
Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Coronary Artery Disease and Atherosclerosis
2.1. Endothelial Dysfunction in Atherosclerosis
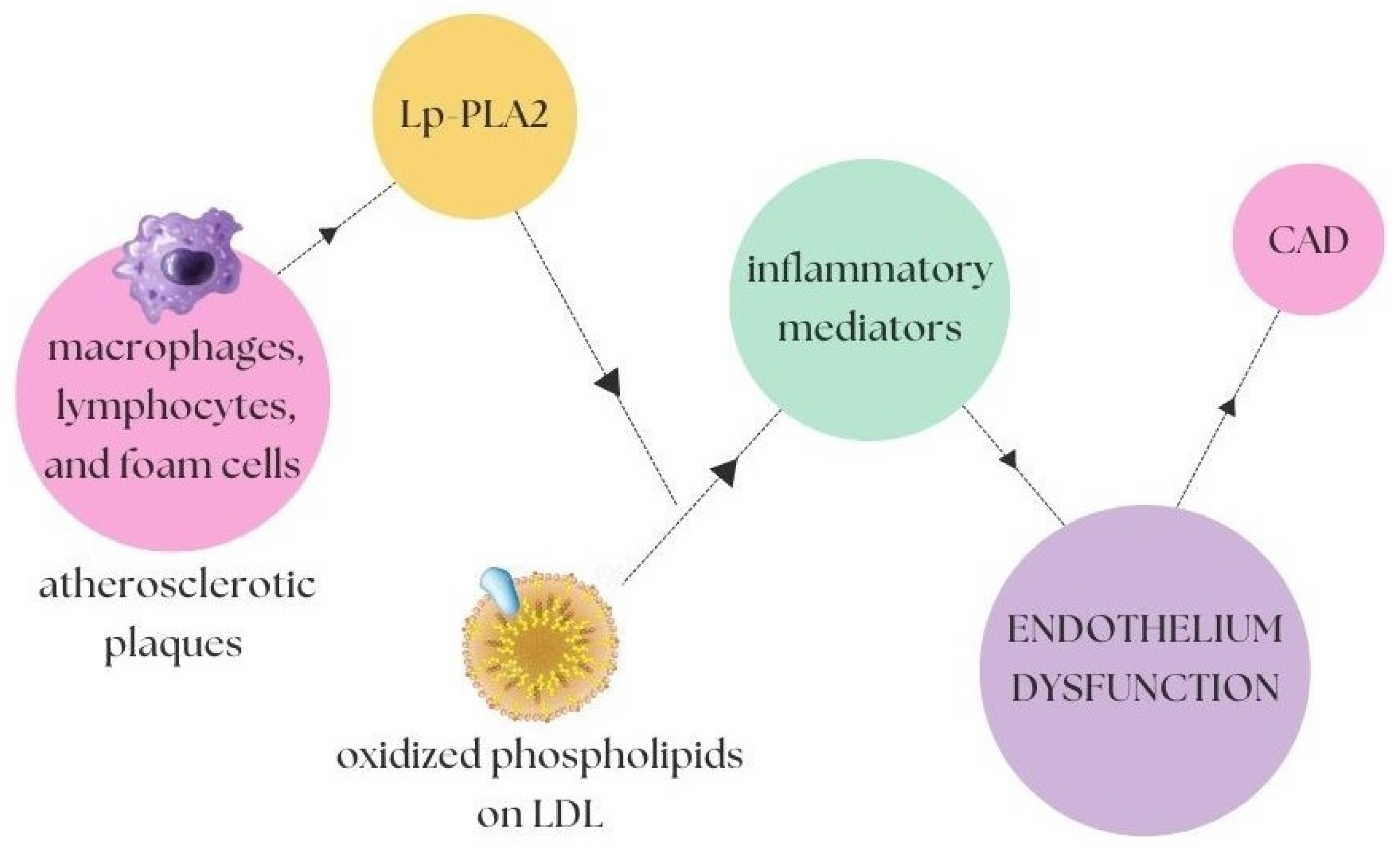
2.2. Inflammatory and Oxidising Factors in Atherosclerosis
2.3. Epigenetic Factors in Atherosclerosis
2.4. Endothelial Dysfunction in CAD
2.5. Inflammation in CAD
2.6. Genetic Background in CAD
3. Arterial Hypertension
3.1. Matrix Metalloproteinases in AH
3.2. Immune System in AH
3.3. Oxidative Stress in AH
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pagidipati, N.J.; Gaziano, T.A. Estimating Deaths from Cardiovascular Disease: A Review of Global Methodologies of Mortality Measurement. Circulation 2013, 127, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.D.; Budoff, M.J.; Ferdinand, K.; Graham, I.M.; Michos, E.D.; Reddy, T.; Shapiro, M.D.; Toth, P.P. Atherosclerotic cardiovascular disease risk assessment: An American Society for Preventive Cardiology clinical practice statement. Am. J. Prev. Cardiol. 2022, 10, 100335. [Google Scholar] [CrossRef] [PubMed]
- Sonja, F.; Nola, I.A. Management of Measurable Variable Cardiovascular Disease’ Risk Factors. Curr. Cardiol. Rev. 2018, 14, 153–163. [Google Scholar] [CrossRef]
- Amini, M.; Zayeri, F.; Salehi, M. Trend analysis of cardiovascular disease mortality, incidence, and mortality-to-incidence ratio: Results from global burden of disease study 2017. BMC Public Health 2021, 21, 401. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Del Castillo, A.M.; Jepsen, S.; Juanatey, J.R.G.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and cardiovascular diseases: Consensus report. J. Clin. Periodontol. 2020, 47, 268–288. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; Sarnak, M.J.; Yan, G.; Dwyer, J.T.; Heyka, R.J.; Rocco, M.V.; Teehan, B.P.; Levey, A.S. Atherosclerotic cardiovascular disease risks in chronic hemodialysis patients. Kidney Int. 2000, 58, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, R.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, Indicators, Risk Factors and New Hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Mitchell, G.F.; Powell, J.T. Arteriosclerosis. Arter. Thromb. Vasc. Biol. 2020, 40, 1025–1027. [Google Scholar] [CrossRef]
- Helvaci, M.R.; Tonyali, O.; Abyad, A. The Safest Value of Plasma Triglycerides. World Fam. Med. J./Middle East J. Fam. Med. 2019, 17, 22–27. [Google Scholar] [CrossRef]
- Shao, C.; Wang, J.; Tian, J.; Tang, Y.-D. Coronary Artery Disease: From Mechanism to Clinical Practice. In Coronary Artery Disease: Therapeutics and Drug Discovery; Advances in Experimental Medicine and Biology; Springer: Singapore, 2020; Volume 1177, pp. 1–36. [Google Scholar] [CrossRef]
