
Nintedanib Reduces Muscle Fibrosis and Improves Muscle Function of the Alpha-Sarcoglycan-Deficient Mice
 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Model
2.2. In Vivo Muscle Function
2.3. Echocardiography
2.4. Histology and Immunofluorescence
2.5. Fast Green-Sirius Red
2.6. Cytokine and Chemokine Arrays
2.7. Gene Expression Profiling
2.8. Statistical Analysis
3. Results
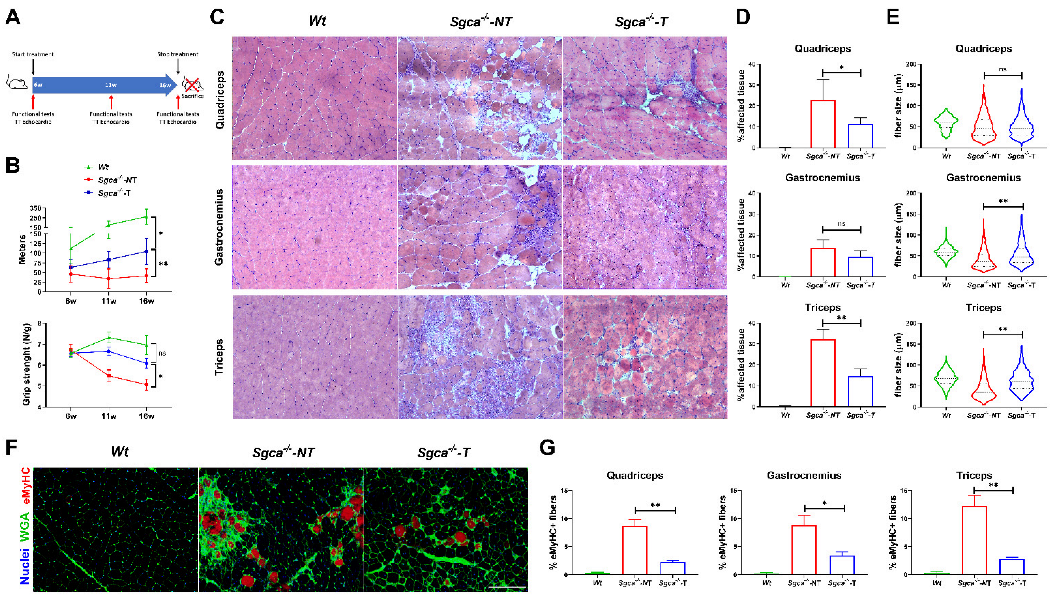
3.1. Nintedanib Improves Muscle Endurance and Strength in Sgca-/- Mice

3.2. Effects of Nintedanib on Cardiac Function and Ventricular Remodeling in Wt and Sgca-/- Mice
3.3. Nintedanib Improves Skeletal Muscle Architecture
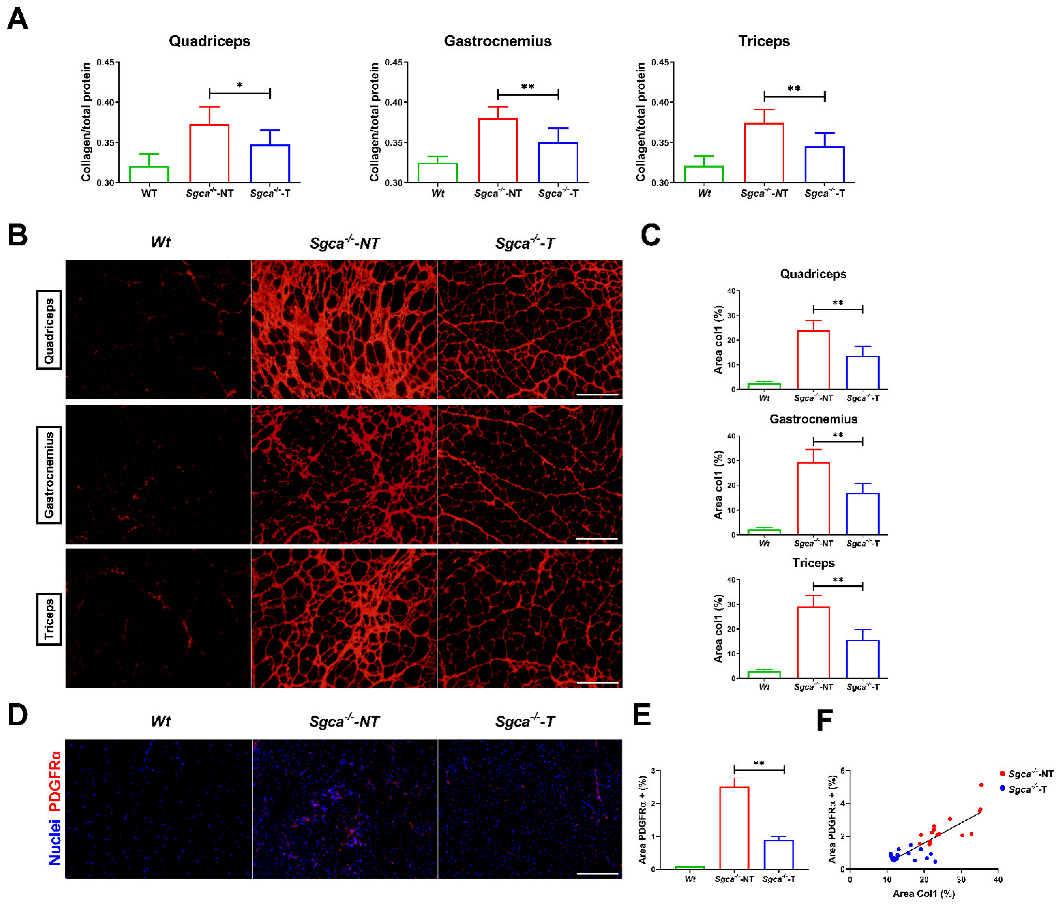
3.4. Nintedanib Reduces Muscle Fibrosis in Sgca-/- Mice
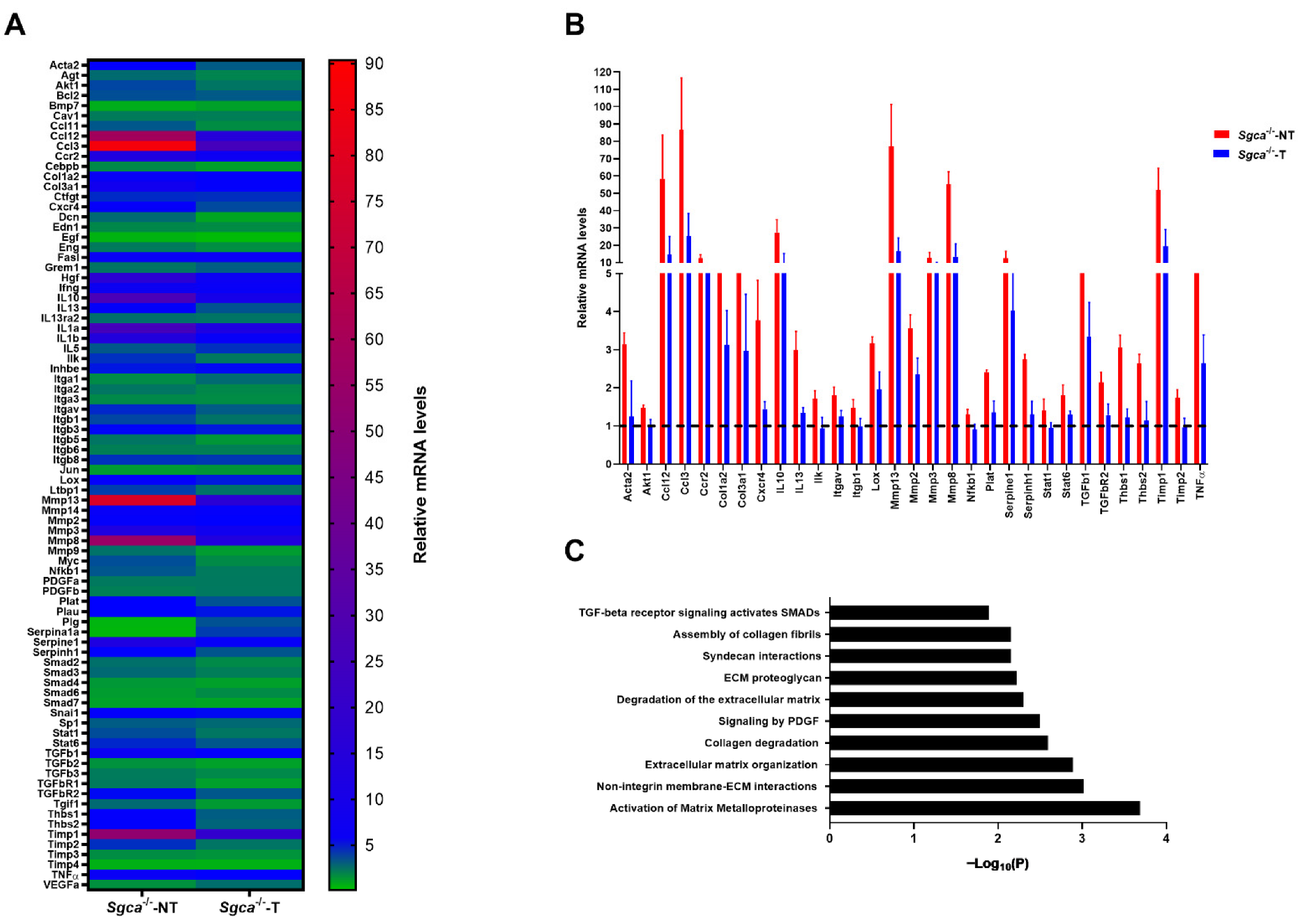
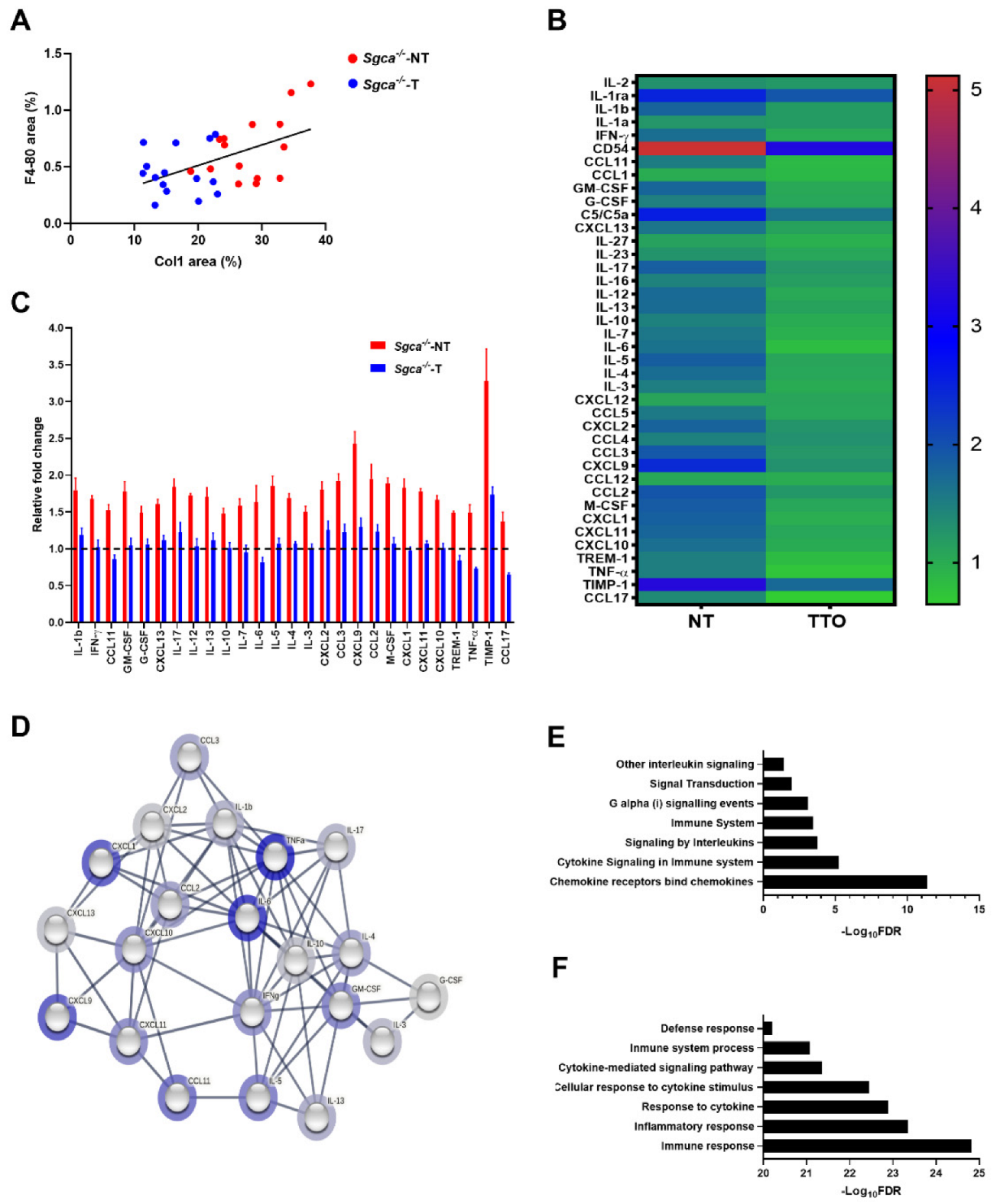
3.5. Nintedanib Modulates Chronic Muscle Inflammation in the Sgca-/- Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclosures
References
- Straub, V.; Murphy, A.; Udd, B. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainzof, M.; Passos-Bueno, M.R.; Pavanello, R.C.M.; Marie, S.K.; Oliveira, A.S.B.; Zatz, M. Sarcoglycanopathies are responsible for 68% of severe autosomal recessive limb-girdle muscular dystrophy in the Brazilian population. J. Neurol. Sci. 1999, 164, 44–49. [Google Scholar] [CrossRef]
- Winckler, P.B.; da Silva, A.M.S.; Coimbra-Neto, A.R.; Carvalho, E.; Cavalcanti, E.B.U.; Sobreira, C.F.R.; Marrone, C.D.; Machado-Costa, M.C.; Carvalho, A.A.S.; Feio, R.H.F.; et al. Clinicogenetic lessons from 370 patients with autosomal recessive limb-girdle muscular dystrophy. Clin. Genet. 2019, 96, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Pérez, J.; González-Quereda, L.; Bello, L.; Guglieri, M.; Straub, V.; Gallano, P.; Semplicini, C.; Pegoraro, E.; Zangaro, V.; Nascimento, A.; et al. New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain 2020, 143, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Pérez, J.; González-Quereda, L.; Bruno, C.; Panicucci, C.; Alavi, A.; Nafissi, S.; Nilipour, Y.; Zanoteli, E.; Isihi, L.M.D.A.; Melegh, B.; et al. Clinical and genetic spectrum of a large cohort of patients with δ-sarcoglycan muscular dystrophy. Brain 2022, 145, 596–606. [Google Scholar] [CrossRef]
