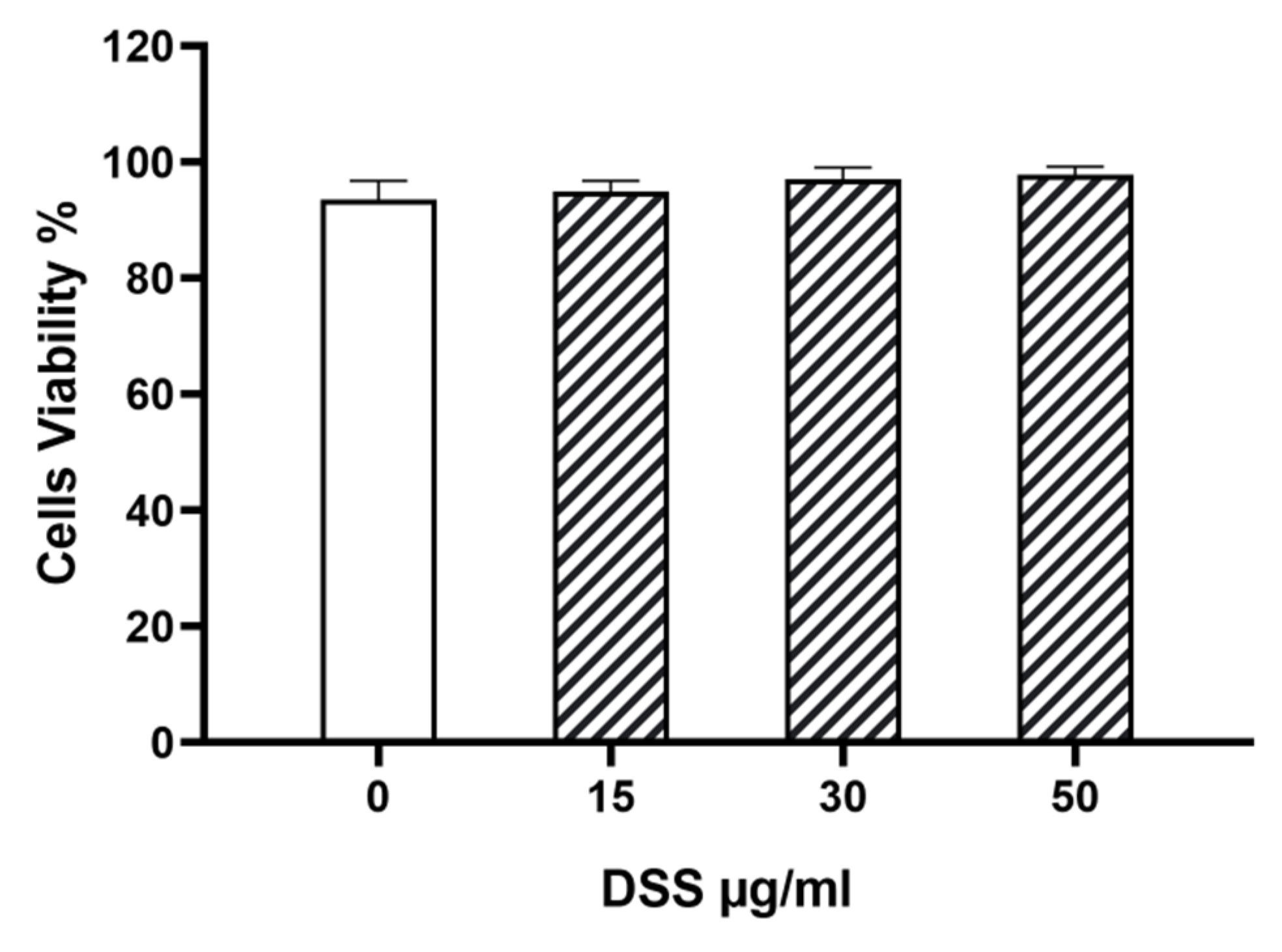
3.8. DSS Effect on Cardiomyocytes In Vitro
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUCs | Areas under the curve |
| Ca | Candida albicans |
| CAD | Coronary artery disease |
| CD | Crohn’s disease |
| CFU | Colony-forming unit |
| cTnI | Cardiac troponin I |
| CVD | Cardiovascular disease |
| DAI | Disease activity index |
| DSS | Dextran sulfate sodium |
| GI | Gastrointestinal |
| HR | Heart rate |
| IBD | Inflammatory bowel disease |
| IHD | Ischemic heart disease |
| IL | Interleukin |
| INFγ | Interferon gamma |
| IR | Ischemia–reperfusion |
| IS | Infarct size |
| Lcn2 | Fecal lipocalin 2 |
| MI | Myocardial infarction |
| TNF-α | Tumor necrosis factor alpha |
| UC | Ulcerative colitis |
References
- Barquera, S.; Pedroza-Tobias, A.; Medina, C.; Hernandez-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch. Med. Res. 2015, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- WHO. The Top 10 Causes of Death; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Wang, F.; Yu, Y.; Mubarik, S. Global burden of ischemic heart disease and attributable risk factors, 1990–2017: A secondary analysis based on the global burden of disease study 2017. Clin. Epidemiol. 2021, 13, 859–870. [Google Scholar] [CrossRef]
- Mason, J.C.; Libby, P. Cardiovascular disease in patients with chronic inflammation: Mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur. Heart J. 2015, 36, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, E.; Atzeni, F.; Sarzi-Puttini, P.; Turiel, M.; Lopez, L.R.; Nurmohamed, M.T. Is atherosclerosis an autoimmune disease? BMC Med. 2014, 12, 47. [Google Scholar] [CrossRef]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune effector mechanisms implicated in atherosclerosis: From mice to humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef]
- Prasad, M.; Hermann, J.; Gabriel, S.E.; Weyand, C.M.; Mulvagh, S.; Mankad, R.; Oh, J.K.; Matteson, E.L.; Lerman, A. Cardiorheumatology: Cardiac involvement in systemic rheumatic disease. Nat. Rev. Cardiol. 2015, 12, 168–176. [Google Scholar] [CrossRef]
- Chen, B.; Collen, L.V.; Mowat, C.; Isaacs, K.L.; Singh, S.; Kane, S.V.; Farraye, F.A.; Snapper, S.; Jneid, H.; Lavie, C.J.; et al. Inflammatory Bowel Disease and Cardiovascular Diseases. Am. J. Med. 2022, 135, 1453–1460. [Google Scholar] [CrossRef]
- Wu, P.; Jia, F.; Zhang, B.; Zhang, P. Risk of cardiovascular disease in inflammatory bowel disease. Exp. Ther. Med. 2017, 13, 395–400. [Google Scholar] [CrossRef]
- Wu, H.; Hu, T.; Hao, H.; Hill, M.A.; Xu, C.; Liu, Z. Inflammatory bowel disease and cardiovascular diseases: A concise review. Eur. Heart J. Open 2021, 2, oeab029. [Google Scholar] [CrossRef]
- Hodges, P.; Kelly, P. Inflammatory bowel disease in Africa: What is the current state of knowledge? Int. Health 2020, 12, 222–230. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Wajda, A.; Svenson, L.W.; MacKenzie, A.; Koehoorn, M.; Jackson, M.; Fedorak, R.; Israel, D.; Blanchard, J.F. The epidemiology of inflammatory bowel disease in Canada: A population-based study. Am. J. Gastroenterol. 2006, 101, 1559–1568. [Google Scholar] [CrossRef]
- Burisch, J.; Jess, T.; Martinato, M.; Lakatos, P.L.; EpiCom, E. The burden of inflammatory bowel disease in Europe. J. Crohn’s Colitis 2013, 7, 322–337. [Google Scholar] [CrossRef]
- Ng, S.C. Epidemiology of inflammatory bowel disease: Focus on Asia. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 363–372. [Google Scholar] [CrossRef]
- De Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Fiocchi, C. Ulcerative colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [PubMed]
- Conrad, K.; Roggenbuck, D.; Laass, M.W. Diagnosis and classification of ulcerative colitis. Autoimmun. Rev. 2014, 13, 463–466. [Google Scholar] [CrossRef]