- Badimon, L.; Padró, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zannad, F.; Bauersachs, R. Rivaroxaban: A New Treatment Paradigm in the Setting of Vascular Protection? Thromb. Haemost. 2018, 118, S12–S22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, S.; Sudano, I.; Kokubo, Y.; Sulaica, E.M. Arterial hypertension. Lancet 2021, 398, 249–261. [Google Scholar] [CrossRef]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; et al. 2020 International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef]
- Kopp, W. How Western Diet and Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2221–2236. [Google Scholar] [CrossRef] [Green Version]
- Medina-Leyte, D.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- VanderLaan, P.; Reardon, C.A.; Getz, G.S. Site Specificity of Atherosclerosis: Site-Selective Responses to Atherosclerotic Modulators. Arter. Thromb. Vasc. Biol. 2004, 24, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Mussbacher, M.; Schossleitner, K.; Kral-Pointner, J.B.; Salzmann, M.; Schrammel, A.; Schmid, J.A. More than Just a Monolayer: The Multifaceted Role of Endothelial Cells in the Pathophysiology of Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Gutiérrez, E.; Flammer, A.J.; Lerman, L.O.; Elízaga, J.; Lerman, A.; Fernández-Avilés, F. Endothelial dysfunction over the course of coronary artery disease. Eur. Heart J. 2013, 34, 3175–3181. [Google Scholar] [CrossRef] [Green Version]
- Bäck, M.; Yurdagul, A., Jr.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Gu, M.; Jia, X.; Wang, X.; Wu, C.; Guo, J.; Zhang, L.; Du, Y.; Wang, J. Integrated DNA methylation and gene expression analysis identifies SLAMF7 as a key regulator of atherosclerosis. Aging 2018, 10, 1324–1337. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Du, W.; Xu, A.; Huang, Y.; Cao, J.; Zhu, H.; Yang, B.; Shao, X.; He, Q.; Ying, M. The role of autophagy in targeted therapy for acute myeloid leukemia. Autophagy 2021, 17, 2665–2679. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Asada, S.; Kitamura, T. Clonal hematopoiesis and associated diseases: A review of recent findings. Cancer Sci. 2021, 112, 3962–3971. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Natarajan, P. Completing the genetic spectrum influencing coronary artery disease: From germline to somatic variation. Cardiovasc. Res. 2019, 115, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Gresele, P.; Momi, S.; Guglielmini, G. Nitric oxide-enhancing or -releasing agents as antithrombotic drugs. Biochem. Pharmacol. 2019, 166, 300–312. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, X.-M.; Tarnawski, L.; Peleli, M.; Zhuge, Z.; Terrando, N.; Harris, R.A.; Olofsson, P.S.; Larsson, E.; Persson, A.E.G.; et al. Dietary nitrate attenuates renal ischemia-reperfusion injuries by modulation of immune responses and reduction of oxidative stress. Redox Biol. 2017, 13, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hu, H.L.; Feng, X.; Wang, S. Nitrate and Nitrite in Health and Disease. Aging Dis. 2018, 9, 938–945. [Google Scholar] [CrossRef] [Green Version]
