
Post-Transcriptional Control of mRNA Metabolism and Protein Secretion: The Third Level of Regulation within the NF-κB System


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. The NF-κB System
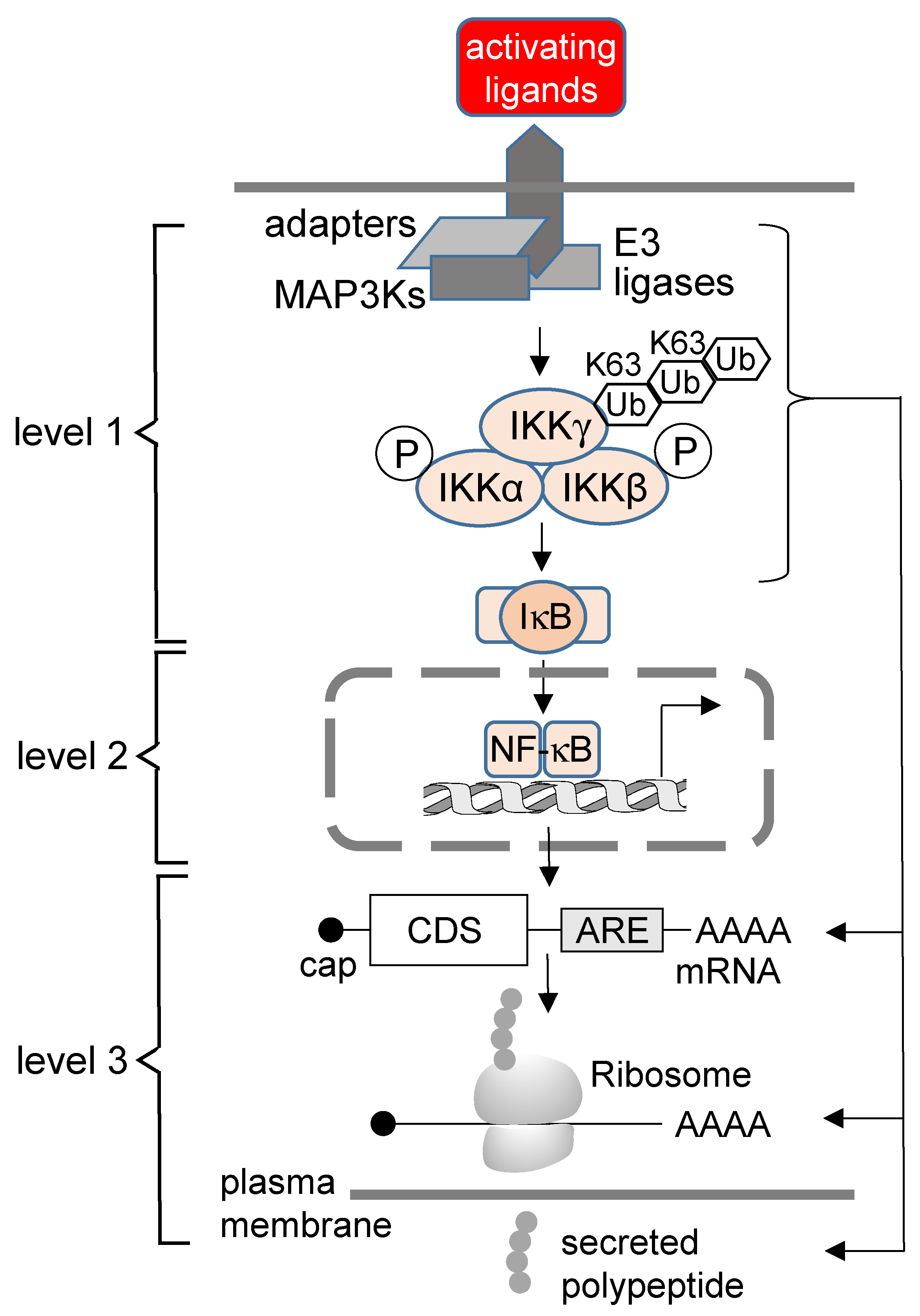
2. The Concept of the Third Level of NF-κB Regulation
3. The Role of the NF-κB System in Posttranscriptional Gene Regulation
3.1. Key Aspects of the Post-Transcriptional Regulation Relevant to Regulation of NF-κB Target Genes
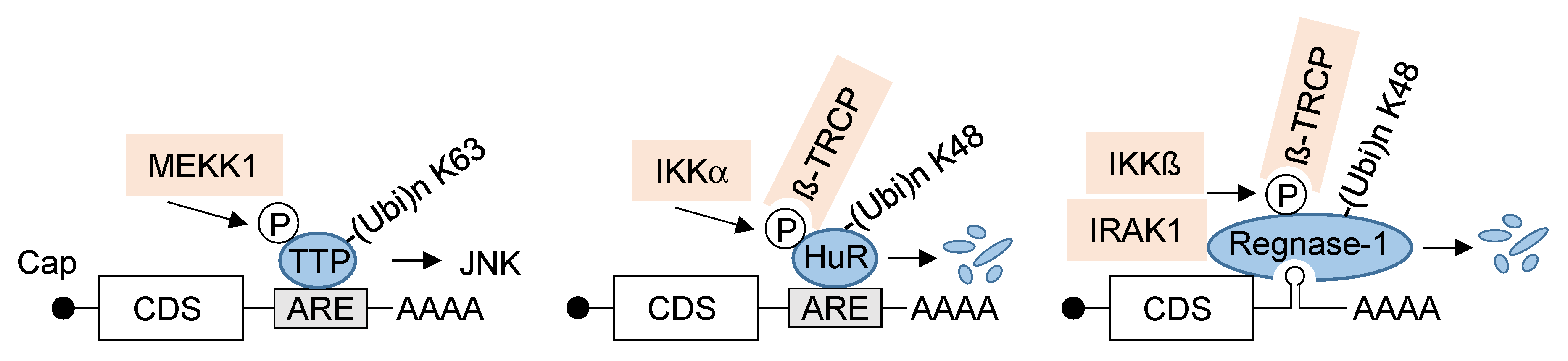
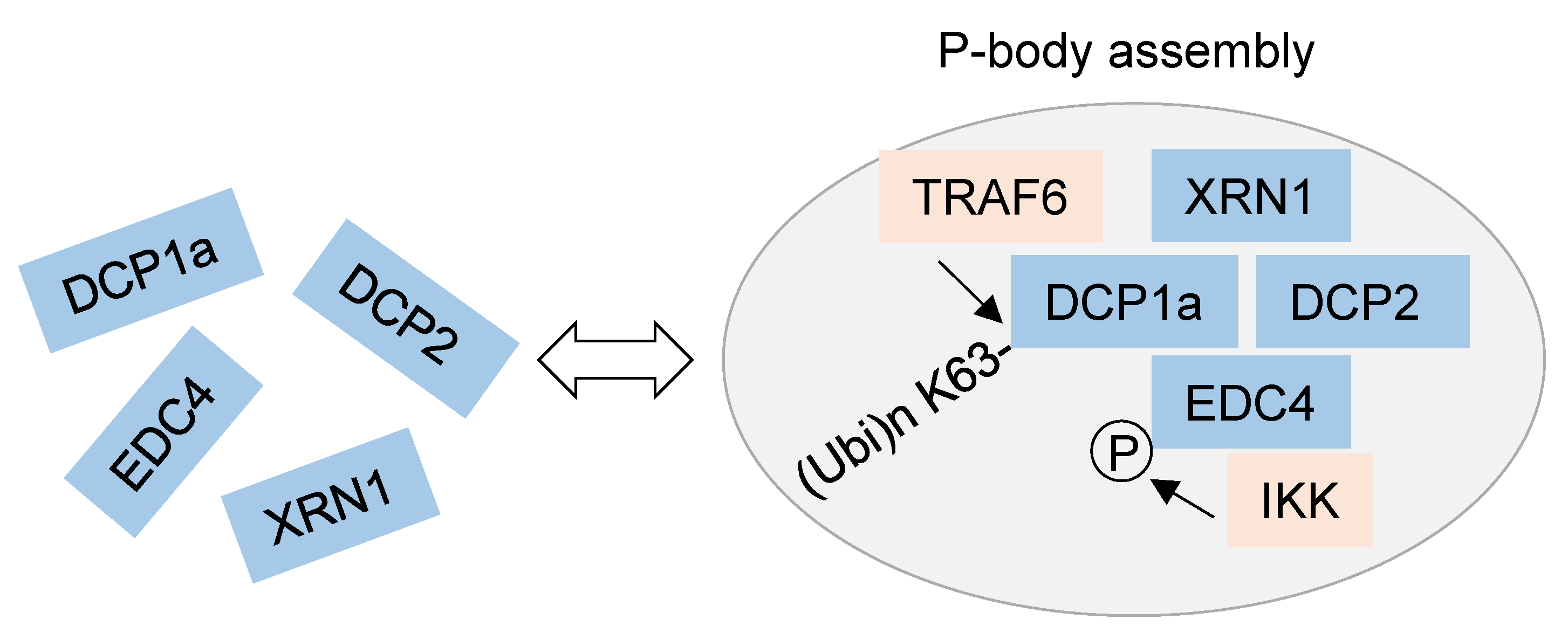
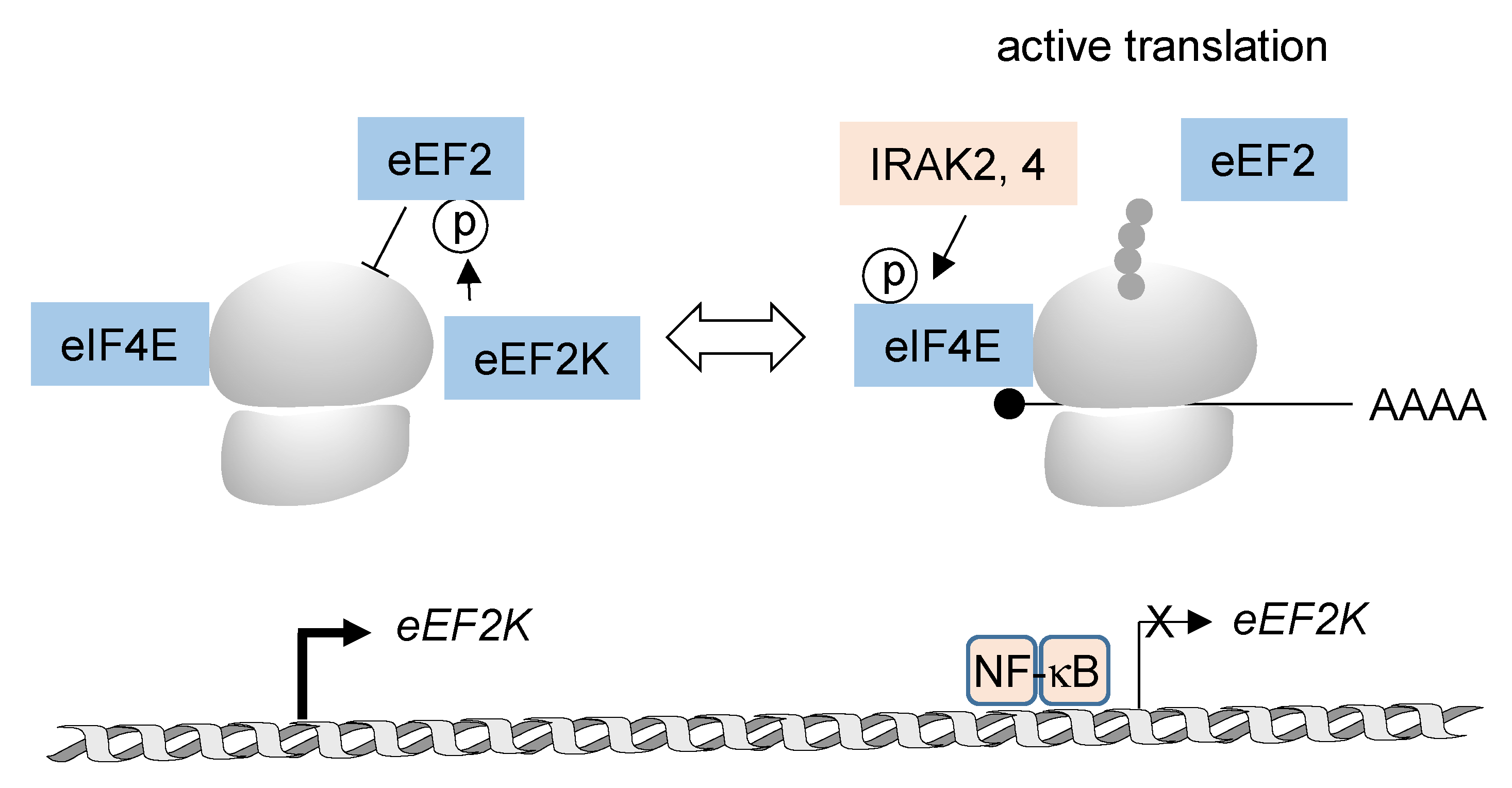
3.2. NF-κB-Mediated Regulation of Post-Transcriptional RNA Processing and Protein Translation
4. The Role of the NF-κB System in Protein Secretion
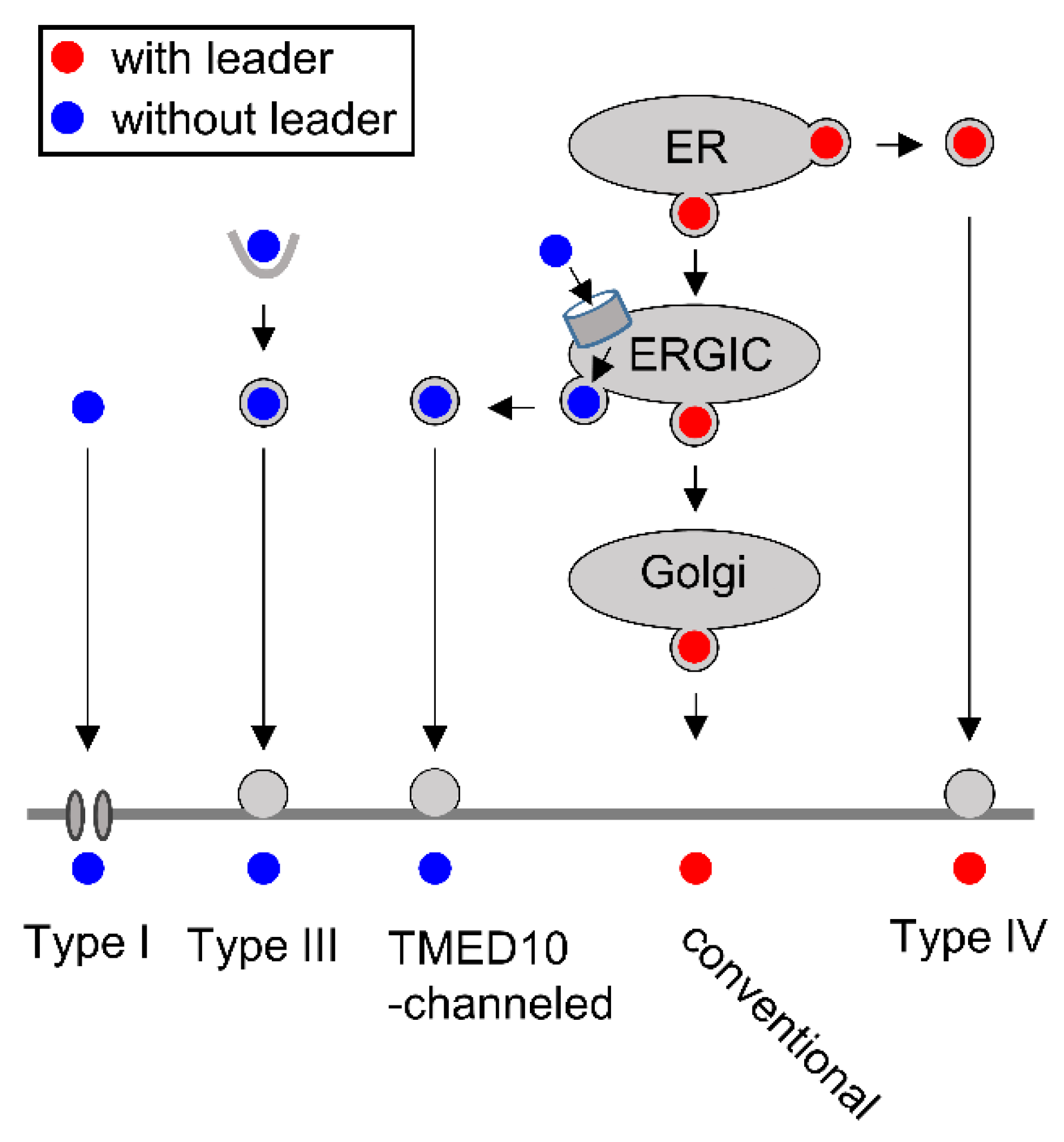
4.1. Key Aspects of Conventional and Nonconventional Protein Secretion
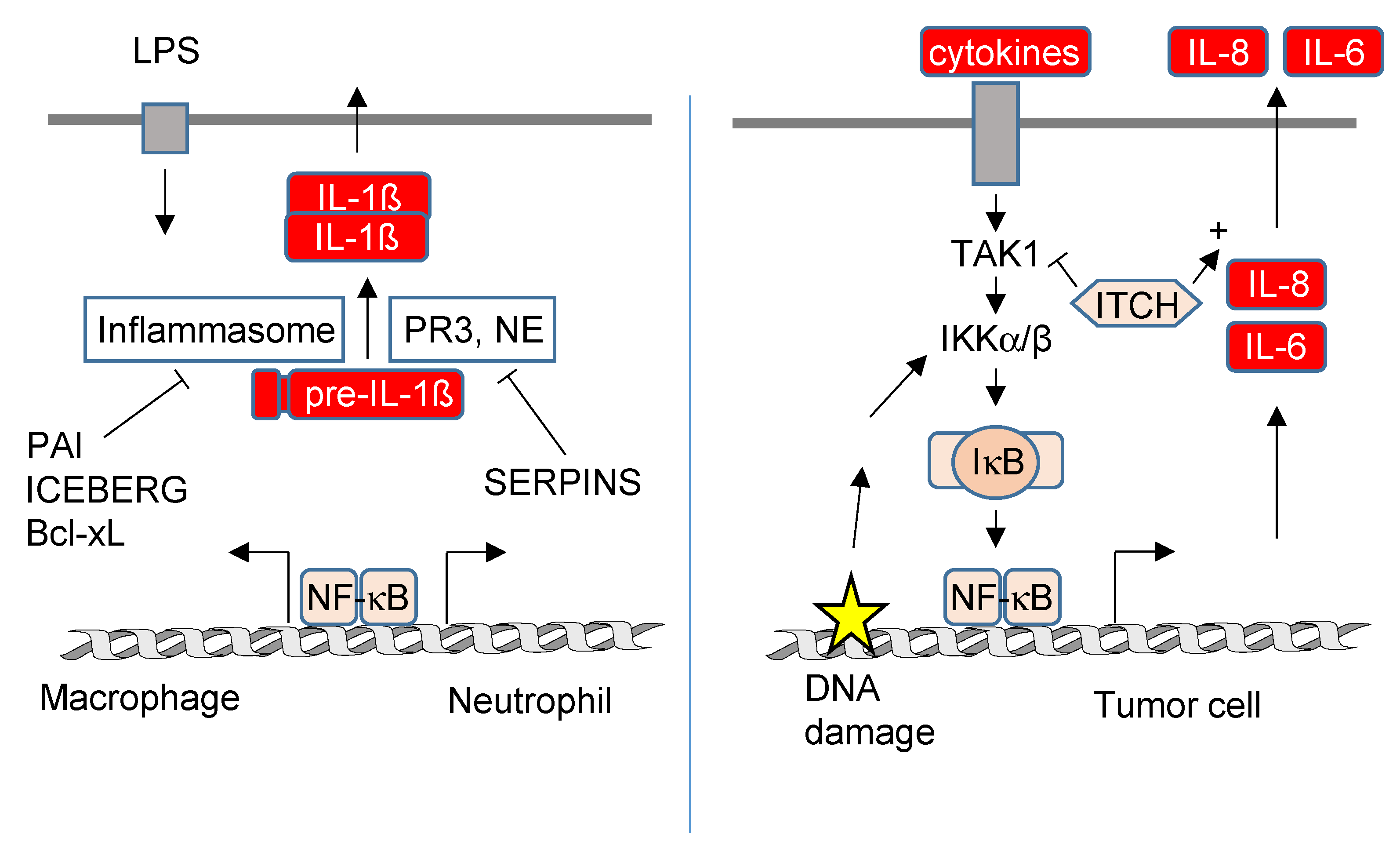
4.2. NF-κB-Mediated Regulation of Protein Secretion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Williams, L.M.; Gilmore, T.D. Looking Down on NF-kappaB. Mol. Cell Biol. 2020, 40, e00104–e00120. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Z.B.; Kofahl, B.; Beaudette, P.; Baum, K.; Ipenberg, I.; Weih, F.; Wolf, J.; Dittmar, G.; Scheidereit, C. Quantitative dissection and modeling of the NF-kappaB p100-p105 module reveals interdependent precursor proteolysis. Cell Rep. 2014, 9, 1756–1769. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Kondylis, V.; Kumari, S.; Vlantis, K.; Pasparakis, M. The interplay of IKK, NF-kappaB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev. 2017, 277, 113–127. [Google Scholar] [CrossRef]
- Hinz, M.; Scheidereit, C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Lucas, R.M.; Liu, L.; Stow, J.L. Signalling, sorting and scaffolding adaptors for Toll-like receptors. J. Cell Sci. 2019, 133, 239194. [Google Scholar] [CrossRef] [PubMed]
- Busch, J.; Moreno, R.; de la Vega, L.; Saul, V.V.; Bacher, S.; von Zweydorf, F.; Ueffing, M.; Weber, A.; Gloeckner, C.J.; Linne, U.; et al. TRAF6 Phosphorylation Prevents Its Autophagic Degradation and Re-Shapes LPS-Triggered Signaling Networks. Cancers 2021, 13, 3618. [Google Scholar] [CrossRef] [PubMed]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef]
- Rothwarf, D.M.; Zandi, E.; Natoli, G.; Karin, M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998, 395, 297–300. [Google Scholar] [CrossRef]
- Yamaoka, S.; Courtois, G.; Bessia, C.; Whiteside, S.T.; Weil, R.; Agou, F.; Kirk, H.E.; Kay, R.J.; Israel, A. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 1998, 93, 1231–1240. [Google Scholar] [CrossRef]
- Alkalay, I.; Yaron, A.; Hatzubai, A.; Orian, A.; Ciechanover, A.; Ben-Neriah, Y. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1995, 92, 10599–10603. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Hinz, M.; Stilmann, M.; Arslan, S.C.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef]