- ten Dam, L.; Frankhuizen, W.S.; Linssen, W.H.J.P.; Straathof, C.S.; Niks, E.H.; Faber, K.; Fock, A.; Kuks, J.B.; Brusse, E.; de Coo, R.; et al. Autosomal recessive limb-girdle and Miyoshi muscular dystrophies in the Netherlands: The clinical and molecular spectrum of 244 patients. Clin. Genet. 2019, 96, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Hou, Y.; Yu, M.; Liu, Y.; Fan, Y.; Zhang, W.; Wang, Z.; Xiong, H.; Yuan, Y. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J. Rare Dis. 2019, 14, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginjaar, H.B.; Van Der Kooi, A.J.; Ceelie, H.; Kneppers, A.L.J.; Van Meegen, M.; Barth, P.G.; Busch, H.F.M.; Wokke, J.H.J.; Anderson, L.V.B.; Bönnemann, C.G.; et al. Sarcoglycanopathies in Dutch patients with autosomal recessive limb girdle muscular dystrophy. J. Neurol. 2000, 247, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Alavi, A.; Esmaeili, S.; Nilipour, Y.; Nafissi, S.; Tonekaboni, S.H.; Zamani, G.; Ashrafi, M.R.; Kahrizi, K.; Najmabadi, H.; Jazayeri, F. LGMD2E is the most common type of sarcoglycanopathies in the Iranian population. J. Neurogenet. 2017, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef]
- Chan, Y.M.; Bönnemann, C.G.; Lidov, H.G.W.; Kunkel, L.M. Molecular organization of sarcoglycan complex in mouse myotubes in culture. J. Cell Biol. 1998, 143, 2033–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarakci, H.; Berger, J. The sarcoglycan complex in skeletal muscle. Front. Biosci.-Landmark 2016, 21, 744–756. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, E.; Mizuno, Y.; Hagiwara, Y.; Sasaoka, T.; Yoshida, M. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 2005, 32, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.E.; Campbell, K.P. The sarcoglycan complex in limb–girdle muscular dystrophy. Curr. Opin. Neurol. 1998, 11, 443–452. [Google Scholar] [CrossRef]
- Wallace, G.Q.; McNally, E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009, 71, 37–57. [Google Scholar] [CrossRef]