- Meza, C.A.; La Favor, J.D.; Kim, D.-H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Carlstrom, M.; Montenegro, M.F. Therapeutic value of stimulating the nitrate-nitrite-nitric oxide pathway to attenuate oxidative stress and restore nitric oxide bioavailability in cardiorenal disease. J. Intern. Med. 2019, 285, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Mourouzis, K.; Siasos, G.; Oikonomou, E.; Zaromitidou, M.; Tsigkou, V.; Antonopoulos, A.; Bletsa, E.; Stampouloglou, P.; Vlasis, K.; Vavuranakis, M.; et al. Lipoprotein-associated phospholipase A2 levels, endothelial dysfunction and arterial stiffness in patients with stable coronary artery disease. Lipids Health Dis. 2021, 20, 12. [Google Scholar] [CrossRef]
- Franzén, O.; Ermel, R.; Cohain, A.; Akers, N.K.; Di Narzo, A.; Talukdar, H.A.; Foroughi-Asl, H.; Giambartolomei, C.; Fullard, J.F.; Sukhavasi, K.; et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science 2016, 353, 827–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillevin, L.; Dörner, T. Vasculitis: Mechanisms involved and clinical manifestations. Arthritis Res. Ther. 2007, 9, S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennette, J.C.; Thomas, D.B.; Falk, R.J. Microscopic polyangiitis (microscopic polyarteritis). Semin. Diagn. Pathol. 2001, 18, 3–13. [Google Scholar] [PubMed]
- Maugeri, N.; Rovere-Querini, P.; Baldini, M.; Sabbadini, M.G.; Manfredi, A.A. Translational Mini-Review Series on Immunology of Vascular Disease: Mechanisms of vascular inflammation and remodelling in systemic vasculitis. Clin. Exp. Immunol. 2009, 156, 395–404. [Google Scholar] [CrossRef]
- Khera, A.V.; Won, H.-H.; Peloso, G.M.; O’Dushlaine, C.; Liu, D.; Stitziel, N.O.; Natarajan, P.; Nomura, A.; Emdin, C.A.; Gupta, N.; et al. Association of Rare and Common Variation in the Lipoprotein Lipase Gene with Coronary Artery Disease. JAMA 2017, 317, 937–946. [Google Scholar] [CrossRef]
- Eric, G.R.; Segrè, A.V.; van de Bunt, M.; Wen, X.; Xi, H.S.; Hormozdiari, F.; Ongen, H. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet. 2018, 50, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Aherrahrou, R.; Guo, L.; Nagraj, V.P.; Aguhob, A.A.; Hinkle, J.; Chen, L.; Soh, J.Y.; Lue, D.; Alencar, G.F.; Boltjes, A.; et al. Genetic Regulation of Atherosclerosis-Relevant Phenotypes in Human Vascular Smooth Muscle Cells. Circ. Res. 2020, 127, 1552–1565. [Google Scholar] [CrossRef]
- Cherepanova, O.A.; Gomez, D.; Shankman, L.S.; Swiatlowska, P.; Williams, J.; Sarmento, O.F.; Alencar, G.F.; Hess, D.L.; Bevard, M.H.; Greene, E.S.; et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat. Med. 2016, 22, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Monguchi, T.; Iwamoto, N.; Akashi, M.; Mori, K.; Oshita, T.; Okano, M.; Toh, R.; Irino, Y.; Shinohara, M.; et al. Targeted Disruption of JCAD (Junctional Protein Associated with Coronary Artery Disease)/KIAA1462, a Coronary Artery Disease–Associated Gene Product, Inhibits Angiogenic Processes In Vitro and In Vivo. Arter. Thromb. Vasc. Biol. 2017, 37, 1667–1673. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.D.; Kaiser, M.A.; Najafabadi, M.G.; Koplev, S.; Zhao, Y.; Douglas, G.; Kyriakou, T.; Andrews, S.; Rajmohan, R.; Watkins, H.; et al. JCAD, a Gene at the 10p11 Coronary Artery Disease Locus, Regulates Hippo Signaling in Endothelial Cells. Arter. Thromb. Vasc. Biol. 2018, 38, 1711–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Xu, Y.; Liu, P.; Zhang, S.; Liu, H.; Slavin, S.; Kumar, S.; Koroleva, M.; Luo, J.; Wu, X.; et al. The novel coronary artery disease risk gene JCAD/KIAA1462 promotes endothelial dysfunction and atherosclerosis. Eur. Heart J. 2019, 40, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Askin, L.; Tibilli, H.; Tanriverdi, O.; Turkmen, S. The Relationship between Coronary Artery Disease and SIRT1 Protein. North. Clin. Istanb. 2020, 7, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Lyu, Q.; Zhao, Q.; Wirka, R.C.; Bagga, J.; Nguyen, T.; Cheng, P.; Kim, J.B.; Pjanic, M.; Miano, J.M.; et al. Coronary Disease-Associated Gene TCF21 Inhibits Smooth Muscle Cell Differentiation by Blocking the Myocardin-Serum Response Factor Pathway. Circ. Res. 2020, 126, 517–529. [Google Scholar] [CrossRef]
- Qi, B.; Chen, J.-H.; Tao, L.; Zhu, C.-M.; Wang, Y.; Deng, G.-X.; Miao, L. Integrated Weighted Gene Co-expression Network Analysis Identified That TLR2 and CD40 Are Related to Coronary Artery Disease. Front. Genet. 2021, 11, 613744. [Google Scholar] [CrossRef]
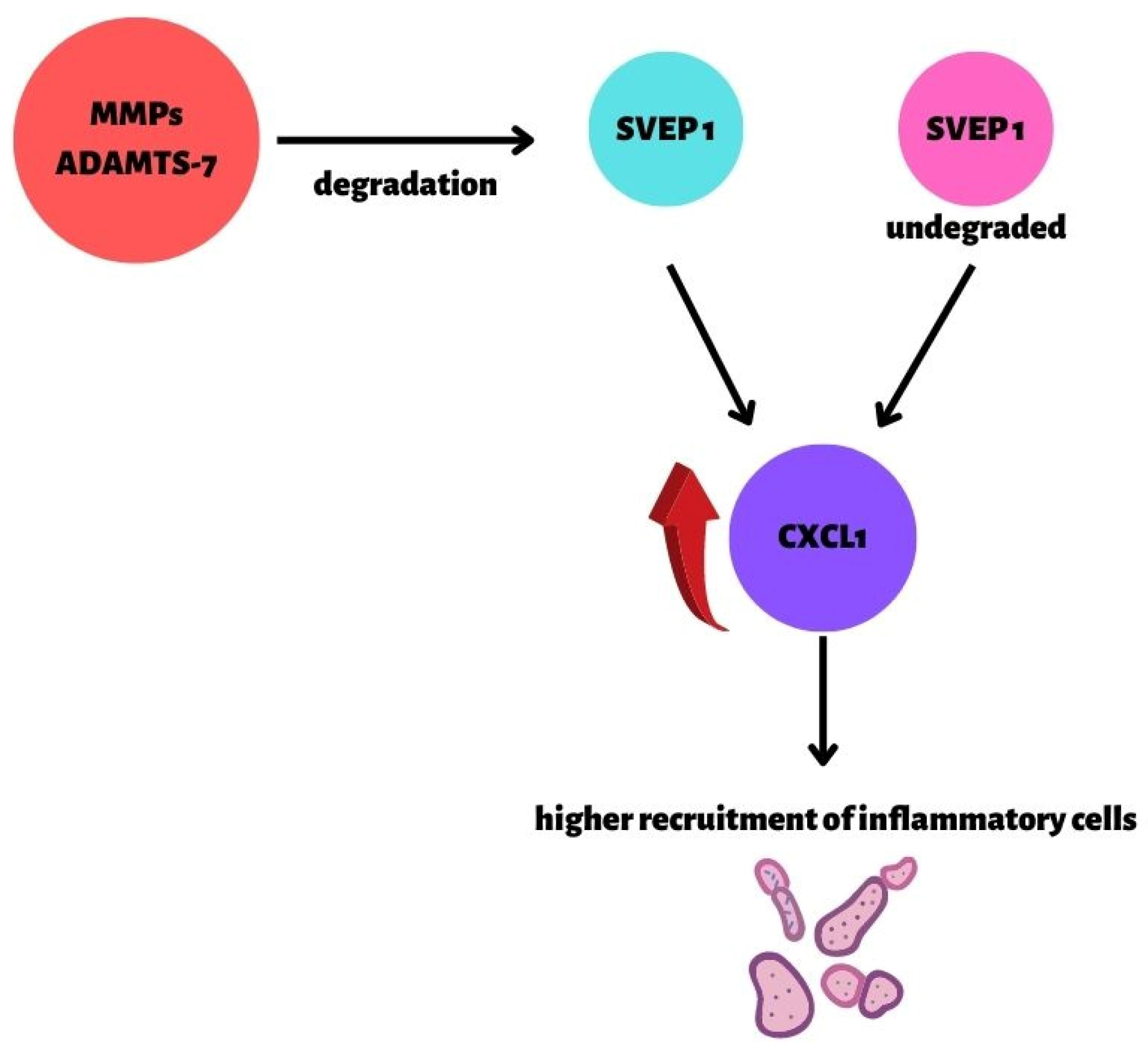
- Winkler, M.J.; Müller, P.; Sharifi, A.M.; Wobst, J.; Winter, H.; Mokry, M.; Ma, L.; Van Der Laan, S.W.; Pang, S.; Miritsch, B.; et al. Functional investigation of the coronary artery disease gene SVEP1. Basic Res. Cardiol. 2020, 115, 67. [Google Scholar] [CrossRef]
- Bengtsson, E.; Hultman, K.; Dunér, P.; Asciutto, G.; Almgren, P.; Orho-Melander, M.; Melander, O.; Nilsson, J.; Hultgårdh-Nilsson, A.; Gonçalves, I. ADAMTS-7 is associated with a high-risk plaque phenotype in human atherosclerosis. Sci. Rep. 2017, 7, 3753. [Google Scholar] [CrossRef] [Green Version]
- Pu, X.; Xiao, Q.; Kiechl, S.; Chan, K.; Ng, F.L.; Gor, S.; Poston, R.N.; Fang, C.; Patel, A.; Senver, E.C.; et al. ADAMTS7 Cleavage and Vascular Smooth Muscle Cell Migration Is Affected by a Coronary-Artery-Disease-Associated Variant. Am. J. Hum. Genet. 2013, 92, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Emdin, C.A.; Khera, A.V.; Klarin, D.; Natarajan, P.; Zekavat, S.; Nomura, A.; Haas, M.E.; Aragam, K.; Ardissino, D.; Wilson, J.G.; et al. Phenotypic Consequences of a Genetic Predisposition to Enhanced Nitric Oxide Signaling. Circulation 2018, 137, 222–232. [Google Scholar] [CrossRef]