- Laass, M.W.; Roggenbuck, D.; Conrad, K. Diagnosis and classification of Crohn’s disease. Autoimmun. Rev. 2014, 13, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Nagao-Kitamoto, H.; Kamada, N. Host-microbial Cross-talk in Inflammatory Bowel Disease. Immune Netw. 2017, 17, 1–12. [Google Scholar] [CrossRef]
- Balderramo, D.C.; Romagnoli, P.A. Fecal Fungal Microbiota (Mycobiome) Study as a Potential Tool for Precision Medicine in Inflammatory Bowel Disease. Gut Liver 2023, 17, 505–515. [Google Scholar] [CrossRef]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell Infect. Microbiol. 2022, 12, 733992. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Tan, V.P.; Chung, A.; Yan, B.P.; Gibson, P.R. Venous and arterial disease in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2013, 28, 1095–1113. [Google Scholar] [CrossRef] [PubMed]
- Zezos, P.; Kouklakis, G.; Saibil, F. Inflammatory bowel disease and thromboembolism. World J. Gastroenterol. 2014, 20, 13863–13878. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, A.R.; Parasa, S.; Navaneethan, U.; Crowell, M.D.; Olden, K. Comprehensive study of cardiovascular morbidity in hospitalized inflammatory bowel disease patients. J. Crohn’s Colitis 2011, 5, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Fumery, M.; Xiaocang, C.; Dauchet, L.; Gower-Rousseau, C.; Peyrin-Biroulet, L.; Colombel, J.F. Thromboembolic events and cardiovascular mortality in inflammatory bowel diseases: A meta-analysis of observational studies. J. Crohn’s Colitis 2014, 8, 469–479. [Google Scholar] [CrossRef]
- Stadnicki, A.; Stadnicka, I. Venous and arterial thromboembolism in patients with inflammatory bowel diseases. World J. Gastroenterol. 2021, 27, 6757–6774. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Ahlehoff, O.; Lindhardsen, J.; Erichsen, R.; Jensen, G.V.; Torp-Pedersen, C.; Nielsen, O.H.; Gislason, G.H.; Hansen, P.R. Disease activity in inflammatory bowel disease is associated with increased risk of myocardial infarction, stroke and cardiovascular death--a Danish nationwide cohort study. PLoS ONE 2013, 8, e56944. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Ahlehoff, O.; Lindhardsen, J.; Erichsen, R.; Lamberts, M.; Khalid, U.; Nielsen, O.H.; Torp-Pedersen, C.; Gislason, G.H.; Hansen, P.R. Prognosis after first-time myocardial infarction in patients with inflammatory bowel disease according to disease activity: Nationwide cohort study. Circulation. Cardiovasc. Qual. Outcomes 2014, 7, 857–862. [Google Scholar] [CrossRef]
- Kuy, S.; Dua, A.; Chappidi, R.; Seabrook, G.; Brown, K.R.; Lewis, B.; Rossi, P.J.; Lee, C.J. The increasing incidence of thromboembolic events among hospitalized patients with inflammatory bowel disease. Vascular 2015, 23, 260–264. [Google Scholar] [CrossRef]
- Panhwar, M.S.; Mansoor, E.; Al-Kindi, S.G.; Sinh, P.; Katz, J.; Oliveira, G.H.; Cooper, G.S.; Ginwalla, M. Risk of Myocardial Infarction in Inflammatory Bowel Disease: A Population-based National Study. Inflamm. Bowel Dis. 2019, 25, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Osterman, M.T.; Yang, Y.X.; Brensinger, C.; Forde, K.A.; Lichtenstein, G.R.; Lewis, J.D. No increased risk of myocardial infarction among patients with ulcerative colitis or Crohn’s disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2011, 9, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.; Magowan, S.; Accortt, N.A.; Chen, J.; Stone, C.D. Risk of arterial thrombotic events in inflammatory bowel disease. Am. J. Gastroenterol. 2009, 104, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.L.; Beery, R.M.; Schulman, A.R.; McCarthy, E.P.; Korzenik, J.R.; Winter, R.W. Hospitalizations for acute myocardial infarction are decreased among patients with inflammatory bowel disease using a nationwide inpatient database. Inflamm. Bowel Dis. 2016, 22, 2229–2237. [Google Scholar] [CrossRef]