- Serrano, A.L.; Muñoz-Cánoves, P. Regulation and dysregulation of fibrosis in skeletal muscle. Exp. Cell Res. 2010, 316, 3050–3058. [Google Scholar] [CrossRef]
- Serrano, A.L.; Muñoz-Cánoves, P. Fibrosis development in early-onset muscular dystrophies: Mechanisms and translational implications. Semin. Cell Dev. Biol. 2017, 64, 181–190. [Google Scholar] [CrossRef]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef]
- Molina, T.; Fabre, P.; Dumont, N.A. Fibro-adipogenic progenitors in skeletal muscle homeostasis, regeneration and diseases. Open Biol. 2021, 11, 210110. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Barton, E.R. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018, 68–69, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Abrigo, J.; Simon, F.; Cabrera, D.; Cordova, G.; Trollet, C.; Cabello-Verrugio, C. Central Role of Transforming Growth Factor Type Beta 1 in Skeletal Muscle Dysfunctions: An Update on Therapeutic Strategies. Curr. Protein Pept. Sci. 2017, 19, 1189–1200. [Google Scholar] [CrossRef]
- Xu, D.; Li, S.; Wang, L.; Jiang, J.; Zhao, L.; Huang, X.; Sun, Z.; Li, C.; Sun, L.; Li, X.; et al. TAK1 inhibition improves myoblast differentiation and alleviates fibrosis in a mouse model of Duchenne muscular dystrophy. J. Cachexia. Sarcopenia Muscle 2021, 12, 192–208. [Google Scholar] [CrossRef]
- Fernández-Simón, E.; Suárez-Calvet, X.; Carrasco-Rozas, A.; Piñol-Jurado, P.; López-Fernández, S.; Pons, G.; Bech Serra, J.J.; de la Torre, C.; de Luna, N.; Gallardo, E.; et al. RhoA/ROCK2 signalling is enhanced by PDGF-AA in fibro-adipogenic progenitor cells: Implications for Duchenne muscular dystrophy. J. Cachexia. Sarcopenia Muscle 2022, 13, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Piñol-Jurado, P.; Suárez-Calvet, X.; Fernández-Simón, E.; Gallardo, E.; De La Oliva, N.; Martínez-Muriana, A.; Gómez-Gálvez, P.; Escudero, L.M.; Pérez-Peiró, M.; Wollin, L.; et al. Nintedanib decreases muscle fibrosis and improves muscle function in a murine model of dystrophinopathy. Cell Death Dis. 2018, 9, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Górecki, D.C. P2X7 purinoceptor as a therapeutic target in muscular dystrophies. Curr. Opin. Pharmacol. 