- Mabb, A.M.; Wuerzberger-Davis, S.M.; Miyamoto, S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat. Cell Biol. 2006, 8, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Kolesnichenko, M.; Mikuda, N.; Hopken, U.E.; Kargel, E.; Uyar, B.; Tufan, A.B.; Milanovic, M.; Sun, W.; Krahn, I.; Schleich, K.; et al. Transcriptional repression of NFKBIA triggers constitutive IKK- and proteasome-independent p65/RelA activation in senescence. EMBO J. 2021, 40, e104296. [Google Scholar] [CrossRef] [PubMed]
- Bacher, S.; Meier-Soelch, J.; Kracht, M.; Schmitz, M.L. Regulation of Transcription Factor NF-kappaB in Its Natural Habitat: The Nucleus. Cells 2021, 10, 753. [Google Scholar] [CrossRef] [PubMed]
- Calderon, D.; Nguyen, M.L.T.; Mezger, A.; Kathiria, A.; Muller, F.; Nguyen, V.; Lescano, N.; Wu, B.; Trombetta, J.; Ribado, J.V.; et al. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet. 2019, 51, 1494–1505. [Google Scholar] [CrossRef]
- Weiterer, S.S.; Meier-Soelch, J.; Georgomanolis, T.; Mizi, A.; Beyerlein, A.; Weiser, H.; Brant, L.; Mayr-Buro, C.; Jurida, L.; Beuerlein, K.; et al. Distinct IL-1alpha-responsive enhancers promote acute and coordinated changes in chromatin topology in a hierarchical manner. EMBO J. 2020, 39, e101533. [Google Scholar] [CrossRef]
- Tartey, S.; Matsushita, K.; Vandenbon, A.; Ori, D.; Imamura, T.; Mino, T.; Standley, D.M.; Hoffmann, J.A.; Reichhart, J.M.; Akira, S.; et al. Akirin2 is critical for inducing inflammatory genes by bridging IkappaB-zeta and the SWI/SNF complex. EMBO J. 2014, 33, 2332–2348. [Google Scholar] [CrossRef]
- Smale, S.T.; Tarakhovsky, A.; Natoli, G. Chromatin contributions to the regulation of innate immunity. Annu. Rev. Immunol. 2014, 32, 489–511. [Google Scholar] [CrossRef]
- Jurida, L.; Soelch, J.; Bartkuhn, M.; Handschick, K.; Muller, H.; Newel, D.; Weber, A.; Dittrich-Breiholz, O.; Schneider, H.; Bhuju, S.; et al. The Activation of IL-1-Induced Enhancers Depends on TAK1 Kinase Activity and NF-kappaB p65. Cell Rep. 2015, 10, 726–739. [Google Scholar] [CrossRef]
- Smale, S.T.; Natoli, G. Transcriptional control of inflammatory responses. Cold Spring Harb. Perspect. Biol. 2014, 6, a016261. [Google Scholar] [CrossRef]
- Rogatsky, I.; Adelman, K. Preparing the First Responders: Building the Inflammatory Transcriptome from the Ground Up. Mol. Cell 2014, 54, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef]
- Carpenter, S.; Ricci, E.P.; Mercier, B.C.; Moore, M.J.; Fitzgerald, K.A. Post-transcriptional regulation of gene expression in innate immunity. Nat. Rev. Immunol. 2014, 14, 361–376. [Google Scholar] [CrossRef]
- Lacy, P.; Stow, J.L. Cytokine release from innate immune cells: Association with diverse membrane trafficking pathways. Blood 2011, 118, 9–18. [Google Scholar] [CrossRef]
- Ziesche, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Muller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-kappaB. Nucleic Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef]