- Mehrpooya, M.; Larti, F.; Nozari, Y.; Sattarzadeh-Badkoobeh, R.; Zand Parsa, A.F.; Zebardast, J.; Tavoosi, A.; Shahbazi, F. Study of serum uric acid levels in myocardial infarction and its association with killip class. Acta Medica Iran. 2017, 55, 97–102. [Google Scholar]
- Karbach, S.; Croxford, A.L.; Oelze, M.; Schuler, R.; Minwegen, D.; Wegner, J.; Koukes, L.; Yogev, N.; Nikolaev, A.; Reissig, S.; et al. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2658–2668. [Google Scholar] [CrossRef]
- Hot, A.; Lenief, V.; Miossec, P. Combination of IL-17 and TNFalpha induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann. Rheum. Dis. 2012, 71, 768–776. [Google Scholar] [CrossRef]
- Messadi, E.; Aloui, Z.; Belaidi, E.; Vincent, M.P.; Couture-Lepetit, E.; Waeckel, L.; Decorps, J.; Bouby, N.; Gasmi, A.; Karoui, H.; et al. Cardioprotective effect of VEGF and venom VEGF-like protein in acute myocardial ischemia in mice: Effect on mitochondrial function. J. Cardiovasc. Pharmacol. 2014, 63, 274–281. [Google Scholar] [CrossRef]
- Messadi, E.; Vincent, M.P.; Griol-Charhbili, V.; Mandet, C.; Colucci, J.; Krege, J.H.; Bruneval, P.; Bouby, N.; Smithies, O.; Alhenc-Gelas, F.; et al. Genetically determined angiotensin converting enzyme level and myocardial tolerance to ischemia. Faseb J. 2010, 24, 4691–4700. [Google Scholar] [CrossRef]
- Messadi-Laribi, E.; Griol-Charhbili, V.; Bascands, J.L.; Heudes, D.; Meneton, P.; Giudicelli, J.F.; Alhenc-Gelas, F.; Richer, C. Role of tissue kallikrein in the cardioprotective effects of ischemic and pharmacological preconditioning in myocardial ischemia. Faseb J. 2005, 19, 1172–1174. [Google Scholar]
- Messadi-Laribi, E.; Griol-Charhbili, V.; Pizard, A.; Vincent, M.P.; Heudes, D.; Meneton, P.; Alhenc-Gelas, F.; Richer, C. Tissue kallikrein is involved in the cardioprotective effect of AT1-receptor blockade in acute myocardial ischemia. J. Pharmacol. Exp. Ther. 2007, 323, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Tourki, B.; Dumesnil, A.; Belaidi, E.; Ghrir, S.; Godin-Ribuot, D.; Marrakchi, N.; Richard, V.; Mulder, P.; Messadi, E. Lebetin 2, a snake venom-derived B-type natriuretic peptide, provides immediate and prolonged protection against myocardial ischemia-reperfusion injury via modulation of post-ischemic inflammatory response. Toxins 2019, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Tourki, B.; Mateo, P.; Morand, J.; Elayeb, M.; Godin-Ribuot, D.; Marrakchi, N.; Belaidi, E.; Messadi, E. Lebetin 2, a snake venom-derived natriuretic peptide, attenuates acute myocardial ischemic injury through the modulation of mitochondrial permeability transition pore at the time of reperfusion. PLoS ONE 2016, 11, e0162632. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Ahlehoff, O.; Lindhardsen, J.; Erichsen, R.; Lamberts, M.; Khalid, U.; Nielsen, O.H.; Torp-Pedersen, C.; Gislason, G.H.; Hansen, P.R. Inflammatory bowel disease is associated with an increased risk of hospitalization for heart failure: A Danish Nationwide Cohort study. Circulation. Heart Fail. 2014, 7, 717–722. [Google Scholar] [CrossRef]
- Kumamoto, C.A. Inflammation and gastrointestinal Candida colonization. Curr. Opin. Microbiol. 2011, 14, 386–391. [Google Scholar] [CrossRef]
- Gillum, A.M.; Tsay, E.Y.; Kirsch, D.R. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet. MGG 1984, 198, 179–182. [Google Scholar] [CrossRef]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef]
- Bauer, C.; Loher, F.; Dauer, M.; Mayer, C.; Lehr, H.A.; Schonharting, M.; Hallwachs, R.; Endres, S.; Eigler, A. The ICE inhibitor pralnacasan prevents DSS-induced colitis in C57BL/6 mice and suppresses IP-10 mRNA but not TNF-alpha mRNA expression. Dig. Dis. Sci. 2007, 52, 1642–1652. [Google Scholar] [CrossRef]
- Randhawa, P.K.; Singh, K.; Singh, N.; Jaggi, A.S. A review on chemical-induced inflammatory bowel disease models in rodents. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2014, 18, 279–288. [Google Scholar] [CrossRef]