2019, 47, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Xin, C.; Chu, X.; Wei, W.; Kuang, B.; Wang, Y.; Tang, Y.; Chen, J.; You, H.; Li, C.; Wang, B. Combined gene therapy via VEGF and mini-dystrophin synergistically improves pathologies in temporalis muscle of dystrophin/utrophin double knockout mice. Hum. Mol. Genet. 2021, 30, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; Fallon, K.S.; Oosterbaan, C.C.; Vaught, L.A.; Reiser, N.L.; Bogdanovic, E.; Velez, M.P.; Salamone, I.M.; Page, P.G.T.; Hadhazy, M.; et al. Anti-latent TGFβ binding protein 4 antibody improves muscle function and reduces muscle fibrosis in muscular dystrophy. Sci. Transl. Med. 2021, 13, eabf0376. [Google Scholar] [CrossRef]
- Hamoudi, D.; Marcadet, L.; Piette Boulanger, A.; Yagita, H.; Bouredji, Z.; Argaw, A.; Frenette, J. An anti-RANKL treatment reduces muscle inflammation and dysfunction and strengthens bone in dystrophic mice. Hum. Mol. Genet. 2019, 28, 3101–3112. [Google Scholar] [CrossRef] [PubMed]
- Tulangekar, A.; Sztal, T.E. Inflammation in duchenne muscular dystrophy–exploring the role of neutrophils in muscle damage and regeneration. Biomedicines 2021, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Gazzerro, E.; Baldassari, S.; Assereto, S.; Fruscione, F.; Pistorio, A.; Panicucci, C.; Volpi, S.; Perruzza, L.; Fiorillo, C.; Minetti, C.; et al. Enhancement of muscle T regulatory cells and improvement of muscular dystrophic process in mdx mice by blockade of extracellular ATP/P2X axis. Am. J. Pathol. 2015, 185, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.M.; Salhany, R.M.; Deighton, A.; Harwood, M.; Mah, J.; Gooch, K.L. The clinical course of Duchenne muscular dystrophy in the corticosteroid treatment era: A systematic literature review. Orphanet J. Rare Dis. 2021, 16, 237. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Principi, E.; Baratto, S.; Panicucci, C.; Pintus, S.; Antonini, F.; Del Zotto, G.; Benzi, A.; Bruzzone, S.; Scudieri, P.; et al. P2X7 Receptor Antagonist Reduces Fibrosis and Inflammation in a Mouse Model of Alpha-Sarcoglycan Muscular Dystrophy. Pharmaceuticals 2022, 15, 89. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zhao, X.S.; Fields, M.; Ransohoff, R.M.; Zhou, L. Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J. 2009, 23, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Ieronimakis, N.; Hays, A.; Prasad, A.; Janebodin, K.; Duffield, J.S.; Reyes, M. PDGFRα signalling promotes fibrogenic responses in collagen-producing cells in Duchenne muscular dystrophy. J. Pathol. 2016, 240, 410–424. [Google Scholar] [CrossRef] [Green Version]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostettler, K.E.; Zhong, J.; Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Seidel, P.; Sun, Q.; Mandal, J.; Lardinois, D.; Lambers, C.; et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir. Res. 2014, 15, 157. [Google Scholar] [CrossRef]
- Huang, J.; Beyer, C.; Palumbo-Zerr, K.; Zhang, Y.; Ramming, A.; Distler, A.; Gelse, K.; Distler, O.; Schett, G.; Wollin, L.; et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann. Rheum. Dis. 2016, 75, 883–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [Green Version]
- Yoon, H.Y.; Park, S.; Kim, D.S.; Song, J.W. Efficacy and safety of nintedanib in advanced idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richeldi, L.; Kolb, M.; Jouneau, S.; Wuyts, W.A.; Schinzel, B.; Stowasser, S.; Quaresma, M.; Raghu, G. Efficacy and safety of nintedanib in patients with advanced idiopathic pulmonary fibrosis. BMC Pulm. Med. 2020, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Kobuke, K.; Piccolo, F.; Garringer, K.W.; Moore, S.A.; Sweezer, E.; Yang, B.; Campbell, K.P. A common disease-associated missense mutation in alpha-sarcoglycan fails to cause muscular dystrophy in mice. Hum. Mol. Genet. 2008, 17, 1201–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Maier, C.; Zhang, Y.; Soare, A.; Dees, C.; Beyer, C.; Harre, U.; Chen, C.W.; Distler, O.; Schett, G.; et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1941–1948. [Google Scholar] [CrossRef]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press (US): Washington, DC, USA, 2011; ISBN 978-0-309-15400-0. [Google Scholar]
