
Understanding the Role of GLUT2 in Dysglycemia Associated with Fanconi–Bickel Syndrome
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
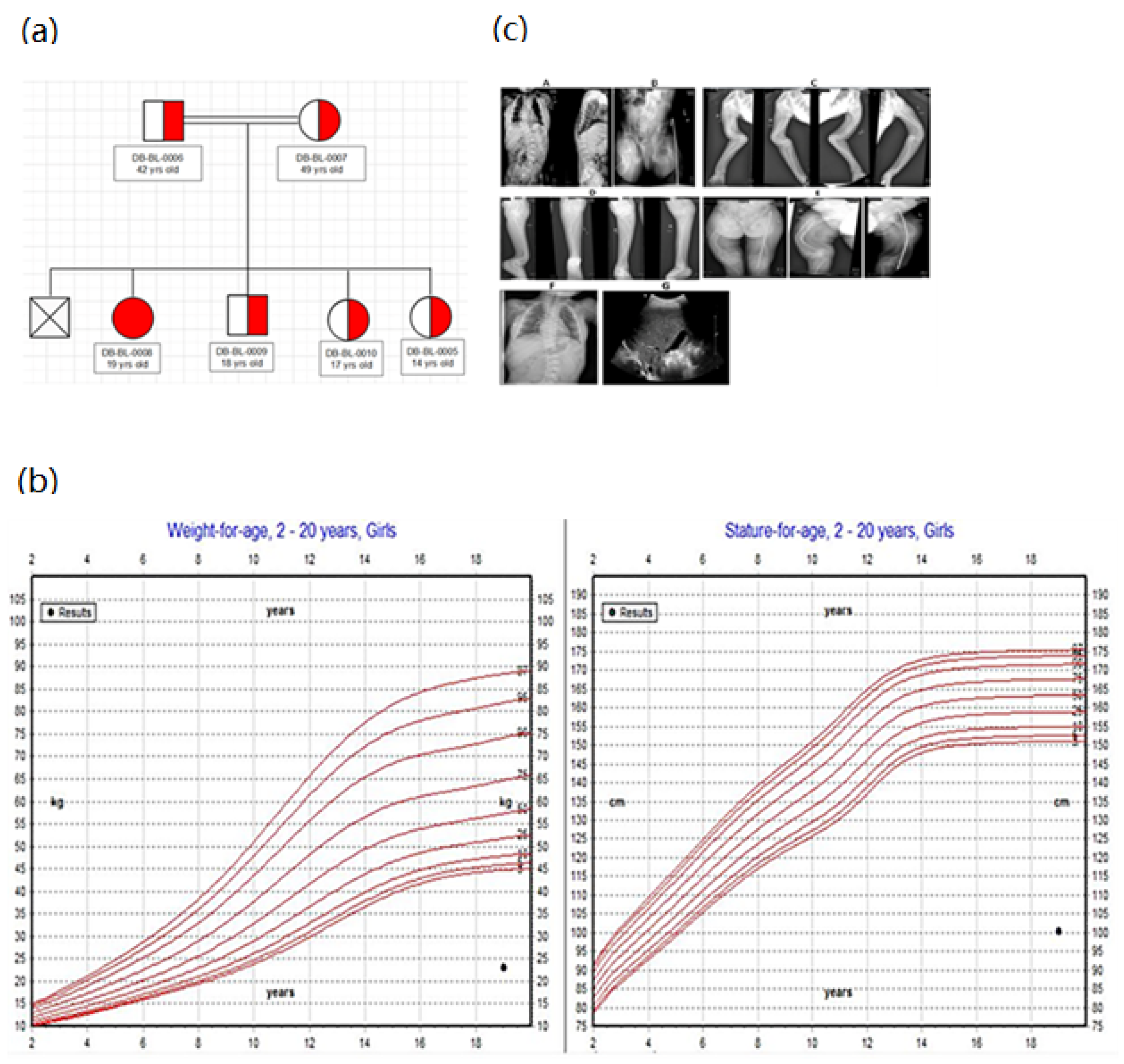
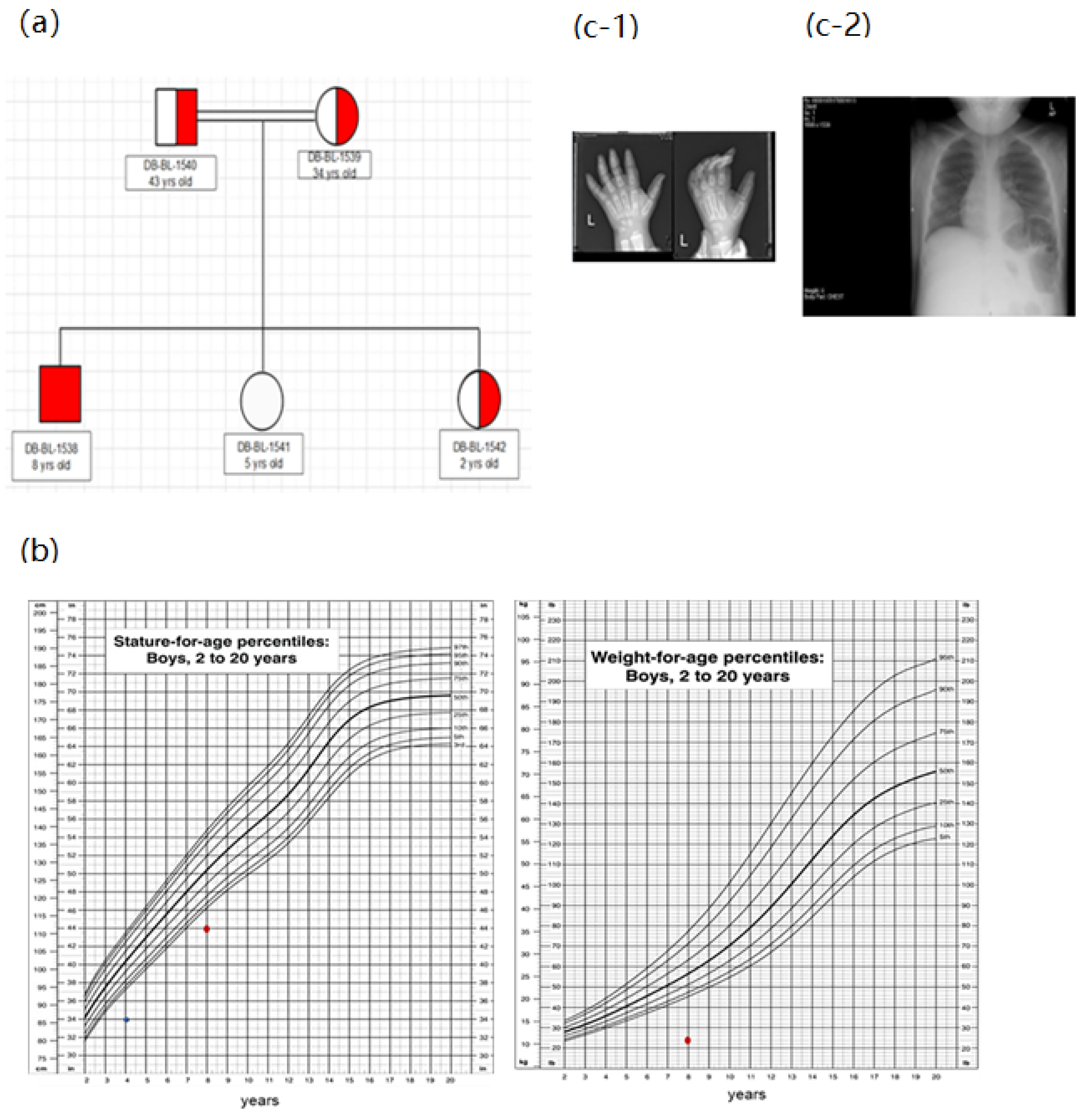
2.1. Patient Recruitment and Clinical Information
2.2. DNA, miRNA, RNA, and PBMCs Isolation
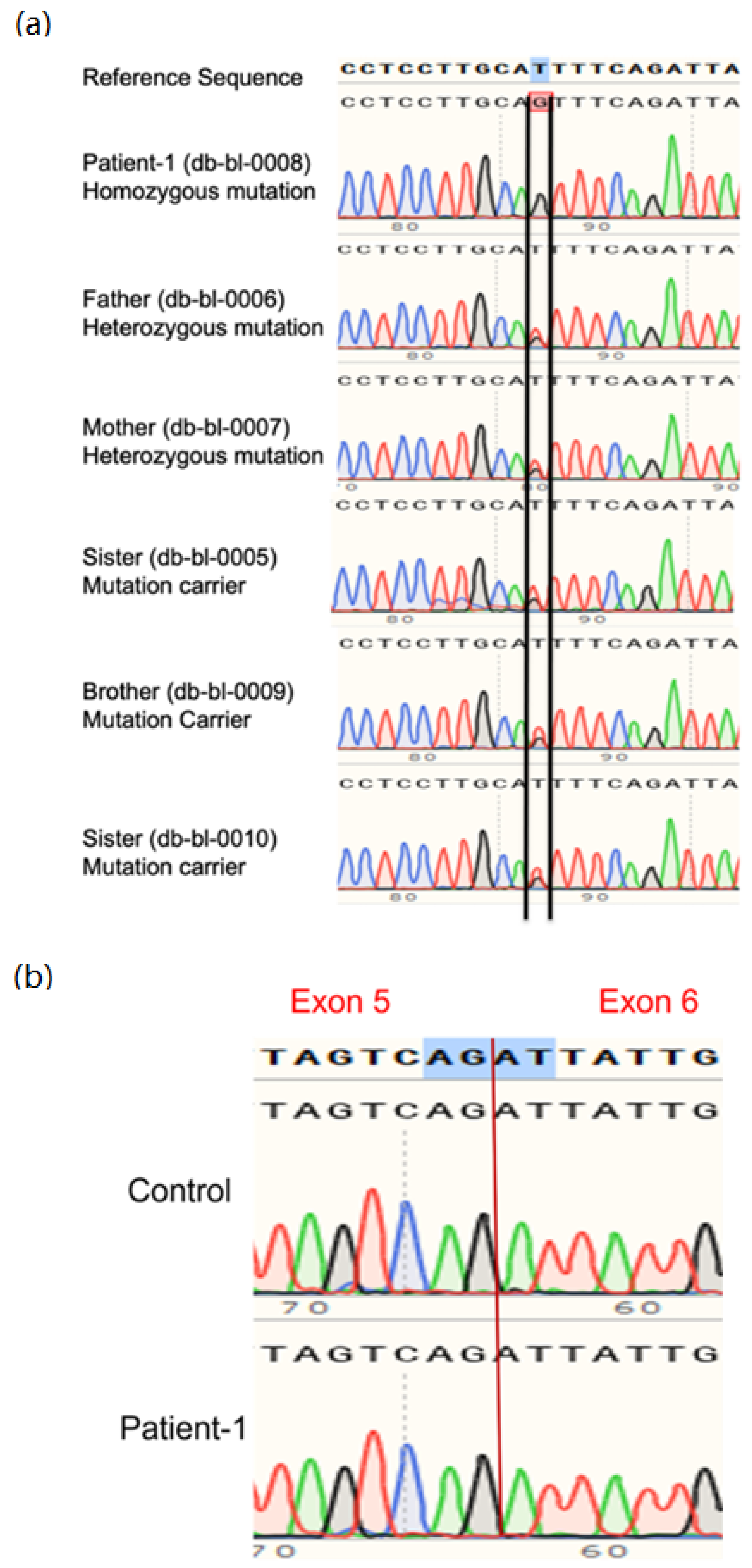
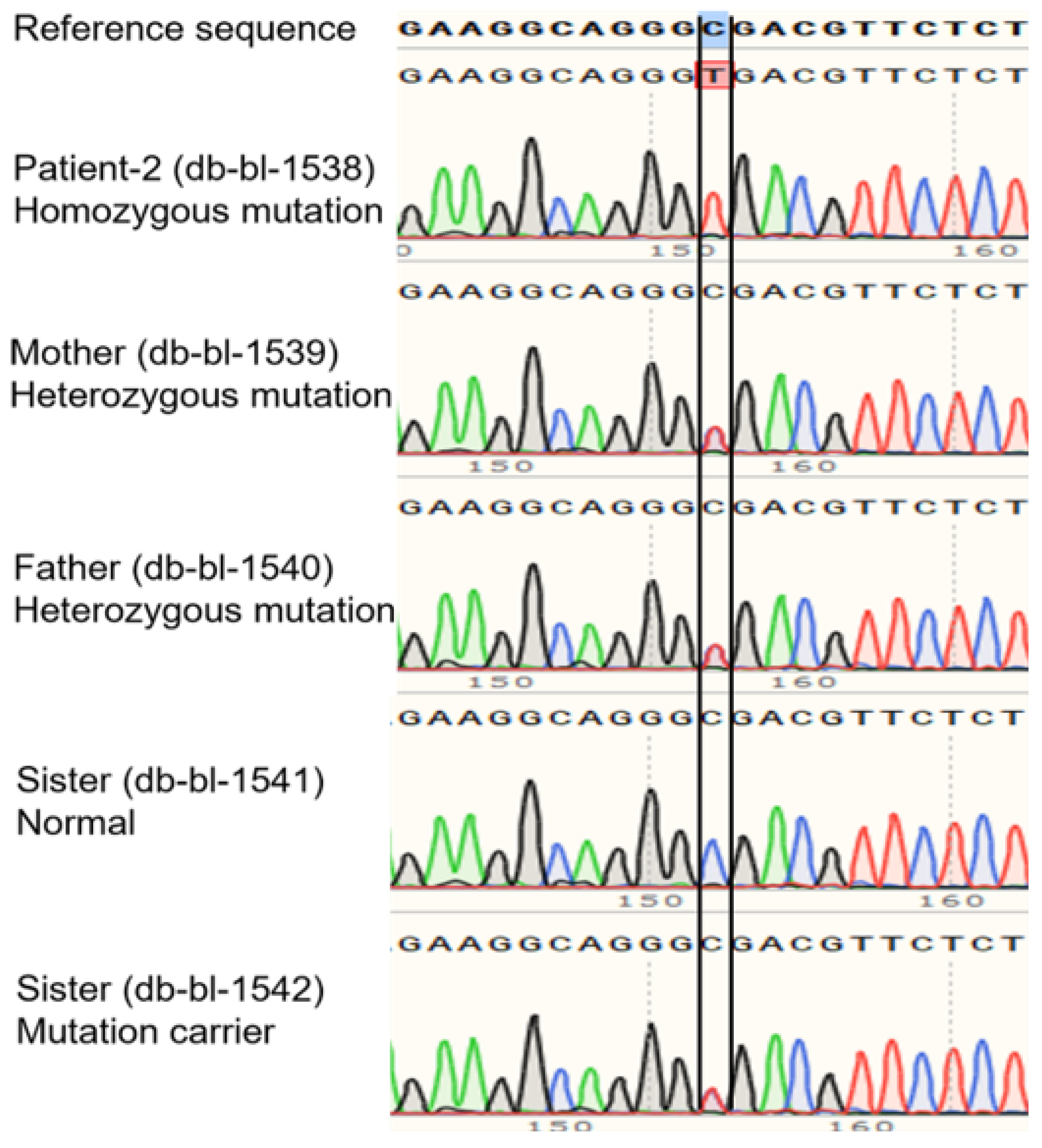
2.3. Whole-Genome and Sanger Sequencing
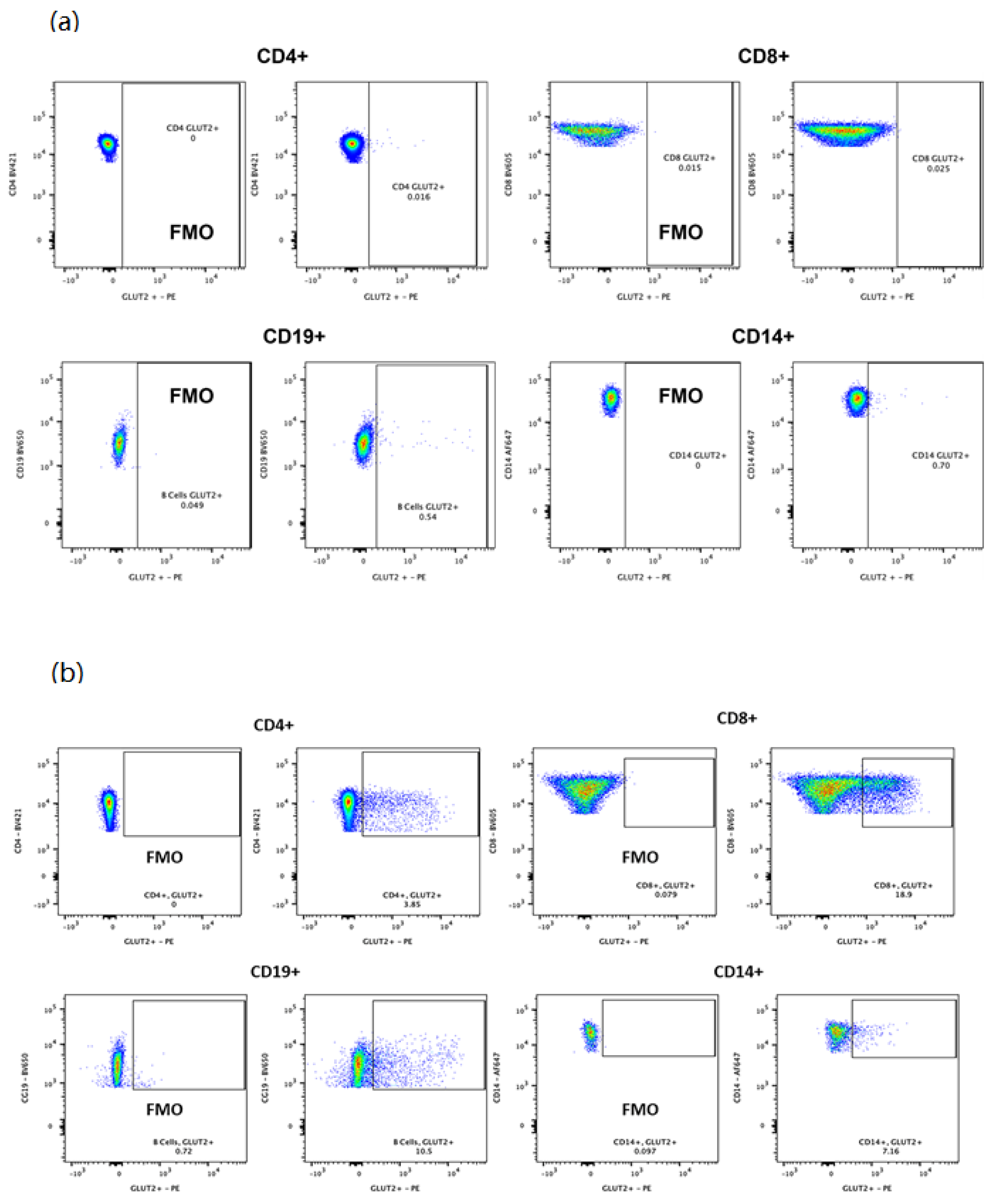
2.4. Flow Cytometry
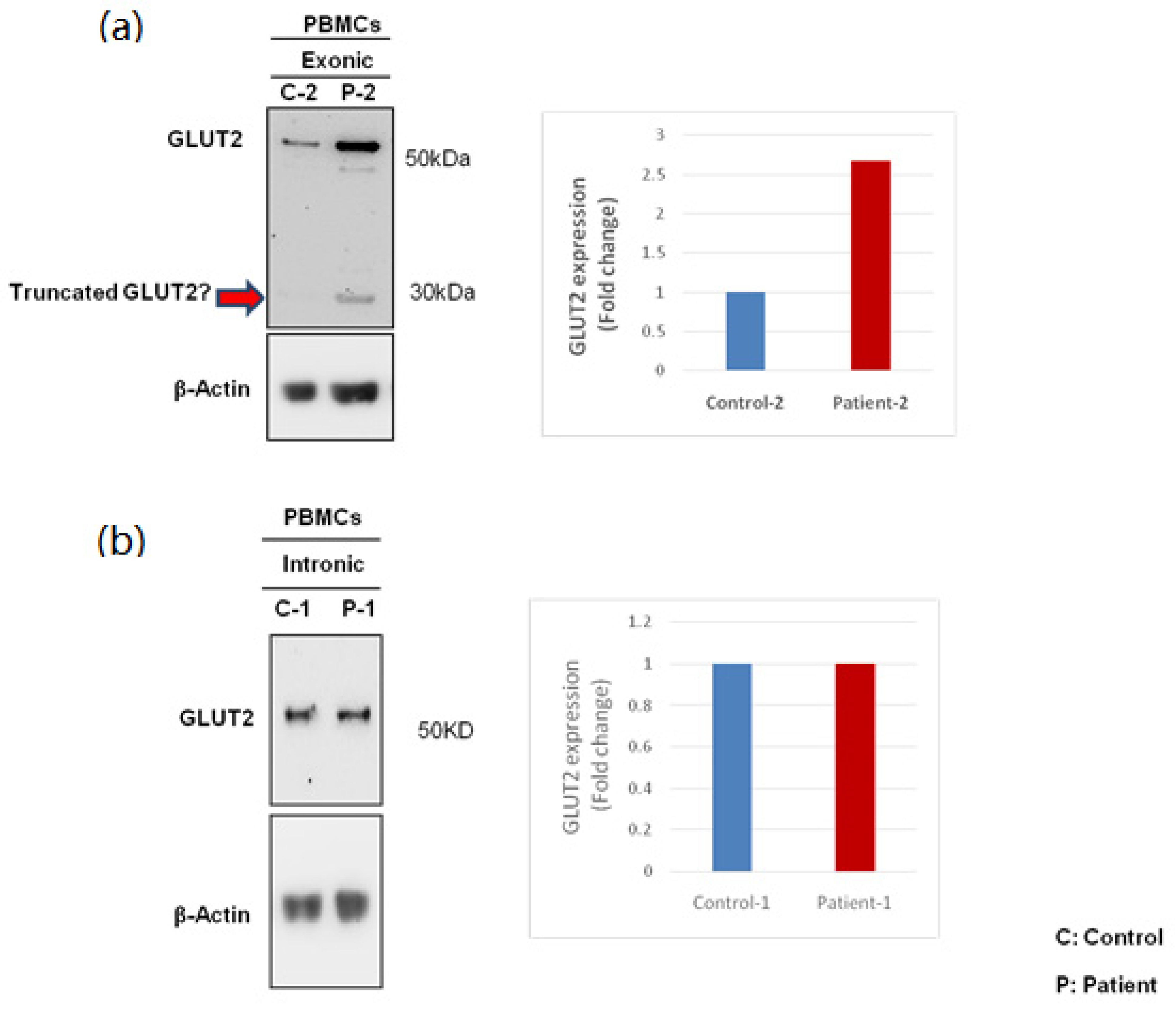
2.5. Western Blot
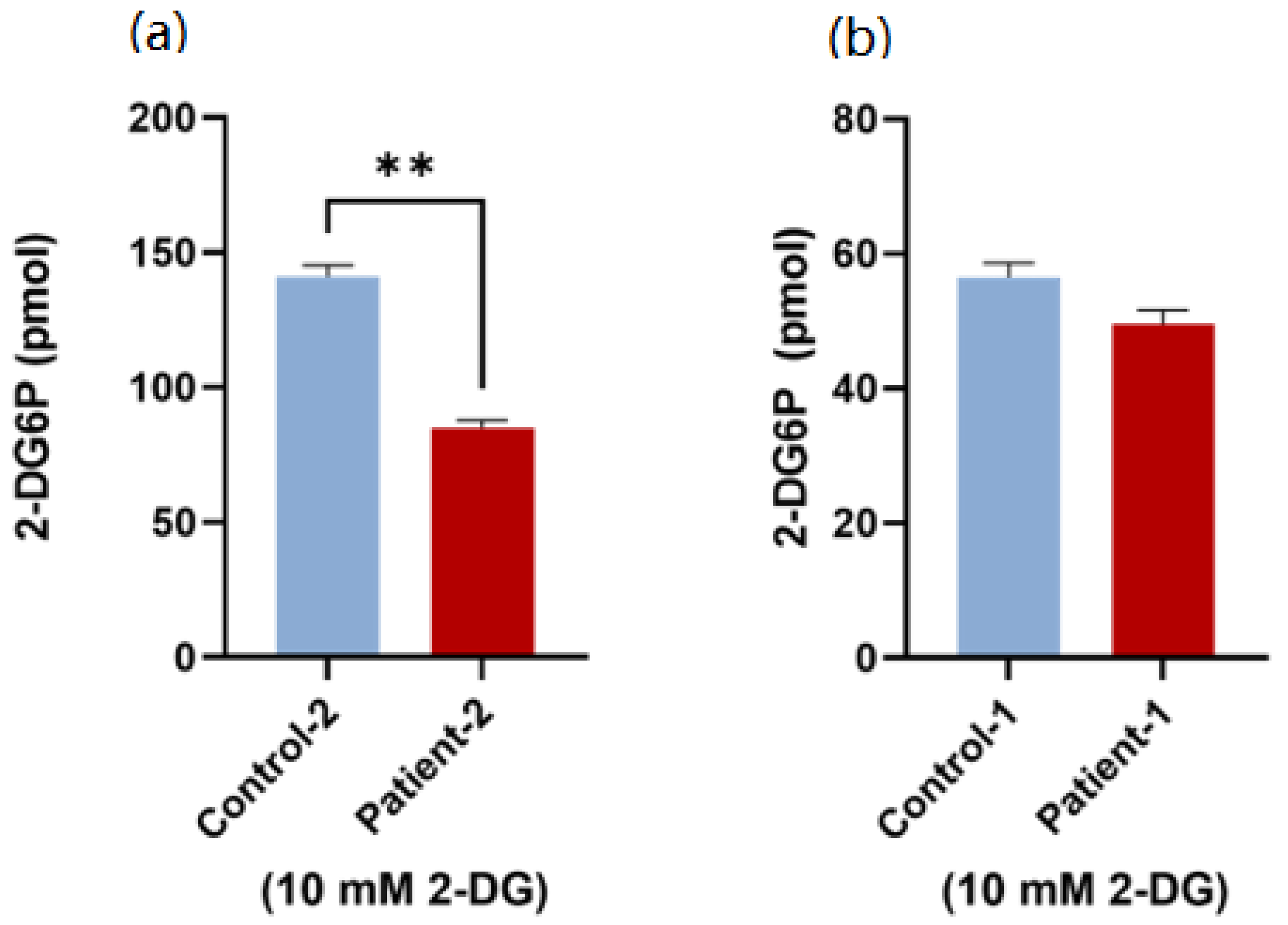
2.6. Glucose Uptake Assay
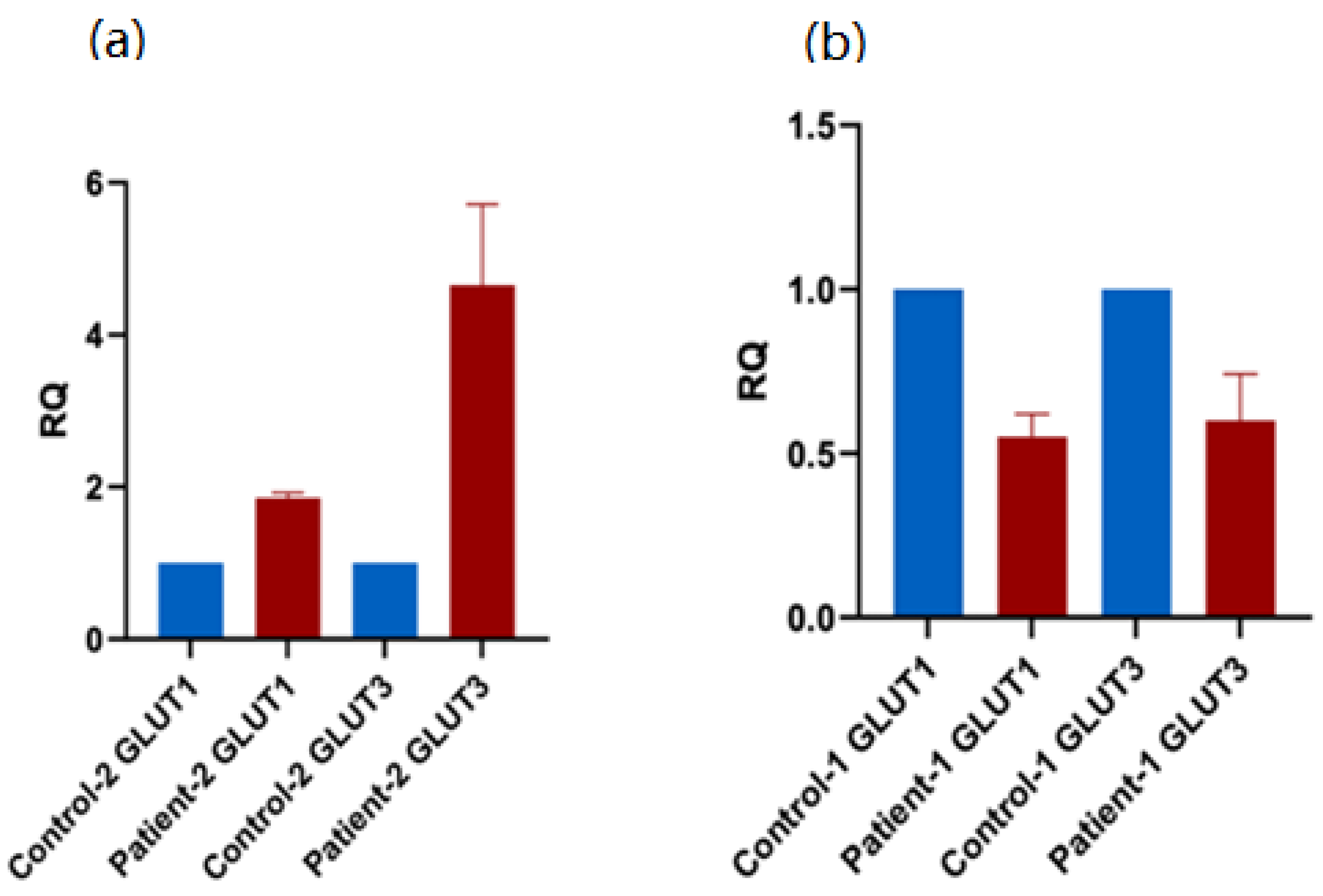
2.7. qRT-PCR
2.8. RNASeq Analysis
2.9. miRNA Analysis
2.10. Statistical Analysis
3. Results
3.1. Clinical and Genetic Information of Patients
3.2. GLUT2 Expression in PBMCs
3.3. Expression of GLUT2 in Patient and Control PBMCs
3.4. Glucose Uptake Activity Using Patient PBMCs
3.5. Expression of of Alternative Glucose Transporters in Patient PBMCs
3.6. RNA-Seq Analysis of Patient PBMCs
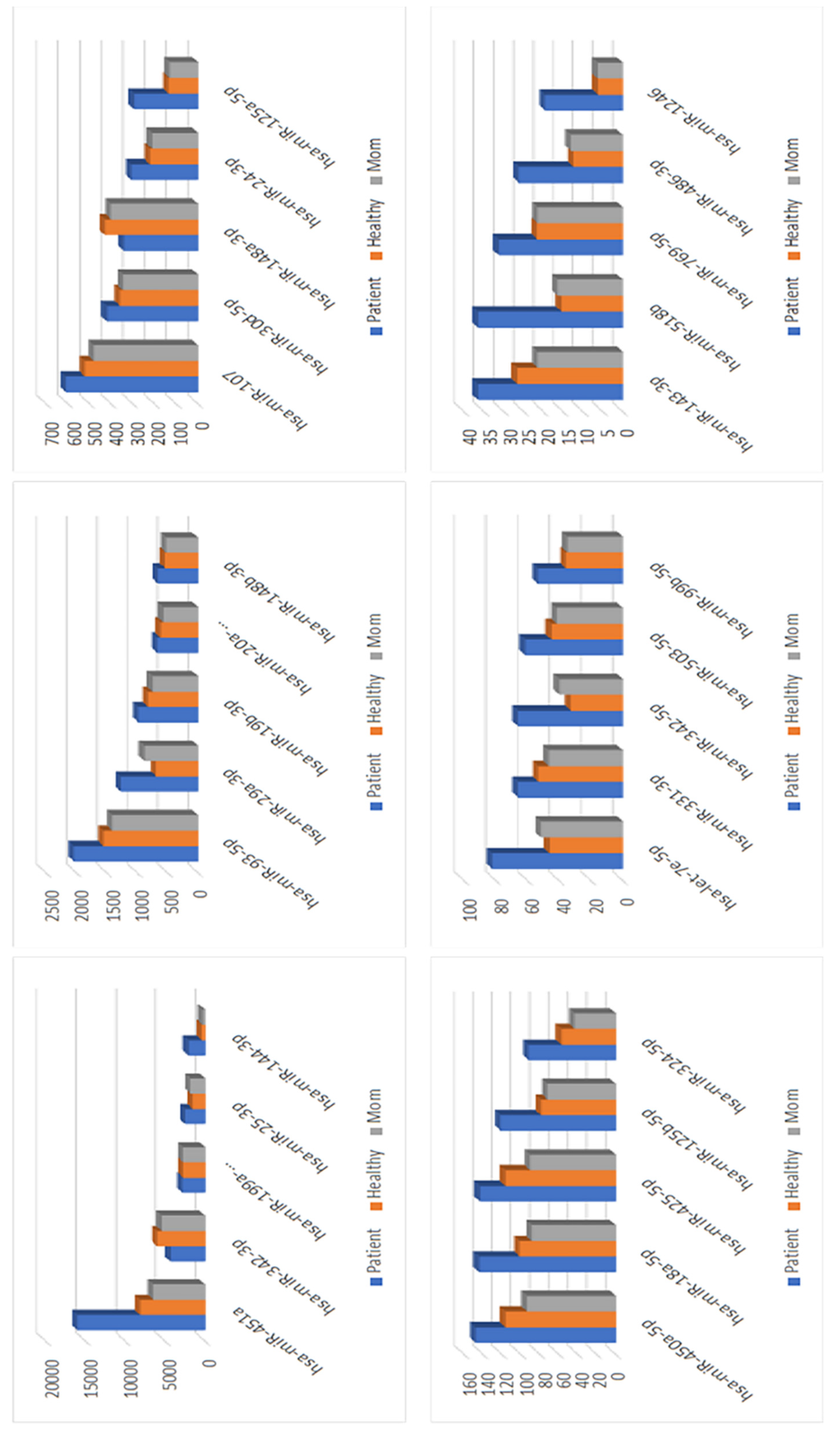
3.7. miRNA Analysis for the Patient with an Intronic Mutation
3.8. Expressions of Genes Regulated by miR-29a-3b
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fanconi, G.; Bickel, H. Die chronische Aminoacidurie (Aminosäurediabetes oder nephrotisch-glukosurischer Zwergwuchs) bei der Glykogenose und der Cystinkrankheit. Helv. Paediatr. Acta 1949, 4, 359–396. [Google Scholar] [PubMed]
- Santer, R.; Schneppenheim, R.; Dombrowski, A.; Götze, H.; Steinmann, B.; Schaub, J. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nat. Genet. 1997, 17, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Sharari, S.; Abou-Alloul, M.; Hussain, K.; Khan, F.A. Fanconi–bickel syndrome: A review of the mechanisms that lead to dysglycaemia. Int. J. Mol. Sci. 2020, 21, 6286. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.-J.; Jiang, M.-L.; Du, L.-N.; Yuan, L.; Xie, X.-L. Fanconi-Bickel syndrome in an infant with cytomegalovirus infection: A case report and review of the literature. World J. Clin. Cases 2020, 8, 5467–5473. [Google Scholar] [CrossRef] [PubMed]
- Eghbali, M.; Fatemi, K.S.; Salehpour, S.; Abiri, M.; Saei, H.; Talebi, S.; Olyaei, N.A.; Yassaee, V.R.; Modarressi, M.H. Whole-Exome Sequencing Uncovers Novel Causative Variants and Additional Findings in Three Patients Affected by Glycogen Storage Disease Type VI and Fanconi-Bickel Syndrome. Front. Genet. 2021, 11, 601566. [Google Scholar] [CrossRef]
- Musa, S.A.; Ibrahim, A.A.; Hassan, S.S.; Johnson, M.B.; Basheer, A.T.; Arabi, A.M.; Abdullah, M.A. Fanconi Bickel syndrome: Clinical phenotypes and genetics in a cohort of Sudanese children. Int. J. Pediatr. Endocrinol. 2020, 2020, 21. [Google Scholar] [CrossRef]
- Ibrahim, M.N.; Laghari, T.M.; Riaz, M.; Khoso, Z.; Khan, Y.N.; Yasir, M.; Hanif, M.I.; Flanagan, S.E.; De Franco, E.; Raza, J. Monogenic diabetes in Pakistani infants and children: Challenges in a resource poor country. J. Pediatr. Endocrinol. Metab. 2021, 34, 1095–1103. [Google Scholar] [CrossRef]
- Ustkoyuncu, P.S.; Bastug, F.; Kiraz, A.; Erdogan, M.; Eren, E.; Yıldız, G. Tubulopathy and hepatomegaly in a 2-year-old boy: Answers. Pediatr. Nephrol. 2021, 36, 2083–2084. [Google Scholar] [CrossRef]
- Scully, K.J.; Wolfsdorf, J.; Dedekian, M. Acquired growth hormone deficiency in Fanconi-Bickel syndrome. BMJ Case Rep. 2021, 14, e246212. [Google Scholar] [CrossRef]
- Kentrup, H.; Altmüller, J.; Pfäffle, R.; Heimann, G. Neonatal diabetes mellitus with hypergalactosemia. Eur. J. Endocrinol. 1999, 141, 379–381. [Google Scholar] [CrossRef] [Green Version]