- Cardiff, R.D.; Miller, C.H.; Munn, R.J. Manual hematoxylin and eosin staining of mouse tissue sections. Cold Spring Harb. Protoc. 2014, 2014, 655–658. [Google Scholar] [CrossRef]
- Erben, U.; Loddenkemper, C.; Doerfel, K.; Spieckermann, S.; Haller, D.; Heimesaat, M.M.; Zeitz, M.; Siegmund, B.; Kuhl, A.A. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 2014, 7, 4557–4576. [Google Scholar] [PubMed]
- Chassaing, B.; Srinivasan, G.; Delgado, M.A.; Young, A.N.; Gewirtz, A.T.; Vijay-Kumar, M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS ONE 2012, 7, e44328. [Google Scholar] [CrossRef] [PubMed]
- Pons, S.; Griol-Charhbili, V.; Heymes, C.; Fornes, P.; Heudes, D.; Hagege, A.; Loyer, X.; Meneton, P.; Giudicelli, J.F.; Samuel, J.L.; et al. Tissue kallikrein deficiency aggravates cardiac remodelling and decreases survival after myocardial infarction in mice. Eur. J. Heart Fail. 2008, 10, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Bamba, T.; Mukaisho, K.; Kanauchi, O.; Ban, H.; Bamba, S.; Andoh, A.; Fujiyama, Y.; Hattori, T.; Sugihara, H. Dextran sulfate sodium administered orally is depolymerized in the stomach and induces cell cycle arrest plus apoptosis in the colon in early mouse colitis. Oncol. Rep. 2012, 28, 1597–1605. [Google Scholar] [CrossRef]
- Kitajima, S.; Takuma, S.; Morimoto, M. Tissue distribution of dextran sulfate sodium (DSS) in the acute phase of murine DSS-induced colitis. J. Vet. Med. Sci. 1999, 61, 67–70. [Google Scholar] [CrossRef]
- Lillo, R.; Graziani, F.; Franceschi, F.; Iannaccone, G.; Massetti, M.; Olivotto, I.; Crea, F.; Liuzzo, G. Inflammation across the spectrum of hypertrophic cardiac phenotypes. Heart Fail. Rev. 2023, 28, 1065–1075. [Google Scholar] [CrossRef]
- Bigeh, A.; Sanchez, A.; Maestas, C.; Gulati, M. Inflammatory bowel disease and the risk for cardiovascular disease: Does all inflammation lead to heart disease? Trends Cardiovasc. Med. 2020, 30, 463–469. [Google Scholar] [CrossRef]
- Twig, G.; Zandman-Goddard, G.; Szyper-Kravitz, M.; Shoenfeld, Y. Systemic thromboembolism in inflammatory bowel disease: Mechanisms and clinical applications. Ann. N. Y. Acad. Sci. 2005, 1051, 166–173. [Google Scholar] [CrossRef]
- Miehsler, W.; Reinisch, W.; Valic, E.; Osterode, W.; Tillinger, W.; Feichtenschlager, T.; Grisar, J.; Machold, K.; Scholz, S.; Vogelsang, H.; et al. Is inflammatory bowel disease an independent and disease specific risk factor for thromboembolism? Gut 2004, 53, 542–548. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Blanchard, J.F.; Houston, D.S.; Wajda, A. The incidence of deep venous thrombosis and pulmonary embolism among patients with inflammatory bowel disease: A population-based cohort study. Thromb. Haemost. 2001, 85, 430–434. [Google Scholar]
- Koutroubakis, I.E. Therapy insight: Vascular complications in patients with inflammatory bowel disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Redel, A.; Jazbutyte, V.; Smul, T.M.; Lange, M.; Eckle, T.; Eltzschig, H.; Roewer, N.; Kehl, F. Impact of ischemia and reperfusion times on myocardial infarct size in mice in vivo. Exp. Biol. Med. 2008, 233, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Redfors, B.; Shao, Y.; Omerovic, E. Myocardial infarct size and area at risk assessment in mice. Exp. Clin. Cardiol. 2012, 17, 268–272. [Google Scholar] [PubMed]
- Matthia, E.L.; Setteducato, M.L.; Elzeneini, M.; Vernace, N.; Salerno, M.; Kramer, C.M. Circulating biomarkers in hypertrophic cardiomyopathy. J. Am. Heart Assoc. 2022, 11, e027618. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, F.; Liu, H.; Chen, H.; Cheng, W.; Dong, S.; Xiong, W. Elevated serum IL-39 in patients with ST-segment elevation myocardial infarction was related with left ventricular systolic dysfunction. Biomark. Med. 2017, 11, 419–426. [Google Scholar] [CrossRef]