- Jeanette, E.; Stark, K.; Esslinger, U.B.; Rumpf, P.M.; Koesling, D.; de Wit, C.; Kaiser, F.J.; Braunholz, D.; Medack, A. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 2013, 504, 432–436. [Google Scholar] [CrossRef]
- Do, R.; Project, N.E.S.; Stitziel, N.; Won, H.-H.; Jørgensen, A.B.; Duga, S.; Merlini, P.A.; Kiezun, A.; Farrall, M.; Goel, A.; et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015, 518, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, A.B.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Loss-of-Function Mutations in APOC3 and Risk of Ischemic Vascular Disease. N. Engl. J. Med. 2014, 371, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewey, F.E.; Gusarova, V.; O’Dushlaine, C.; Gottesman, O.; Trejos, J.; Hunt, C.; Van Hout, C.V.; Habegger, L.; Buckler, D.; Lai, K.-M.V.; et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
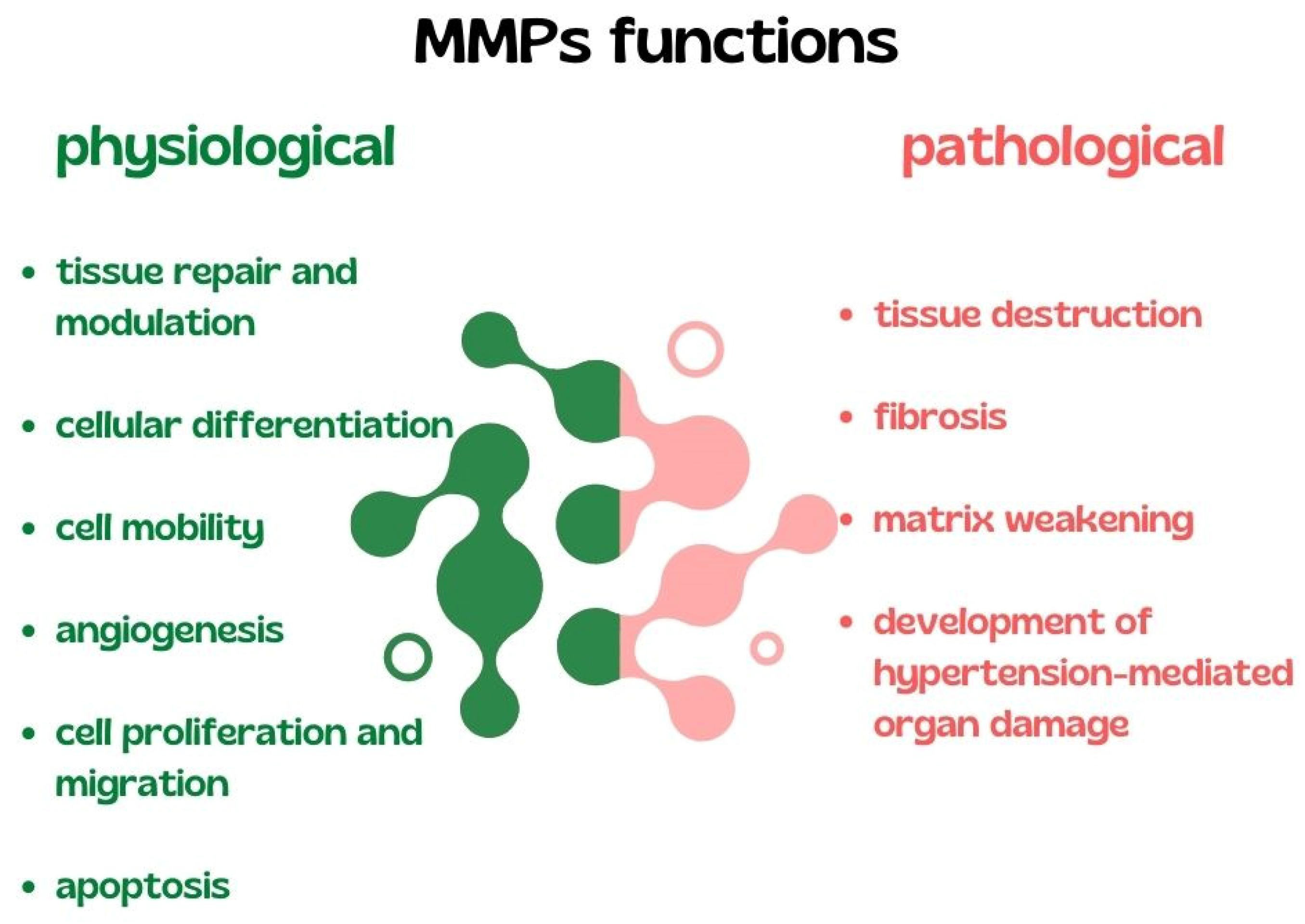
- Laronha, H.; Caldeira, J. Structure and Function of Human Matrix Metalloproteinases. Cells 2020, 9, 1076. [Google Scholar] [CrossRef] [PubMed]
- Bisogni, V.; Cerasari, A.; Pucci, G.; Vaudo, G. Matrix Metalloproteinases and Hypertension-Mediated Organ Damage: Current Insights. Integr. Blood Press. Control 2020, 13, 157–169. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Khalil, R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharmacol. 2008, 75, 346–359. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, C.; Dentali, F.; Nicolini, E.; Maresca, A.M.; Tayebjee, M.H.; Franz, M.; Guasti, L.; Venco, A.; Schiffrin, E.; Lip, G.Y.; et al. Plasma levels of matrix metalloproteinases and their inhibitors in hypertension: A Systematic Review and Meta-Analysis. J. Hypertens. 2012, 30, 3–16. [Google Scholar] [CrossRef]
- Kostov, K.; Blazhev, A. Changes in Serum Levels of Matrix Metalloproteinase-1 and Tissue Inhibitor of Metalloproteinases-1 in Patients with Essential Hypertension. Bioengineering 2022, 9, 119. [Google Scholar] [CrossRef]
- Basu, R.; Lee, J.; Morton, J.S.; Takawale, A.; Fan, D.; Kandalam, V.; Wang, X.; Davidge, S.T.; Kassiri, Z. TIMP3 is the primary TIMP to regulate agonist-induced vascular remodelling and hypertension. Cardiovasc. Res. 2013, 98, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Pushpakumar, S.; Kundu, S.; Pryor, T.; Givvimani, S.; Lederer, E.; Tyagi, S.C.; Sen, U. Angiotensin-II induced hypertension and renovascular remodelling in tissue inhibitor of metalloproteinase 2 knockout mice. J. Hypertens. 2013, 31, 2270–2281. [Google Scholar] [CrossRef] [Green Version]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; La Rosa, C.C.-D.; Ramirez-Acuña, J.M.; Perez-Romero, A.B.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Gong, Z.; Li, Z.; Li, L.; Kong, W. Vascular Extracellular Matrix Remodeling and Hypertension. Antioxid. Redox Signal. 2021, 34, 765–783. [Google Scholar] [CrossRef]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [Green Version]