- Li, H.; Wittwer, T.; Weber, A.; Schneider, H.; Moreno, R.; Maine, G.N.; Kracht, M.; Schmitz, M.L.; Burstein, E. Regulation of NF-kappaB activity by competition between RelA acetylation and ubiquitination. Oncogene 2012, 31, 611–623. [Google Scholar] [CrossRef]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef]
- Pope, S.D.; Medzhitov, R. Emerging Principles of Gene Expression Programs and Their Regulation. Mol. Cell 2018, 71, 389–397. [Google Scholar] [CrossRef]
- Anderson, P. Intrinsic mRNA stability helps compose the inflammatory symphony. Nat. Immunol. 2009, 10, 233–234. [Google Scholar] [CrossRef]
- Schmid, M.; Jensen, T.H. Controlling nuclear RNA levels. Nat. Rev. Genet. 2018, 19, 518–529. [Google Scholar] [CrossRef]
- Hartenian, E.; Glaunsinger, B.A. Feedback to the central dogma: Cytoplasmic mRNA decay and transcription are interdependent processes. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 385–398. [Google Scholar] [CrossRef]
- Berry, S.; Pelkmans, L. Mechanisms of cellular mRNA transcript homeostasis. Trends Cell Biol. 2022, 32, 655–668. [Google Scholar] [CrossRef]
- Tuck, A.C.; Rankova, A.; Arpat, A.B.; Liechti, L.A.; Hess, D.; Iesmantavicius, V.; Castelo-Szekely, V.; Gatfield, D.; Buhler, M. Mammalian RNA Decay Pathways Are Highly Specialized and Widely Linked to Translation. Mol. Cell 2020, 77, 1222–1236.e13. [Google Scholar] [CrossRef]
- Hao, S.; Baltimore, D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 2009, 10, 281–288. [Google Scholar] [CrossRef]
- Gordon, J.M.; Phizicky, D.V.; Neugebauer, K.M. Nuclear mechanisms of gene expression control: Pre-mRNA splicing as a life or death decision. Curr. Opin Genet. Dev. 2021, 67, 67–76. [Google Scholar] [CrossRef]
- Stewart, M. Polyadenylation and nuclear export of mRNAs. J. Biol. Chem. 2019, 294, 2977–2987. [Google Scholar] [CrossRef]
- Zarnack, K.; Balasubramanian, S.; Gantier, M.P.; Kunetsky, V.; Kracht, M.; Schmitz, M.L.; Sträßer, K. Dynamic mRNP Remodeling in Response to Internal and External Stimuli. Biomolecules 2020, 10, 1310. [Google Scholar] [CrossRef]
- Schott, J.; Stoecklin, G. Networks controlling mRNA decay in the immune system. Wiley Interdiscip Rev. RNA 2010, 1, 432–456. [Google Scholar] [CrossRef]
- Anderson, P. Post-transcriptional control of cytokine production. Nat. Immunol. 2008, 9, 353–359. [Google Scholar] [CrossRef]
- Rabouille, C.; Malhotra, V.; Nickel, W. Diversity in unconventional protein secretion. J. Cell Sci. 2012, 125, 5251–5255. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, J.; Stierhof, Y.D.; Robinson, D.G.; Jiang, L. Unconventional protein secretion. Trends Plant Sci. 2012, 17, 606–615. [Google Scholar] [CrossRef]
- Zhao, W.; Erle, D.J. Widespread Effects of Chemokine 3’ Untranslated Regions on mRNA Degradation and Protein Production in Human Cells. J. Immunol. 2018, 201, 1053–1061. [Google Scholar] [CrossRef]
- Akira, S.; Maeda, K. Control of RNA Stability in Immunity. Annu. Rev. Immunol. 2021, 39, 481–509. [Google Scholar] [CrossRef]
- Nagarajan, V.K.; Jones, C.I.; Newbury, S.F.; Green, P.J. XRN 5’-->3’ exoribonucleases: Structure, mechanisms and functions. Biochim. Biophys. Acta 2013, 1829, 590–603. [Google Scholar] [CrossRef]
- Schoenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012, 13, 246–259. [Google Scholar] [CrossRef]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 2007, 8, 9–22. [Google Scholar] [CrossRef]
- Hubstenberger, A.; Courel, M.; Benard, M.; Souquere, S.; Ernoult-Lange, M.; Chouaib, R.; Yi, Z.; Morlot, J.B.; Munier, A.; Fradet, M.; et al. P-Body Purification Reveals the Condensation of Repressed mRNA Regulons. Mol. Cell 2017, 68, 144–157.e5. [Google Scholar] [CrossRef]
- Youn, J.Y.; Dyakov, B.J.A.; Zhang, J.; Knight, J.D.R.; Vernon, R.M.; Forman-Kay, J.D.; Gingras, A.C. Properties of Stress Granule and P-Body Proteomes. Mol. Cell 2019, 76, 286–294. [Google Scholar] [CrossRef]
- Jain, S.; Parker, R. The discovery and analysis of p bodies. Adv. Exp. Med. Biol. 2013, 768, 23–43. [Google Scholar]
- Standart, N.; Weil, D. P-Bodies: Cytosolic Droplets for Coordinated mRNA Storage. Trends Genet. 2018, 34, 612–626. [Google Scholar] [CrossRef]
- Van Treeck, B.; Parker, R. Emerging Roles for Intermolecular RNA-RNA Interactions in RNP Assemblies. Cell 2018, 174, 791–802. [Google Scholar] [CrossRef]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Stress-induced reversal of microRNA repression and mRNA P-body localization in human cells. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 513–521. [Google Scholar] [CrossRef]
- Brengues, M.; Teixeira, D.; Parker, R. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 2005, 310, 486–489. [Google Scholar] [CrossRef]
- Khong, A.; Parker, R. The landscape of eukaryotic mRNPs. RNA 2020, 26, 229–239. [Google Scholar] [CrossRef]
- Franks, T.M.; Lykke-Andersen, J. The control of mRNA decapping and P-body formation. Mol. Cell 2008, 32, 605–615. [Google Scholar] [CrossRef]
- Stoecklin, G.; Mayo, T.; Anderson, P. ARE-mRNA degradation requires the 5’-3’ decay pathway. EMBO Rep. 2006, 7, 72–77. [Google Scholar] [CrossRef]
- Valkov, E.; Muthukumar, S.; Chang, C.T.; Jonas, S.; Weichenrieder, O.; Izaurralde, E. Structure of the Dcp2-Dcp1 mRNA-decapping complex in the activated conformation. Nat. Struct. Mol. Biol. 2016, 23, 574–579. [Google Scholar] [CrossRef]
- Chang, C.T.; Bercovich, N.; Loh, B.; Jonas, S.; Izaurralde, E. The activation of the decapping enzyme DCP2 by DCP1 occurs on the EDC4 scaffold and involves a conserved loop in DCP1. Nucleic Acids Res. 2014, 42, 5217–5233. [Google Scholar] [CrossRef]
- Braun, J.E.; Truffault, V.; Boland, A.; Huntzinger, E.; Chang, C.T.; Haas, G.; Weichenrieder, O.; Coles, M.; Izaurralde, E. A direct interaction between DCP1 and XRN1 couples mRNA decapping to 5’ exonucleolytic degradation. Nat. Struct. Mol. Biol. 2012, 19, 1324–1331. [Google Scholar] [CrossRef]
- Corbet, G.A.; Parker, R. RNP Granule Formation: Lessons from P-Bodies and Stress Granules. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 203–215. [Google Scholar] [CrossRef]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida-Sugitani, R.; Kobayashi, T.; Toyama-Sorimachi, N. The Assembly of EDC4 and Dcp1a into Processing Bodies Is Critical for the Translational Regulation of IL-6. PLoS ONE 2015, 10, e0123223. [Google Scholar] [CrossRef]
