
Aβ and Tau Regulate Microglia Metabolism via Exosomes in Alzheimer’s Disease


Key Laboratory of Pathobiology, Department of Pathophysiology, Ministry of Education, College of Basic Medical Sciences, Jilin University, 126 Xinmin Street, Changchun 130021, China
*
Authors to whom correspondence should be addressed.
Biomedicines 2022, 10(8), 1800; https://doi.org/10.3390/biomedicines10081800
Submission received: 17 May 2022
/
Revised: 4 July 2022
/
Accepted: 11 July 2022
/
Published: 27 July 2022
(This article belongs to the Special Issue Regulation and Deregulation of Cell Metabolism in the Brain: Molecular Aspects, Functional Outcomes)
Abstract
:One of the most striking hallmarks shared by various neurodegenerative diseases, including Alzheimer’s disease (AD), is microglia-mediated neuroinflammation. The main pathological features of AD are extracellular amyloid-β (Aβ) plaques and intracellular tau-containing neurofibrillary tangles in the brain. Amyloid-β (Aβ) peptide and tau protein are the primary components of the plaques and tangles. The crosstalk between microglia and neurons helps maintain brain homeostasis, and the metabolic phenotype of microglia determines its polarizing phenotype. There are currently many research and development efforts to provide disease-modifying therapies for AD treatment. The main targets are Aβ and tau, but whether there is a causal relationship between neurodegenerative proteins, including Aβ oligomer and tau oligomer, and regulation of microglia metabolism in neuroinflammation is still controversial. Currently, the accumulation of Aβ and tau by exosomes or other means of propagation is proposed as a regulator in neurological disorders, leading to metabolic disorders of microglia that can play a key role in the regulation of immune cells. In this review, we propose that the accumulation of Aβ oligomer and tau oligomer can propagate to adjacent microglia through exosomes and change the neuroinflammatory microenvironment by microglia metabolic reprogramming. Clarifying the relationship between harmful proteins and microglia metabolism will help people to better understand the mechanism of crosstalk between neurons and microglia, and provide new ideas for the development of AD drugs.
1. Introduction
AD is a progressive chronic neurodegenerative disease characterized by cognitive dysfunction that affects the cognitive and emotional behavior of patients, generally 65 years and older [1]. AD is characterized by two major pathological lesions in the brain, amyloid plaques composed mainly of amyloid-β (Aβ) peptides and hyperphosphorylated tau, and neurofibrillary tangles (NFTs) [2]. NFTs are described as one of the leading brain injuries in AD [3] and AD is the most common type of amyloidosis in which amyloid and insoluble neurofibrillary proteins are abnormally deposited in neurons and glia [4]. This may result in the dysfunction of specific brain subregions or neuronal populations, leading to motor or psychiatric disorders with associated behavioral deficits.
Microglia, as the resident immune cells in the brain, dynamically monitor the microenvironment through the movement process of constantly interacting with neurons [5,6]. This crosstalk between microglia and neurons helps maintain brain homeostasis. In the large-scale genome-wide association studies, more than 20 loci associated with AD have been identified, many of which are expressed or only expressed in microglia or myeloid cells [7,8]. Before the formation of plaques in AD, microglia play a role in promoting early synaptic loss and dysfunction [9]. For example, adenosine, the catabolite of ATP, primarily acts on microglia through adenosine A2a receptor (A2AR) and adenosine A3 receptor [10,11]. The combination of adenosine and microglia A2AR promotes neuroprotection by releasing nerve growth factor, but it also induces the expression of prostaglandin E2 and cyclooxygenase-2 and the release of nitric oxide (NO), causing damage to neurons [12]. In clinical studies, the polymorphism of the microglia fractalkine receptor (CX3CR1) has been found. CX3CR1 is associated with the faster progression of disease symptoms and shorter survival time of patients with late-onset AD, which may be caused by the reduction of microglia neuron communication due to the reduction in the signals of CX3CR1 and its ligand chemokine CX3CL1 [13,14]. In addition, fragments of dead neurons are thought to trigger glial cell-mediated neuroinflammation in AD, increasing neuronal death [15]. Microglia can often be reprogrammed by metabolism to change their phenotype and slow the process of neuronal damage [16].
The crosstalk between neurons and microglia may be the key to induce neuroinflammation, as mitochondrial energy metabolism in neurons is crucial for neuronal homeostasis in AD. Recent studies have shown that microglia monitor and protect neuronal function through special purinergic connections, and the activity of neuronal mitochondria is related to the formation of microglia connections [14]. Furthermore, microglia activation-mediated activation of astrocytes can aggravate neuroinflammation and induce neuronal damage in the AD mouse model [17]. Recent evidence suggests that extracellular vesicles (EVs) secreted by neurons and glia including exosomes play a key role in intercellular communication and neuroinflammation by transporting messenger RNA (mRNA), microRNA (miRNA) and proteins [18,19,20]. Similarly, microglia can secrete extracellular vesicles for cell-to-cell communication [21], and the composition of its content depends on the state of the cells and the nature of the priming stimulus [22,23]. This has further deepened the complexity of the cross-talk between microglia and neurons in neurodegenerative diseases. In the case of bidirectional communication between microglia and neurons, microglia phagocytosis and the production of cytokines and free radicals can also be regulated by the neuronal exosomes [24]. In conclusion, the role of microglia in multiple neuronal pathways is crucial in AD.