- Stelmańskan, E. The important role of GLUT2 in intestinal sugar transport and absorption. Postepy Biochem. 2009, 55, 385–387. [Google Scholar]
- Koepsell, H. Glucose transporters in the small intestine in health and disease. Pflug. Arch. Eur. J. Physiol. 2020, 472, 1207–1248. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Adams, D.H.; Lalor, P.F. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol. 2012, 18, 6771–6781. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef]
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflugers Arch. Eur. J. Physiol. 2020, 472, 1273–1298. [Google Scholar] [CrossRef] [PubMed]
- De Souza Cordeiro, L.M.; Devisetty, N.; McDougal, D.; Peters, D.J.M.; Chhabra, K.H. Renal GLUT2 is Essential in Regulating Systemic Glucose Homeostasis by Glycosuria. J. Endocr. Soc. 2021, 5, A324. [Google Scholar] [CrossRef]
- Sędzikowska, A.; Szablewski, L. Human Glucose Transporters in Renal Glucose Homeostasis. Int. J. Mol. Sci. 2021, 22, 13522. [Google Scholar] [CrossRef]
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653. [Google Scholar] [CrossRef]
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Investig. 1995, 96, 2489–2495. [Google Scholar] [CrossRef]
- Berger, C.; Zdzieblo, D. Glucose transporters in pancreatic islets. Pflug. Arch. Eur. J. Physiol. 2020, 472, 1249–1272. [Google Scholar] [CrossRef]
- Enogieru, O.J.; Ung, P.M.U.; Yee, S.W.; Schlessinger, A.; Giacomini, K.M. Functional and structural analysis of rare SLC2A2 variants associated with Fanconi-Bickel syndrome and metabolic traits. Hum. Mutat. 2019, 40, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Wangen, J.R.; Green, R. Stop codon context influences genome-wide stimulation of termination codon readthrough by aminoglycosides. eLife 2020, 9, e52611. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Dimitriadis, G.; Newsholme, P. Glucose metabolism in lymphoid and inflammatory cells and tissues. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 531–540. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.J.; Contreras, G.A.; Mattmiller, S.A.; Sordillo, L.M. Changes in glucose transporter expression in monocytes of periparturient dairy cows. J. Dairy Sci. 2012, 95, 5709–5719. [Google Scholar] [CrossRef] [PubMed]
- Von Ah Morano, A.E.; Dorneles, G.P.; Peres, A.; Lira, F.S. The role of glucose homeostasis on immune function in response to exercise: The impact of low or higher energetic conditions. J. Cell. Physiol. 2020, 235, 3169–3188. [Google Scholar] [CrossRef]
- Fu, Y.; Maianu, L.; Melbert, B.R.; Garvey, W.T. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: A role for GLUT isoforms 1, 3, and 5 in the immune response and foam cell formation. Blood Cells Mol. Dis. 2004, 32, 182–190. [Google Scholar] [CrossRef]
- Palmer, C.S.; Anzinger, J.J.; Zhou, J.; Gouillou, M.; Landay, A.; Jaworowski, A.; McCune, J.M.; Crowe, S.M. Glucose Transporter 1–Expressing Proinflammatory Monocytes Are Elevated in Combination Antiretroviral Therapy–Treated and Untreated HIV + Subjects. J. Immunol. 2014, 193, 5595–5603. [Google Scholar] [CrossRef]
- Sticka, K.D.; Schnurr, T.M.; Jerome, S.P.; Dajles, A.; Reynolds, A.J.; Duffy, L.K.; Knall, C.M.; Dunlap, K.L. Exercise Increases Glucose Transporter-4 Levels on Peripheral Blood Mononuclear Cells. Med. Sci. Sports Exerc. 2018, 50, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate regulates metabolic and proinflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef]