- Rzeczkowski, K.; Beuerlein, K.; Muller, H.; Dittrich-Breiholz, O.; Schneider, H.; Kettner-Buhrow, D.; Holtmann, H.; Kracht, M. c-Jun N-terminal kinase phosphorylates DCP1a to control formation of P bodies. J. Cell Biol. 2011, 194, 581–596. [Google Scholar] [CrossRef]
- Youn, J.Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.R.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.M.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef]
- Protter, D.S.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef]
- Matheny, T.; Van Treeck, B.; Huynh, T.N.; Parker, R. RNA partitioning into stress granules is based on the summation of multiple interactions. RNA 2021, 27, 174–189. [Google Scholar] [CrossRef]
- Franks, T.M.; Lykke-Andersen, J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev. 2007, 21, 719–735. [Google Scholar] [CrossRef]
- Fan, X.C.; Steitz, J.A. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998, 17, 3448–3460. [Google Scholar] [CrossRef]
- Peng, S.S.; Chen, C.Y.; Xu, N.; Shyu, A.B. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 1998, 17, 3461–3470. [Google Scholar] [CrossRef]
- Galban, S.; Kuwano, Y.; Pullmann, R., Jr.; Martindale, J.L.; Kim, H.H.; Lal, A.; Abdelmohsen, K.; Yang, X.; Dang, Y.; Liu, J.O.; et al. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1alpha. Mol. Cell Biol. 2008, 28, 93–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Poria, D.K.; Ray, P.S. RNA-binding proteins La and HuR cooperatively modulate translation repression of PDCD4 mRNA. J. Biol. Chem. 2021, 296, 100154. [Google Scholar] [CrossRef]
- Katsanou, V.; Papadaki, O.; Milatos, S.; Blackshear, P.J.; Anderson, P.; Kollias, G.; Kontoyiannis, D.L. HuR as a negative posttranscriptional modulator in inflammation. Mol. Cell 2005, 19, 777–789. [Google Scholar] [CrossRef]
- Mino, T.; Takeuchi, O. Regnase-1-related endoribonucleases in health and immunological diseases. Immunol. Rev. 2021, 304, 97–110. [Google Scholar] [CrossRef]
- Iwasaki, H.; Takeuchi, O.; Teraguchi, S.; Matsushita, K.; Uehata, T.; Kuniyoshi, K.; Satoh, T.; Saitoh, T.; Matsushita, M.; Standley, D.M.; et al. The IkappaB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR-IL-1R by controlling degradation of regnase-1. Nat. Immunol. 2011, 12, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Naeli, P.; Winter, T.; Hackett, A.P.; Alboushi, L.; Jafarnejad, S.M. The intricate balance between microRNA-induced mRNA decay and translational repression. FEBS J. 2022. [Google Scholar] [CrossRef]
- Mortazavi-Jahromi, S.S.; Aslani, M.; Mirshafiey, A. A comprehensive review on miR-146a molecular mechanisms in a wide spectrum of immune and non-immune inflammatory diseases. Immunol. Lett. 2020, 227, 8–27. [Google Scholar] [CrossRef]