- Castro, B.; Kuang, S. Evaluation of Muscle Performance in Mice by Treadmill Exhaustion Test and Whole-limb Grip Strength Assay. Bio-Protocol 2017, 7, e2237. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; van Putten, M. Assessing functional performance in the Mdx mouse model. J. Vis. Exp. 2014, 85, e51303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasteuning-Vuhman, S.; Putker, K.; Tanganyika-De Winter, C.L.; Boertje-Van Der Meulen, J.W.; Van Vliet, L.; Overzier, M.; Plomp, J.J.; Aartsma-Rus, A.; Van Putten, M. Natural disease history of mouse models for limb girdle muscular dystrophy types 2D and 2F. PLoS ONE 2017, 12, e182704. [Google Scholar] [CrossRef] [Green Version]
- Verhaart, I.E.C.; Putker, K.; van de Vijver, D.; Tanganyika-De Winter, C.L.; Pasteuning-Vuhman, S.; Plomp, J.J.; Aartsma-Rus, A.M.; Van Putten, M. Cross-sectional study into age-related pathology of mouse models for limb girdle muscular dystrophy types 2D and 2F. PLoS ONE 2019, 14, e220665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carrera, I.; Frise, E.; Verena, K.; Mark, L.; Tobias, P.; Stephan, P.; Curtis, R.; Stephan, S.; Benjamin, S.; et al. Fiji—An Open platform for biological image analysis. Nat. Methods 2009, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Houghton, P.E.; Keeper, K.A.; Diegelmann, R.; Krummel, T.M. A simple method to assess the relative amount of collagen deposition in wounded fetal mouse limbs. Wound Repair Regen. 1996, 4, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Ardite, E.; Perdiguero, E.; Vidal, B.; Gutarra, S.; Serrano, A.L.; Muñoz-Cánoves, P. PAI-1-regulated miR-21 defines a novel age-associated fibrogenic pathway in muscular dystrophy. J. Cell Biol. 2012, 196, 163–175. [Google Scholar] [CrossRef]