- Sharari, S.; Aouida, M.; Mohammed, I.; Haris, B.; Bhat, A.A.; Hawari, I.; Nisar, S.; Pavlovski, I.; Biswas, K.H.; Syed, N.; et al. Understanding the Mechanism of Dysglycemia in a Fanconi-Bickel Syndrome Patient. Front. Endocrinol. 2022, 13, 841788. [Google Scholar] [CrossRef]
- Aghaei, M.; Khodadadian, A.; Elham, K.N.; Nazari, M.; Babakhanzadeh, E. Major miRNA involved in insulin secretion and production in beta-cells. Int. J. Gen. Med. 2020, 13, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Qian, X.L.; Li, J.; Xiao, X.H.; Lu, X.T.; Lv, L.C.; Huang, Q.Y.; Ding, W.; Zhang, H.Y.; Xiong, L.X.; et al. MIR-29a negatively affects glucose-stimulated insulin secretion and MIN6 cell proliferation via Cdc42/β-catenin signaling. Int. J. Endocrinol. 2019, 2019, 5219782. [Google Scholar] [CrossRef] [PubMed]
- Filios, S.R.; Shalev, A. β-cell microRNAs: Small but powerful. Diabetes 2015, 64, 3631–3644. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Guo, F. MicroRNAs and type 2 diabetes. ExRNA 2019, 1, 36. [Google Scholar] [CrossRef]
- Zhou, Y.; Gu, P.; Shi, W.; Li, J.; Hao, Q.; Cao, X.; Lu, Q.; Zeng, Y. MicroRNA-29a induces insulin resistance by targeting PPARδ in skeletal muscle cells. Int. J. Mol. Med. 2016, 37, 931–938. [Google Scholar] [CrossRef]
- Hromadnikova, I.; Kotlabova, K.; Dvorakova, L.; Krofta, L.; Sirc, J. Substantially Altered Expression Profile of Diabetes/Cardiovascular/Cerebrovascular Disease Associated microRNAs in Children Descending from Pregnancy Complicated by Gestational Diabetes Mellitus-One of Several Possible Reasons for an Increased Cardiovasc. Cells 2020, 9, 1557. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, Y.; Shi, Y.; Zhang, Y.; Liu, K.; Liang, R.; Sun, P.; Chang, X.; Tang, W.; Zhang, Y.; et al. Expression of miRNA-29 in Pancreatic β Cells Promotes Inflammation and Diabetes via TRAF3. Cell Rep. 2021, 34, 108576. [Google Scholar] [CrossRef]