- Bacchiega, B.C.; Bacchiega, A.B.; Usnayo, M.J.; Bedirian, R.; Singh, G.; Pinheiro, G.D. Interleukin 6 inhibition and coronary artery disease in a high-risk population: A prospective community-based clinical study. J. Am. Heart Assoc. 2017, 6, 38. [Google Scholar] [CrossRef]
- Leppkes, M.; Neurath, M.F. Cytokines in inflammatory bowel diseases—Update 2020. Pharmacol. Res. 2020, 158, 104835. [Google Scholar] [CrossRef]
- Kamperidis, N.; Kamperidis, V.; Zegkos, T.; Kostourou, I.; Nikolaidou, O.; Arebi, N.; Karvounis, H. Atherosclerosis and Inflammatory Bowel Disease-Shared Pathogenesis and Implications for Treatment. Angiology 2021, 72, 303–314. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Heusch, G.; Schulz, R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol. Ther. 2010, 127, 295–314. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Ito, H. Novel therapy for Crohn’s disease targeting IL-6 signalling. Expert Opin. Ther. Targets 2004, 8, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Finotto, S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011, 22, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Neurath, M.F. New therapeutic strategies for treatment of inflammatory bowel disease. Mucosal Immunol. 2008, 1, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, T.; Kusugami, K.; Ina, K.; Ando, T.; Shinoda, M.; Imada, A.; Ohsuga, M.; Sakai, T.; Matsuura, T.; Ito, K.; et al. Interleukin-6 and soluble interleukin-6 receptor in the colonic mucosa of inflammatory bowel disease. J. Gastroenterol. Hepatol. 1999, 14, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Pai, J.K.; Pischon, T.; Ma, J.; Manson, J.E.; Hankinson, S.E.; Joshipura, K.; Curhan, G.C.; Rifai, N.; Cannuscio, C.C.; Stampfer, M.J.; et al. Inflammatory markers and the risk of coronary heart disease in men and women. N. Engl. J. Med. 2004, 351, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Nevulis, M.G.; Baker, C.; Lebovics, E.; Frishman, W.H. Overview of link between inflammatory bowel disease and cardiovascular disease. Cardiol. Rev. 2018, 26, 287–293. [Google Scholar] [CrossRef]
- Esteve, E.; Castro, A.; Lopez-Bermejo, A.; Vendrell, J.; Ricart, W.; Fernandez-Real, J.M. Serum interleukin-6 correlates with endothelial dysfunction in healthy men independently of insulin sensitivity. Diabetes Care 2007, 30, 939–945. [Google Scholar] [CrossRef]
- Huber, S.A.; Sakkinen, P.; Conze, D.; Hardin, N.; Tracy, R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2364–2367. [Google Scholar] [CrossRef]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Bohm, M.; Nickenig, G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef]
- Schuett, H.; Luchtefeld, M.; Grothusen, C.; Grote, K.; Schieffer, B. How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb. Haemost. 2009, 102, 215–222. [Google Scholar] [CrossRef]
- Su, J.H.; Luo, M.Y.; Liang, N.; Gong, S.X.; Chen, W.; Huang, W.Q.; Tian, Y.; Wang, A.P. Interleukin-6: A Novel Target for Cardio-Cerebrovascular Diseases. Front. Pharmacol. 2021, 12, 745061. [Google Scholar] [CrossRef] [PubMed]
- Groot, H.E.; Al Ali, L.; van der Horst, I.C.C.; Schurer, R.A.J.; van der Werf, H.W.; Lipsic, E.; van Veldhuisen, D.J.; Karper, J.C.; van der Harst, P. Plasma interleukin 6 levels are associated with cardiac function after ST-elevation myocardial infarction. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2019, 108, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Lindmark, E.; Diderholm, E.; Wallentin, L.; Siegbahn, A. Relationship between interleukin 6 and mortality in patients with unstable coronary artery disease: Effects of an early invasive or noninvasive strategy. JAMA 2001, 286, 2107–2113. [Google Scholar] [CrossRef] [PubMed]
- Fisman, E.Z.; Benderly, M.; Esper, R.J.; Behar, S.; Boyko, V.; Adler, Y.; Tanne, D.; Matas, Z.; Tenenbaum, A. Interleukin-6 and the risk of future cardiovascular events in patients with angina pectoris and/or healed myocardial infarction. Am. J. Cardiol. 2006, 98, 14–18. [Google Scholar] [CrossRef]