- von Mering, C.; Jensen, L.J.; Kuhn, M.; Chaffron, S.; Doerks, T.; Krüger, B.; Snel, B.; Bork, P. STRING 7—Recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007, 35, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Chicoine, L.G.; Al-Zaidy, S.A.; Sahenk, Z.; Lehman, K.; Lowes, L.; Miller, N.; Alfano, L.; Galliers, B.; Lewis, S.; et al. Gene Delivery for Limb-Girdle Muscular Dystrophy Type 2D by Isolated Limb Infusion. Hum. Gene Ther. 2019, 30, 794–801. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-klapac, L.R.; Rosales-quintero, X.; Kota, J.; Coley, B.D.; Galloway, G.; Josepha, M.; Lewis, S.; Malik, V.; Shilling, C.; et al. LGMD 2D gene therapy restores alpha-sarcoglycan and associated proteins. Ann. Neurol. 2018, 66, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.A.; Pozsgai, E.R.; Heller, K.N.; Potter, R.A.; Peterson, E.L.; Rodino-Klapac, L.R. Preclinical Systemic Delivery of Adeno-Associated a-Sarcoglycan Gene Transfer for Limb-Girdle Muscular Dystrophy. Hum. Gene Ther. 2021, 32, 390–404. [Google Scholar] [CrossRef]
- Joe, A.W.B.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. V Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Contreras, O.; Rossi, F.M.; Theret, M. Origins, Potency and Heterogeneity of Skeletal Muscle Fibro-Adipogenic Progenitors—Time for new definitions. Skelet. Muscle 2021, 11, 16. [Google Scholar] [CrossRef]
- Contreras, O.; Rebolledo, D.L.; Oyarzún, J.E.; Olguín, H.C.; Brandan, E. Connective tissue cells expressing fibro/adipogenic progenitor markers increase under chronic damage: Relevance in fibroblast-myofibroblast differentiation and skeletal muscle fibrosis. Cell Tissue Res. 2016, 364, 647–660. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Haginoya, K.; Chiba, Y.; Uematsu, M.; Hino-Fukuyo, N.; Tanaka, S.; Onuma, A.; Iinuma, K.; Tsuchiya, S. Elevated plasma levels of tissue inhibitors of metalloproteinase-1 and their overexpression in muscle in human and mouse muscular dystrophy. J. Neurol. Sci. 2010, 297, 19–28. [Google Scholar] [CrossRef]
- Giuliani, G.; Rosina, M.; Reggio, A. Signaling pathways regulating the fate of fibro/adipogenic progenitors (FAPs) in skeletal muscle regeneration and disease. FEBS J. 2021, 11, 16. [Google Scholar] [CrossRef]