- Shen, L.; Li, Q.; Wang, J.; Zhao, Y.; Niu, L.; Bai, L.; Shuai, S.; Li, X.; Zhang, S.; Zhu, L. miR-144-3p Promotes Adipogenesis Through Releasing C/EBPα From Klf3 and CtBP2. Front. Genet. 2018, 9, 677. [Google Scholar] [CrossRef]
- Demirsoy, I.H.; Ertural, D.Y.; Balci, Ş.; Çlnklr, Ü.; Sezer, K.; Tamer, L.; Aras, N. Profiles of Circulating miRNAs Following Metformin Treatment in Patients with Type 2 Diabetes. J. Med. Biochem. 2018, 37, 499–506. [Google Scholar] [CrossRef]
- Jordan, S.D.; Krüger, M.; Willmes, D.M.; Redemann, N.; Wunderlich, F.T.; Brönneke, H.S.; Merkwirth, C.; Kashkar, H.; Olkkonen, V.M.; Böttger, T.; et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol. 2011, 13, 434–446. [Google Scholar] [CrossRef]
- Guo, J.; Yang, C.; Lin, Y.; Hu, G.; Wei, J.; Zhang, X.; Chen, X.; Li, J. Enhanced peripheral blood miR-324-5p is associated with the risk of metabolic syndrome by suppressing ROCK1. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2020, 1865, 158727. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Yang, C.Y.; Rui, Z.L. MicroRNA-125b-5p improves pancreatic β-cell function through inhibiting JNK signaling pathway by targeting DACT1 in mice with type 2 diabetes mellitus. Life Sci. 2019, 224, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Tavano, F.; Fontana, A.; Mazza, T.; Gioffreda, D.; Biagini, T.; Palumbo, O.; Carella, M.; Andriulli, A. Early-Onset Diabetes as Risk Factor for Pancreatic Cancer: miRNA Expression Profiling in Plasma Uncovers a Role for miR-20b-5p, miR-29a, and miR-18a-5p in Diabetes of Recent Diagnosis. Front. Oncol. 2020, 10, 1567. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, Y.; Yin, L.; Qi, Y.; Sun, H.; Sun, P.; Xu, M.; Tang, Z.; Peng, J. miR-125a-5p ameliorates hepatic glycolipid metabolism disorder in type 2 diabetes mellitus through targeting of STAT3. Theranostics 2018, 8, 5593–5609. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Investigation | Test Value | Normal Range |
|---|---|---|
| Electrolyte level | ||
| Serum phosphorus (mmol/L) | 0.43 | 0.93–1.64 |
| Serum calcium (mmol/L) | 2.15 | 2.2–2.7 |
| Serum Sodium (mmol/L) | 136 | 134–146 |
| Serum Potassium (mmol/L) | 4 | 3.5–5.0 |
| Liver function test | ||
| Alanine amino transferase (ALT) (IU/L) | 56 | 8–22 |
| Aspartate transaminase (AST) (IU/L) | 74 | 0–30 |
| Alkaline phosphatase (IU/L) | 1108 | 48–95 |
| Blood glucose test | ||
| Fasting glucose (mmol/L) | 2.5 | 3.5–5.5 |
| 2 h glucose tolerance (mmol/L) | 28.2 | 7.8–11.1 |
| C peptide (ng/mL) | 0.68 | 0.78–5.19 |
| Hemoglobin A1c (HbA1c)% | 6.4 | 4.8–6.0 |
| Diabetes mellitus type 1 evaluation (GAD65, ICA, ZnT8, IAA) | Negative | - |
| Growth hormone test | ||
| Thyroid stimulating hormone (TSH) (mIU/L) | 3.82 | 0.4–4.0 |
| Lipid test | ||
| Cholesterol (mmol/L) | 6.53 | <5.18 |
| Triglyceride (mmol/L) | 1.3 | <1.7 |
| High density lipoprotein (HDL-C) (mmol/L) | 0.9 | >1.17 |
| Low density lipoprotein LDL (mmol/L) | 4.72 | <2.6 |
| Kidney test | ||
| Blood urea nitrogen (BUN) (mmol/L) | 1.8 | 1.9–6.7 |
| Creatinine (µmol/L) | 35 | 54–95 |
| Urinalysis | Generalized aminoaciduria ++, phosphaturia, glucosuria (+2) | |
| Investigation | Test Value | Normal Range |
|---|---|---|
| Electrolyte level | ||
| Serum phosphorus (mmol/L) | 0.79 | 0.93–1.64 |
| Serum calcium (mmol/L) | 2.17 | 2.2–2.7 |
| Serum Sodium (mmol/L) | 135 | 134–146 |
| Serum potassium (mmol/L) | 3.3 | 3.5–5.0 |
| Liver function test | ||