- Singh, R.P.; Massachi, I.; Manickavel, S.; Singh, S.; Rao, N.P.; Hasan, S.; Mc Curdy, D.K.; Sharma, S.; Wong, D.; Hahn, B.H.; et al. The role of miRNA in inflammation and autoimmunity. Autoimmun. Rev. 2013, 12, 1160–1165. [Google Scholar] [CrossRef]
- Liao, K.; Xu, J.; Yang, W.; You, X.; Zhong, Q.; Wang, X. The research progress of LncRNA involved in the regulation of inflammatory diseases. Mol. Immunol. 2018, 101, 182–188. [Google Scholar] [CrossRef]
- Bai, R.; Cui, Z.; Ma, Y.; Wu, Y.; Wang, N.; Huang, L.; Yao, Q.; Sun, J. The NF-kappaB-modulated miR-19a-3p enhances malignancy of human ovarian cancer cells through inhibition of IGFBP-3 expression. Mol. Carcinog. 2019, 58, 2254–2265. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Zhao, G.; Su, Z.; Song, D.; Mao, Y.; Mao, X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-kappaB. FEBS Lett. 2016, 590, 2884–2895. [Google Scholar] [CrossRef]
- Mann, M.; Mehta, A.; Zhao, J.L.; Lee, K.; Marinov, G.K.; Garcia-Flores, Y.; Lu, L.F.; Rudensky, A.Y.; Baltimore, D. An NF-kappaB-microRNA regulatory network tunes macrophage inflammatory responses. Nat. Commun. 2017, 8, 851. [Google Scholar] [CrossRef] [PubMed]
- Rezcallah, M.C.; Al-Mazi, T.; Ammit, A.J. Cataloguing the phosphorylation sites of tristetraprolin (TTP): Functional implications for inflammatory diseases. Cell Signal. 2021, 78, 109868. [Google Scholar] [CrossRef]
- Schichl, Y.M.; Resch, U.; Lemberger, C.E.; Stichlberger, D.; de Martin, R. Novel phosphorylation-dependent ubiquitination of tristetraprolin by mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 1 (MEKK1) and tumor necrosis factor receptor-associated factor 2 (TRAF2). J. Biol. Chem. 2011, 286, 38466–38477. [Google Scholar] [CrossRef]
- Chu, P.C.; Chuang, H.C.; Kulp, S.K.; Chen, C.S. The mRNA-stabilizing factor HuR protein is targeted by beta-TrCP protein for degradation in response to glycolysis inhibition. J. Biol. Chem. 2012, 287, 43639–43650. [Google Scholar] [CrossRef]
- Mikuda, N.; Kolesnichenko, M.; Beaudette, P.; Popp, O.; Uyar, B.; Sun, W.; Tufan, A.B.; Perder, B.; Akalin, A.; Chen, W.; et al. The IkappaB kinase complex is a regulator of mRNA stability. EMBO J. 2018, 37, e98658. [Google Scholar] [CrossRef]
- Tenekeci, U.; Poppe, M.; Beuerlein, K.; Buro, C.; Muller, H.; Weiser, H.; Kettner-Buhrow, D.; Porada, K.; Newel, D.; Xu, M.; et al. K63-Ubiquitylation and TRAF6 Pathways Regulate Mammalian P-Body Formation and mRNA Decapping. Mol. Cell 2016, 62, 943–957. [Google Scholar] [CrossRef]
- Miller, C.J.; Turk, B.E. Homing in: Mechanisms of Substrate Targeting by Protein Kinases. Trends Biochem. Sci. 2018, 43, 380–394. [Google Scholar] [CrossRef]