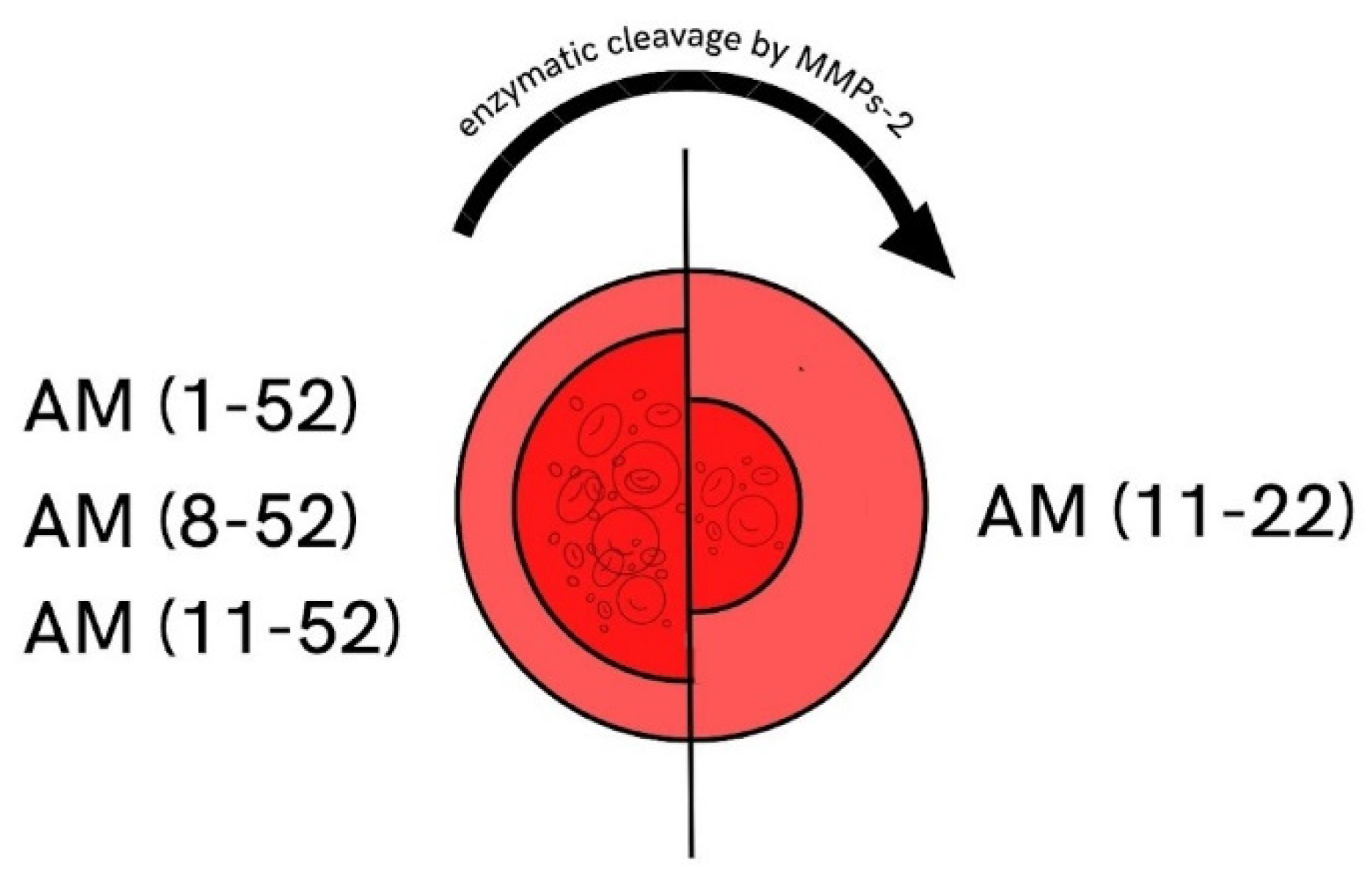
- Martínez, A.; Oh, H.-R.; Unsworth, E.J.; Bregonzio, C.; Saavedra, J.M.; Stetler-Stevenson, W.; Cuttitta, F. Matrix metalloproteinase-2 cleavage of adrenomedullin produces a vasoconstrictor out of a vasodilator. Biochem. J. 2004, 383, 413–418. [Google Scholar] [CrossRef]
- Wakisaka, Y.; Chu, Y.; Miller, J.; Rosenberg, G.A.; Heistad, D.D. Spontaneous Intracerebral Hemorrhage during Acute and Chronic Hypertension in Mice. J. Cereb. Blood Flow Metab. 2010, 30, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Mirhafez, S.R.; Mohebati, M.; Disfani, M.F.; Karimian, M.S.; Ebrahimi, M.; Avan, A.; Eslami, S.; Pasdar, A.; Rooki, H.; Esmaeili, H.; et al. An imbalance in serum concentrations of inflammatory and anti-inflammatory cytokines in hypertension. J. Am. Soc. Hypertens. 2014, 8, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Sesso, H.D.; Wang, L.; Buring, J.E.; Ridker, P.M.; Gaziano, J.M. Comparison of Interleukin-6 and C-Reactive Protein for the Risk of Developing Hypertension in Women. Hypertension 2007, 49, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Hage, F. C-reactive protein and Hypertension. J. Hum. Hypertens. 2014, 28, 410–415. [Google Scholar] [CrossRef]
- Kong, H.; Qian, Y.-S.; Tang, X.-F.; Zhang, J.; Gao, P.-J.; Zhang, Y.; Zhu, D.-L. C-reactive protein (CRP) gene polymorphisms, CRP levels and risk of incident essential hypertension: Findings from an observational cohort of Han Chinese. Hypertens. Res. 2012, 35, 1019–1023. [Google Scholar] [CrossRef] [Green Version]
- Lima, V.V.; Zemse, S.M.; Chiao, C.-W.; Bomfim, G.F.; Tostes, R.C.; Webb, R.C.; Giachini, F.R. Interleukin-10 limits increased blood pressure and vascular RhoA/Rho-kinase signaling in angiotensin II-infused mice. Life Sci. 2016, 145, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Sarwar, Z.; Yang, X.-P.; Peterson, E.L.; Xu, J.; Janic, B.; Rhaleb, N.; Carretero, O.A.; Rhaleb, N.-E. Profibrotic Role for Interleukin-4 in Cardiac Remodeling and Dysfunction. Hypertension 2015, 66, 582–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parissis, J.T.; Korovesis, S.; Giazitzoglou, E.; Kalivas, P.; Katritsis, D. Plasma profiles of peripheral monocyte-related inflammatory markers in patients with arterial hypertension. Correlations with plasma endothelin-1. Int. J. Cardiol. 2002, 83, 13–21. [Google Scholar] [CrossRef]
- Dörffel, Y.; Lätsch, C.; Stuhlmüller, B.; Schreiber, S.; Scholze, S.; Burmester, G.R.; Scholze, J. Preactivated Peripheral Blood Monocytes in Patients with Essential Hypertension. Hypertension 1999, 34, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraco, G.; Sugiyama, Y.; Lane, D.; Garcia-Bonilla, L.; Chang, H.; Santisteban, M.; Racchumi, G.; Murphy, M.; Van Rooijen, N.; Anrather, J.; et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J. Clin. Investig. 