- Zhang, M.; Bai, Y.; Wang, Y.; Cui, H.; Tang, M.; Wang, L.; Wang, X.; Gu, D. Cumulative Evidence for Associations Between Genetic Variants in Interleukin 6 Receptor Gene and Human Diseases and Phenotypes. Front. Immunol. 2022, 13, 860703. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yoshizaki, K.; Kishimoto, T.; Ito, H. IL-6 is required for the development of Th1 cell-mediated murine colitis. J. Immunol. 2000, 164, 4878–4882. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Aden, K.; Bernardes, J.P.; Conrad, C.; Tran, F.; Hoper, H.; Volk, V.; Mishra, N.; Blase, J.I.; Nikolaus, S.; et al. Therapeutic Interleukin-6 Trans-signaling Inhibition by Olamkicept (sgp130Fc) in Patients with Active Inflammatory Bowel Disease. Gastroenterology 2021, 160, 2354–2366. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, M.; Zhong, C.; Xu, B.; Kang, L. M2-like macrophages transplantation protects against the doxorubicin-induced heart failure via mitochondrial transfer. Biomater. Res. 2022, 26, 14. [Google Scholar] [CrossRef]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef]
- Kumar, V.; Prabhu, S.D.; Bansal, S.S. CD4(+) T-lymphocytes exhibit biphasic kinetics post-myocardial infarction. Front. Cardiovasc. Med. 2022, 9, 992653. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; D’Onofrio, N.; Mauro, C.; Balestrieri, M.L.; Marfella, R. Thrombus Aspiration in Hyperglycemic Patients with High Inflammation Levels in Coronary Thrombus. J. Am. Coll. Cardiol. 2019, 73, 530–531. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Li, Q.; Zhou, H.; Sun, L.; Lin, C.; Jin, Y.; Wang, D.; Guo, G. The role of major immune cells in myocardial infarction. Front. Immunol. 2022, 13, 1084460. [Google Scholar] [CrossRef]
- Bouzazi, D.; Mami, W.; Mosbah, A. Natriuretic-like peptide lebetin 2 mediates m2 macrophage polarization in lps-activated raw264.7 cells in an il-10-dependent manner. Toxins 2023, 15, 298. [Google Scholar] [CrossRef]
- Barsig, J.; Kusters, S.; Vogt, K.; Volk, H.D.; Tiegs, G.; Wendel, A. Lipopolysaccharide-induced interleukin-10 in mice: Role of endogenous tumor necrosis factor-alpha. Eur. J. Immunol. 1995, 25, 2888–2893. [Google Scholar] [CrossRef]
- Jing, R.; Long, T.Y.; Pan, W.; Li, F.; Xie, Q.Y. IL-6 knockout ameliorates myocardial remodeling after myocardial infarction by regulating activation of M2 macrophages and fibroblast cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6283–6291. [Google Scholar] [CrossRef]
- Scaldaferri, F.; Lancellotti, S.; Pizzoferrato, M.; De Cristofaro, R. Haemostatic system in inflammatory bowel diseases: New players in gut inflammation. World J. Gastroenterol. 2011, 17, 594–608. [Google Scholar] [CrossRef]
- Roman, M.J.; Devereux, R.B.; Schwartz, J.E.; Lockshin, M.D.; Paget, S.A.; Davis, A.; Crow, M.K.; Sammaritano, L.; Levine, D.M.; Shankar, B.A.; et al. Arterial stiffness in chronic inflammatory diseases. Hypertension 2005, 46, 194–199. [Google Scholar] [CrossRef]
- Chiaro, T.R.; Soto, R.; Zac Stephens, W.; Kubinak, J.L.; Petersen, C.; Gogokhia, L.; Bell, R.; Delgado, J.C.; Cox, J.; Voth, W.; et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci. Transl. Med. 2017, 9, eaaf9044. [Google Scholar] [CrossRef]
- Nesci, A.; Carnuccio, C. Gut Microbiota and Cardiovascular Disease: Evidence on the Metabolic and Inflammatory Background of a Complex Relationship. Int. J. Mol. Sci. 2023, 24, 9087. [Google Scholar] [CrossRef]
- Sardu, C.; Consiglia Trotta, M.; Santella, B.; D’Onofrio, N.; Barbieri, M.; Rizzo, M.R.; Sasso, F.C.; Scisciola, L.; Turriziani, F.; Torella, M.; et al. Microbiota thrombus colonization may influence athero-thrombosis in hyperglycemic patients with ST segment elevation myocardialinfarction (STEMI). Marianella study. Diabetes Res. Clin. Pract. 2021, 173, 108670. [Google Scholar] [CrossRef] [PubMed]