- Pfeiffer, C.T.; Paulo, J.A.; Gygi, S.P.; Rockman, H.A. Proximity labeling for investigating protein-protein interactions. Methods Cell Biol. 2022, 169, 237–266. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, B.; Li, X.; Barik, S. Translation control: A multifaceted regulator of inflammatory response. J. Immunol. 2010, 184, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, C.; Holtmann, H.; Gaestel, M. The role of mammalian MAPK signaling in regulation of cytokine mRNA stability and translation. J. Interferon Cytokine Res. 2014, 34, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Pennini, M.E.; Perkins, D.J.; Salazar, A.M.; Lipsky, M.; Vogel, S.N. Complete dependence on IRAK4 kinase activity in TLR2, but not TLR4, signaling pathways underlies decreased cytokine production and increased susceptibility to Streptococcus pneumoniae infection in IRAK4 kinase-inactive mice. J. Immunol. 2013, 190, 307–316. [Google Scholar] [CrossRef]
- Wan, Y.; Xiao, H.; Affolter, J.; Kim, T.W.; Bulek, K.; Chaudhuri, S.; Carlson, D.; Hamilton, T.; Mazumder, B.; Stark, G.R.; et al. Interleukin-1 receptor-associated kinase 2 is critical for lipopolysaccharide-mediated post-transcriptional control. J. Biol. Chem. 2009, 284, 10367–10375. [Google Scholar] [CrossRef]
- Bianco, C.; Thompson, L.; Mohr, I. Repression of eEF2K transcription by NF-kappaB tunes translation elongation to inflammation and dsDNA-sensing. Proc. Natl. Acad. Sci. USA 2019, 116, 22583–22590. [Google Scholar] [CrossRef]
- Klann, K.; Münch, C. Quantitative Translation Proteomics Using mePROD. Methods Mol. Biol. 2022, 2428, 75–87. [Google Scholar] [CrossRef]
- Barrett, R.M.; Liu, H.W.; Jin, H.; Goodman, R.H.; Cohen, M.S. Cell-specific Profiling of Nascent Proteomes Using Orthogonal Enzyme-mediated Puromycin Incorporation. ACS Chem. Biol. 2016, 11, 1532–1536. [Google Scholar] [CrossRef]
- Duerr, T.J.; Comellas, E.; Jeon, E.K.; Farkas, J.E.; Joetzjer, M.; Garnier, J.; Shefelbine, S.J.; Monaghan, J.R. 3D visualization of macromolecule synthesis. eLife 2020, 9, e60354. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Munn, D.H.; Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 2016, 39, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Propper, D.J.; Balkwill, F.R. Harnessing cytokines and chemokines for cancer therapy. Nat. Rev. Clin. Oncol. 2022, 19, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Akopian, D.; Shen, K.; Zhang, X.; Shan, S.O. Signal recognition particle: An essential protein-targeting machine. Annu. Rev. Biochem. 2013, 82, 693–721. [Google Scholar] [CrossRef] [PubMed]
- Arakel, E.C.; Schwappach, B. Looking inside the cell. eLife 2017, 6, e33650. [Google Scholar] [CrossRef]
- Rabouille, C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2017, 27, 230–240. [Google Scholar] [CrossRef]
- Kim, J.; Gee, H.Y.; Lee, M.G. Unconventional protein secretion—New insights into the pathogenesis and therapeutic targets of human diseases. J. Cell Sci. 2018, 131, jcs213686. [Google Scholar] [CrossRef]
- Maricchiolo, E.; Panfili, E.; Pompa, A.; De Marchis, F.; Bellucci, M.; Pallotta, M.T. Unconventional Pathways of Protein Secretion: Mammals vs. Plants. Front. Cell Dev. Biol. 2022, 10, 895853. [Google Scholar] [CrossRef]
- Meissner, F.; Scheltema, R.A.; Mollenkopf, H.J.; Mann, M. Direct proteomic quantification of the secretome of activated immune cells. Science 2013, 340, 475–478. [Google Scholar] [CrossRef]