2016, 126, 4674–4689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.Z.; Li, Y.; Li, L.; Shah, K.H.; Bernstein, K.; Lyden, P.D.; Shi, P. Microglia Participate in Neurogenic Regulation of Hypertension. Hypertension 2015, 66, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Barbaro, N.R.; Foss, J.D.; Kryshtal, D.O.; Tsyba, N.; Kumaresan, S.; Xiao, L.; Mernaugh, R.L.; Itani, H.A.; Loperena, R.; Chen, W.; et al. Dendritic Cell Amiloride-Sensitive Channels Mediate Sodium-Induced Inflammation and Hypertension. Cell Rep. 2017, 21, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Hevia, D.; Araos, P.; Prado, C.; Luppichini, E.F.; Rojas, M.; Alzamora, R.; Cifuentes-Araneda, F.; Gonzalez, A.A.; Amador, C.A.; Pacheco, R.; et al. Myeloid CD11c + Antigen-Presenting Cells Ablation Prevents Hypertension in Response to Angiotensin II Plus High-Salt Diet. Hypertension 2018, 71, 709–718. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Dowling, J.; Ling, Y.H.; Diep, H.; Chan, C.T.; Ferens, D.; Kett, M.M.; Pinar, A.; Samuel, C.S.; Vinh, A.; et al. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. J. Cereb. Blood Flow Metab. 2016, 173, 752–765. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Ling, Y.H.; Huuskes, B.M.; Ferens, D.M.; Saini, N.; Chan, C.T.; Diep, H.; Kett, M.M.; Samuel, C.S.; Kemp-Harper, B.K.; et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc. Res. 2019, 115, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Abboud, F.M.; Harwani, S.; Chapleau, M. Autonomic Neural Regulation of the Immune System: Implications for Hypertension and Cardiovascular Disease. Hypertension 2012, 59, 755–762. [Google Scholar] [CrossRef]
- Harwani, S.C.; Chapleau, M.W.; Legge, K.L.; Ballas, Z.K.; Abboud, F.M. Neurohormonal Modulation of the Innate Immune System Is Proinflammatory in the Prehypertensive Spontaneously Hypertensive Rat, a Genetic Model of Essential Hypertension. Circ. Res. 2012, 111, 1190–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, R.; González, J.; Paoletto, F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens. Res. 2011, 34, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, S.D. The Cooperative Roles of Inflammation and Oxidative Stress in the Pathogenesis of Hypertension. Antioxid. Redox Signal. 2014, 20, 102–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Khotina, V.; Ivanova, E.A.; Orekhov, A.N. NADPH Oxidases and Their Role in Atherosclerosis. Biomedicines 2020, 8, 206. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Reactive oxygen species in vascular biology: Implications in hypertension. Histochem. Cell Biol. 2004, 122, 339–352. [Google Scholar] [CrossRef]
- Dikalova, A.; Clempus, R.; Lassègue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 Overexpression Potentiates Angiotensin II-Induced Hypertension and Vascular Smooth Muscle Hypertrophy in Transgenic Mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef] [Green Version]