- Xiao, W.; Liu, Y.; Luo, B.; Zhao, L.; Liu, X.; Zeng, Z.; Chen, P. Time-dependent gene expression analysis after mouse skeletal muscle contusion. J. Sport Health Sci. 2016, 5, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the Tyrosine Kinase inhibitor Nintedanib in Experimental Models of Lung Fibrosiss. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Hoogaars, W.M.H.; Mouisel, E.; Pasternack, A.; Hulmi, J.J.; Relizani, K.; Schuelke, M.; Schirwis, E.; Garcia, L.; Ritvos, O.; Ferry, A.; et al. Combined effect of AAV-U7-induced dystrophin exon skipping and soluble activin type IIB receptor in mdx mice. Hum. Gene Ther. 2012, 23, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yue, Y.; Li, L.; Hakim, C.H.; Zhang, K.; Thomas, G.D.; Duan, D. Dual AAV therapy ameliorates exercise-induced muscle injury and functional ischemia in murine models of duchenne muscular dystrophy. Hum. Mol. Genet. 2013, 22, 3720–3729. [Google Scholar] [CrossRef] [Green Version]
- Heller, K.N.; Mendell, J.T.; Mendell, J.R.; Rodino-Klapac, L.R. MicroRNA-29 overexpression by adeno-associated virus suppresses fibrosis and restores muscle function in combination with micro-dystrophin. JCI Insight 2017, 2, e93309. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Pérez, J.; Carrasco-Rozas, A.; Borrell-Pages, M.; Fernández-Simón, E.; Piñol-Jurado, P.; Badimon, L.; Wollin, L.; Lleixà, C.; Gallardo, E.; Olivé, M.; et al. Nintedanib Reduces Muscle Fibrosis and Improves Muscle Function of the Alpha-Sarcoglycan-Deficient Mice. Biomedicines 2022, 10, 2629. https://doi.org/10.3390/biomedicines10102629
Alonso-Pérez J, Carrasco-Rozas A, Borrell-Pages M, Fernández-Simón E, Piñol-Jurado P, Badimon L, Wollin L, Lleixà C, Gallardo E, Olivé M, et al. Nintedanib Reduces Muscle Fibrosis and Improves Muscle Function of the Alpha-Sarcoglycan-Deficient Mice. Biomedicines. 2022; 10(10):2629. https://doi.org/10.3390/biomedicines10102629
Chicago/Turabian StyleAlonso-Pérez, Jorge, Ana Carrasco-Rozas, Maria Borrell-Pages, Esther Fernández-Simón, Patricia Piñol-Jurado, Lina Badimon, Lutz Wollin, Cinta Lleixà, Eduard Gallardo, Montse Olivé, and et al. 2022. "Nintedanib Reduces Muscle Fibrosis and Improves Muscle Function of the Alpha-Sarcoglycan-Deficient Mice" Biomedicines 10, no. 10: 2629. https://doi.org/10.3390/biomedicines10102629