- Brun, A.; Nuzzo, A.; Prouvost, B.; Diallo, D.; Hamdan, S.; Meseguer, E.; Guidoux, C.; Lavallée, P.; Amarenco, P.; Lesèche, G.; et al. Oral microbiota and atherothrombotic carotid plaque vulnerability in periodontitis patients. A cross-sectional study. J. Periodontal Res. 2021, 56, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Qiu, X.; Liu, Y.; Yuan, C.; Yang, X. Trimethylamine N-oxide promotes tissue factor expression and activity in vascular endothelial cells: A new link between trimethylamine N-oxide and atherosclerotic thrombosis. Thromb. Res. 2019, 177, 110–116. [Google Scholar] [CrossRef]
- Shankar, J.; Solis, N.V.; Mounaud, S.; Szpakowski, S.; Liu, H.; Losada, L.; Nierman, W.C.; Filler, S.G. Using Bayesian modelling to investigate factors governing antibiotic-induced Candida albicans colonization of the GI tract. Sci. Rep. 2015, 5, 8131. [Google Scholar] [CrossRef] [PubMed]
- Jabra-Rizk, M.A.; Kong, E.F.; Tsui, C.; Nguyen, M.H.; Clancy, C.J.; Fidel, P.L., Jr.; Noverr, M. Candida albicans Pathogenesis: Fitting within the Host-Microbe Damage Response Framework. Infect. Immun. 2016, 84, 2724–2739. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.L.; Wilson, D.; Hube, B. Candida albicans pathogenicity mechanisms. Virulence 2013, 4, 119–128. [Google Scholar] [CrossRef]
- Dassopoulos, T.; Frangakis, C.; Cruz-Correa, M.; Talor, M.V.; Burek, C.L.; Datta, L.; Nouvet, F.; Bayless, T.M.; Brant, S.R. Antibodies to saccharomyces cerevisiae in Crohn’s disease: Higher titers are associated with a greater frequency of mutant NOD2/CARD15 alleles and with a higher probability of complicated disease. Inflamm. Bowel Dis. 2007, 13, 143–151. [Google Scholar] [CrossRef]
- Dassopoulos, T.; Nguyen, G.C.; Talor, M.V.; Datta, L.W.; Isaacs, K.L.; Lewis, J.D.; Gold, M.S.; Valentine, J.F.; Smoot, D.T.; Harris, M.L.; et al. NOD2 mutations and anti-Saccharomyces cerevisiae antibodies are risk factors for Crohn’s disease in African Americans. Am. J. Gastroenterol. 2010, 105, 378–386. [Google Scholar] [CrossRef]
- Standaert-Vitse, A.; Sendid, B.; Joossens, M.; Francois, N.; Vandewalle-El Khoury, P.; Branche, J.; Van Kruiningen, H.; Jouault, T.; Rutgeerts, P.; Gower-Rousseau, C.; et al. Candida albicans colonization and ASCA in familial Crohn’s disease. Am. J. Gastroenterol. 2009, 104, 1745–1753. [Google Scholar] [CrossRef]
- Li, J.; Tian, H.; Jiang, H.J.; Han, B. Interleukin-17 SNPs and serum levels increase ulcerative colitis risk: A meta-analysis. World J. Gastroenterol. 2014, 20, 15899–15909. [Google Scholar] [CrossRef]
- Koh, A.Y. Murine models of Candida gastrointestinal colonization and dissemination. Eukaryot. Cell 2013, 12, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Panpetch, W.; Somboonna, N.; Bulan, D.E.; Issara-Amphorn, J.; Worasilchai, N.; Finkelman, M.; Chindamporn, A.; Palaga, T.; Tumwasorn, S.; Leelahavanichkul, A. Gastrointestinal Colonization of Candida Albicans Increases Serum (1-->3)-beta-D-Glucan, without Candidemia, and Worsens Cecal Ligation and Puncture Sepsis in Murine Model. Shock 2018, 49, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Jawhara, S.; Thuru, X.; Standaert-Vitse, A.; Jouault, T.; Mordon, S.; Sendid, B.; Desreumaux, P.; Poulain, D. Colonization of mice by Candida albicans is promoted by chemically induced colitis and augments inflammatory responses through galectin-3. J. Infect. Dis. 2008, 197, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Strijbis, K.; Yilmaz, O.H.; Dougan, S.K.; Esteban, A.; Grone, A.; Kumamoto, C.A.; Ploegh, H.L. Intestinal colonization by Candida albicans alters inflammatory responses in Bruton’s tyrosine kinase-deficient mice. PLoS ONE 2014, 9, e112472. [Google Scholar] [CrossRef]
- Yan, L.; Wu, C.R.; Wang, C.; Yang, C.H.; Tong, G.Z.; Tang, J.G. Effect of Candida albicans on Intestinal Ischemia-reperfusion Injury in Rats. Chin. Med. J. 2016, 129, 1711–1718. [Google Scholar] [CrossRef]