- Malhotra, V. Unconventional protein secretion: An evolving mechanism. EMBO J. 2013, 32, 1660–1664. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, L.; Lin, X.; Wang, Y.; Li, Y.; Guo, Q.; Li, S.; Sun, Y.; Tao, X.; Zhang, D.; et al. A Translocation Pathway for Vesicle-Mediated Unconventional Protein Secretion. Cell 2020, 181, 637–652.e15. [Google Scholar] [CrossRef] [PubMed]
- Georgilis, A.; Klotz, S.; Hanley, C.J.; Herranz, N.; Weirich, B.; Morancho, B.; Leote, A.C.; D’Artista, L.; Gallage, S.; Seehawer, M.; et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer Cell 2018, 34, 85–102.e9. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Ronacher, L.; Liu-Bryan, R.; Takai, S.; Karin, M.; Corr, M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum 2009, 60, 3642–3650. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef]
- Semino, C.; Carta, S.; Gattorno, M.; Sitia, R.; Rubartelli, A. Progressive waves of IL-1beta release by primary human monocytes via sequential activation of vesicular and gasdermin D-mediated secretory pathways. Cell Death Dis. 2018, 9, 1088. [Google Scholar] [CrossRef]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. eLife 2015, 4, e11205. [Google Scholar] [CrossRef]
- Farhan, H.; Rabouille, C. Signalling to and from the secretory pathway. J. Cell Sci. 2011, 124, 171–180. [Google Scholar] [CrossRef]
- Sitia, R.; Rubartelli, A. Evolution, role in inflammation, and redox control of leaderless secretory proteins. J. Biol. Chem. 2020, 295, 7799–7811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Bruey-Sedano, N.; Luciano, F.; Zhai, D.; Balpai, R.; Xu, C.; Kress, C.L.; Bailly-Maitre, B.; Li, X.; Osterman, A.; et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 2007, 129, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Zeng, M.; Sinha, I.; Polin, L.; Wei, W.Z.; Rathinam, C.; Flavell, R.; Massoumi, R.; Venuprasad, K. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat. Immunol. 2011, 12, 1176–1183. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, N.S.; Parvatiyar, K.; Copeland, N.G.; Jenkins, N.A.; Matesic, L.E.; Harhaj, E.W. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat. Immunol. 2008, 9, 254–262. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Priester, J.; Dreute, J.; Kracht, M.; Schmitz, M.L. Post-Transcriptional Control of mRNA Metabolism and Protein Secretion: The Third Level of Regulation within the NF-κB System. Biomedicines 2022, 10, 2108. https://doi.org/10.3390/biomedicines10092108
Priester J, Dreute J, Kracht M, Schmitz ML. Post-Transcriptional Control of mRNA Metabolism and Protein Secretion: The Third Level of Regulation within the NF-κB System. Biomedicines. 2022; 10(9):2108. https://doi.org/10.3390/biomedicines10092108
Chicago/Turabian StylePriester, Jasmin, Jan Dreute, Michael Kracht, and M. Lienhard Schmitz. 2022. "Post-Transcriptional Control of mRNA Metabolism and Protein Secretion: The Third Level of Regulation within the NF-κB System" Biomedicines 10, no. 9: 2108. https://doi.org/10.3390/biomedicines10092108