- Nosalski, R.; Mikolajczyk, T.; Siedlinski, M.; Saju, B.; Koziol, J.; Maffia, P.; Guzik, T. Nox1/4 inhibition exacerbates age dependent perivascular inflammation and fibrosis in a model of spontaneous hypertension. Pharmacol. Res. 2020, 161, 105235. [Google Scholar] [CrossRef]
- Murdoch, C.E.; Alom-Ruiz, S.P.; Wang, M.; Zhang, M.; Walker, S.; Yu, B.; Brewer, A.; Shah, A.M. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res. Cardiol. 2011, 106, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 Is a Protective Reactive Oxygen Species Generating Vascular NADPH Oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH Oxidase Enhances Vasodilatation and Reduces Blood Pressure In Vivo. Arter. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef] [Green Version]
- Tanase, D.M.; Apostol, A.G.; Costea, C.F.; Tarniceriu, C.C.; Tudorancea, I.; Maranduca, M.A.; Floria, M.; Serban, I.L. Oxidative Stress in Arterial Hypertension (HTN): The Nuclear Factor Erythroid Factor 2-Related Factor 2 (Nrf2) Pathway, Implications and Future Perspectives. Pharmaceutics 2022, 14, 534. [Google Scholar] [CrossRef] [PubMed]
- Dovinova, I.; Kvandová, M.; Balis, P.; Gresova, L.; Majzunova, M.; Horakova, L.; Chan, J.Y.; Barancik, M. The role of Nrf2 and PPARgamma in the improvement of oxidative stress in hypertension and cardiovascular diseases. Physiol. Res. 2020, 69, S541–S553. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, Z.; Mohammad, R.S.; Lokhandwala, M.F.; Banday, A.A. Nrf2 inhibition induces oxidative stress, renal inflammation and hypertension in mice. Clin. Exp. Hypertens. 2021, 43, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Ren, K.; Liu, S.; Li, W.; Huang, C.; Yang, X. MicroRNA-140-5p aggravates hypertension and oxidative stress of atherosclerosis via targeting Nrf2 and Sirt2. Int. J. Mol. Med. 2019, 43, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biernacki, M.; Łuczaj, W.; Jarocka-Karpowicz, I.; Ambrożewicz, E.; Toczek, M.; Skrzydlewska, E. The Effect of Long-Term Administration of Fatty Acid Amide Hydrolase Inhibitor URB597 on Oxidative Metabolism in the Heart of Rats with Primary and Secondary Hypertension. Molecules 2018, 23, 2350. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frąk, W.; Wojtasińska, A.; Lisińska, W.; Młynarska, E.; Franczyk, B.; Rysz, J. Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease. Biomedicines 2022, 10, 1938. https://doi.org/10.3390/biomedicines10081938
Frąk W, Wojtasińska A, Lisińska W, Młynarska E, Franczyk B, Rysz J. Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease. Biomedicines. 2022; 10(8):1938. https://doi.org/10.3390/biomedicines10081938
Chicago/Turabian StyleFrąk, Weronika, Armanda Wojtasińska, Wiktoria Lisińska, Ewelina Młynarska, Beata Franczyk, and Jacek Rysz. 2022. "Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease" Biomedicines 10, no. 8: 1938. https://doi.org/10.3390/biomedicines10081938