- Panpetch, W.; Hiengrach, P.; Nilgate, S.; Tumwasorn, S.; Somboonna, N.; Wilantho, A.; Chatthanathon, P.; Prueksapanich, P.; Leelahavanichkul, A. Additional Candida albicans administration enhances the severity of dextran sulfate solution induced colitis mouse model through leaky gut-enhanced systemic inflammation and gut-dysbiosis but attenuated by Lactobacillus rhamnosus L34. Gut Microbes 2020, 11, 465–480. [Google Scholar] [CrossRef]
- Panpetch, W.; Sawaswong, V.; Chanchaem, P.; Ondee, T.; Dang, C.P.; Payungporn, S.; Leelahavanichkul, A. Candida Administration Worsens Cecal Ligation and Puncture-Induced Sepsis in Obese Mice through Gut Dysbiosis Enhanced Systemic Inflammation, Impact of Pathogen-Associated Molecules from Gut Translocation and Saturated Fatty Acid. Front. Immunol. 2020, 11, 561652. [Google Scholar] [CrossRef]
- Danese, S.; Vermeire, S.; Hellstern, P.; Panaccione, R.; Rogler, G.; Fraser, G.; Kohn, A.; Desreumaux, P.; Leong, R.W.; Comer, G.M.; et al. Randomised trial and open-label extension study of an anti-interleukin-6 antibody in Crohn’s disease (ANDANTE I and II). Gut 2019, 68, 40–48. [Google Scholar] [CrossRef]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tollefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damas, J.K.; et al. Randomized trial of interleukin-6 receptor inhibition in patients with acute st-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef]
- Nuñez, P.; García Mateo, S.; Quera, R.; Gomollón, F. Inflammatory bowel disease and the risk of cardiovascular diseases. Gastroenterol. Y Hepatol. 2021, 44, 236–242. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weight Loss * | Stool Consistency | Blood in Stools | |||
|---|---|---|---|---|---|
| Observation | Score | Observation | Score | Observation | Score |
| ≤2% | 0 | Normal | 0 | No blood | 0 |
| 3–6% | 1 | Semi-solid | 1 | Gross blood in stools | 2 |
| 7–12% | 2 | Loose to pasty | 2 | Rectal bleeding | 4 |
| >12% | 3 | Diarrhea | 3 | ||
| Inflammatory Cell Infiltration | Intestinal Architecture | ||
|---|---|---|---|
| SCORE | 0 | Rare inflammatory cells in the lamina propria | Absence of lesions on the mucosa |
| 1 | Increased number of inflammatory cells, including neutrophils in the lamina propria | Discrete focal lymphoepithelial lesions | |
| 2 | Confluence of inflammatory cells, with extension to the submucosa | Mucosal erosion/ulceration | |
| 3 | Transmural extension of inflammatory cell infiltrate | Extensive mucosal damage and extension to deep structures of the intestinal wall |
| Heart Rate (bpm) | |||||||
|---|---|---|---|---|---|---|---|
| Groups | n | BW (g) | LVW/BW (%) | AAR/LV (%) | Before Ischemia | In Ischemia | In Reperfusion |
| Ctrl | 8 | 27.9 ± 1.5 | 3.3 ± 0.9 | 33.8 ± 1.8 | 468 ± 14 | 461 ± 21 | 464 ± 16 |
| DSS | 7 | 27.0 ± 0.9 | 3.8 ± 0.5 | 29.4 ± 2.7 | 435 ± 21 | 488 ± 21 | 533 ± 16 |
| Ca | 5 | 28.3 ± 0.6 | 3.5 ± 0.04 | 29.9 ± 3.2 | 440 ± 20 | 478 ± 11 | 531 ± 20 |
| DSS + Ca | 5 | 28.5 ± 0.5 | 3.3 ± 0.11 | 32.2 ± 0.2 | 441 ± 41 | 517 ± 30 | 550 ± 19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mami, W.; Znaidi-Marzouki, S.; Doghri, R.; Ben Ahmed, M.; Znaidi, S.; Messadi, E. Inflammatory Bowel Disease Increases the Severity of Myocardial Infarction after Acute Ischemia–Reperfusion Injury in Mice. Biomedicines 2023, 11, 2945. https://doi.org/10.3390/biomedicines11112945
Mami W, Znaidi-Marzouki S, Doghri R, Ben Ahmed M, Znaidi S, Messadi E. Inflammatory Bowel Disease Increases the Severity of Myocardial Infarction after Acute Ischemia–Reperfusion Injury in Mice. Biomedicines. 2023; 11(11):2945. https://doi.org/10.3390/biomedicines11112945
Chicago/Turabian StyleMami, Wael, Soumaya Znaidi-Marzouki, Raoudha Doghri, Melika Ben Ahmed, Sadri Znaidi, and Erij Messadi. 2023. "Inflammatory Bowel Disease Increases the Severity of Myocardial Infarction after Acute Ischemia–Reperfusion Injury in Mice" Biomedicines 11, no. 11: 2945. https://doi.org/10.3390/biomedicines11112945