| Alanine amino transferase (ALT) (IU/L) | 30 | 8–22 |
| Aspartate transaminase (AST) (IU/L) | 40 | 0–30 |
| Alkaline phosphatase (IU/L) | 388 | 48–95 |
| Blood glucose test | ||
| Fasting glucose (mmol/L) | 3.2 | 3.5–5.5 |
| 2 h post-feed glucose (mmol/L) | 18.3 | 3.5–5.5 |
| Insulin (pmol/L) | 6 | 18–48 |
| Hemoglobin A1c (HbA1c)% | 5.7 | 4–5.6 |
| Diabetes mellitus type 1 evaluation (GAD65, Insulin, ZnT8, IA-2) | Negative | - |
| Growth hormone test | ||
| Thyroid stimulating hormone (TSH) (mIU/L) | 1.72 | 0.4–4.0 |
| Insulin-like growth factor-1 (IGF-1) (nmol/L) | 7.8 | 8.2–30.8 |
| Parathyroid hormone (PTH intact) | 10.3 | 2.0–6.8 |
| Kidney test | ||
| Blood urea nitrogen (BUN) (mmol/L) | 3 | 2.5–7.1 |
| Creatinine (µmol/L) | 34 | 60–110 |
| Urinalysis | Proteinuria (+1), glucosuria (+3) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharari, S.; Kabeer, B.; Mohammed, I.; Haris, B.; Pavlovski, I.; Hawari, I.; Bhat, A.A.; Toufiq, M.; Tomei, S.; Mathew, R.; et al. Understanding the Role of GLUT2 in Dysglycemia Associated with Fanconi–Bickel Syndrome. Biomedicines 2022, 10, 2114. https://doi.org/10.3390/biomedicines10092114
Sharari S, Kabeer B, Mohammed I, Haris B, Pavlovski I, Hawari I, Bhat AA, Toufiq M, Tomei S, Mathew R, et al. Understanding the Role of GLUT2 in Dysglycemia Associated with Fanconi–Bickel Syndrome. Biomedicines. 2022; 10(9):2114. https://doi.org/10.3390/biomedicines10092114
Chicago/Turabian StyleSharari, Sanaa, Basirudeen Kabeer, Idris Mohammed, Basma Haris, Igor Pavlovski, Iman Hawari, Ajaz Ahmad Bhat, Mohammed Toufiq, Sara Tomei, Rebecca Mathew, and et al. 2022. "Understanding the Role of GLUT2 in Dysglycemia Associated with Fanconi–Bickel Syndrome" Biomedicines 10, no. 9: 2114. https://doi.org/10.3390/biomedicines10092114