In summary, the crosstalk between microglia and neurons is a key factor affecting the progression of AD, and metabolic reprogramming of microglia may be an important link in preventing the occurrence and development of AD. In this review, we describe the regulation of microglia metabolism by the accumulation of Aβ oligomers and tau oligomers, which in turn aggravate neuroinflammation and aggravate neuronal damage. Further exploration of the mechanism of the effects of harmful proteins on microglia metabolism may provide new strategies for the development of targeted drugs and the treatment of neurodegenerative diseases.
2. Plaques and Tangles Formed by the Accumulation of Aβ Oligomers and Tau Oligomers Play a Key Neuropathogenic Role Leading to Neuronal Damage
Although the cause of AD is not fully understood, two major factors that are often cited in its progression are plaques and tangles. Aβ denotes peptides of 36–43 amino acids that are the main component of the amyloid plaques found in the brains of people with Alzheimer’s disease [25]. The imprecise cleavage of γ-secretase at C-terminus of Aβ sequence results in two major Aβ isoforms: Aβ42 (42 residues long) and Aβ40 (40 residues long). The only difference between them is extra isoleucine and an alanine at the C-terminus of Aβ42 [26]. However, the toxicity of Aβ42 is much more toxic than Aβ40 due to its greater tendency to fibrillise [25,27]. Currently, most people suggested that the mechanism of plaques formation is Aβ accumulated, which was called the “amyloid cascade hypothesis” (Figure 1A,B). Aβ is typically produced by cleavage of amyloid precursor proteins (APP). APP is a type 1 transmembrane protein, which helps the neuron grow and repair itself after an injury [28]. Normally, APP is chopped up by α-secretase and γ-secretase, then formed extracellular soluble products called sAPPα and a secreted fragment called p3, the membrane-bound intracellular domain of APP (AICD). This whole process called the non-amyloidogenic pathway. However, in the amyloidogenic pathway, APP is first cleaved by β-secretase instead of the processing by γ-secretase, producing extracellular products called sAPPβ and Aβ, and the same membrane-bound AICD. The leftover fragment creates a monomer called Aβ, and the monomer tends to be more chemically ‘sticky’. The monomeric Aβ spontaneously assembles into soluble oligomers, and the oligomers are clustered to form insoluble fibrils, which are called β-amyloid plaques [29]. These plaques potentially get between the neurons and make brain functions impaired. In addition, plaques can initiate an immune response and cause inflammation, such as activation of microglia, which could damage surrounding neurons.
Another big part of AD is tangles, which are actually found inside the cell. Neurons are held together by their cytoskeleton, which is partly made up of microtubules. Tau, a normal, unfolded, highly soluble protein, in normal circumstances, tau is a microtubule-associated protein involved in microtubule stabilization [28,30].
Tau is found primarily in axons where it regulates microtubule polymerization and stabilization [31]. The tau protein contains various phosphorylation sites, and under normal conditions, phosphorylation helps to maintain the cytoskeletal structure [32]. Phosphorylation of Tau serine/threonine effectively regulates the binding affinity of Tau to microtubules. However, hyperphosphorylation of tau is known to contribute to the pathology of Alzheimer’s disease, with approximately 45 specific phosphorylation sites identified in the brain of Alzheimer’s disease patients [32,33]. Anormal phosphorylation of the tau protein changes its structure and hyperphosphorylated tau protein becomes dissociated from neuronal microtubules and accumulates in paired helical filaments, and proteolytic processing leads to the formation of tau oligomers, which stimulate the process of aggregation and form NFTs [34].
Some studies suggest that amyloid-β plaques lies upstream of tau oligomers and triggers tau pathology [2,35]. However, there are also studies that have shown that tau can act independently of Aβ, leading to neurodegeneration [36,37,38]. Summarily, tau and Aβ play a role in parallel pathways, leading to AD.
3. Metabolic Reprogramming of Microglia to Aggravate the Neuroinflammatory Microenvironment
Microglia as tissue macrophages of the CNS continuously monitors cerebral parenchyma to detect neuronal activities and alteration of homeostatic processes [39,40,41]. Brain metabolism is tightly controlled to maintain accurate neuronal function [42] that also requires metabolic interaction with microglia [43]. Recent studies have shown that microglia might alter neuronal substrate utilization during neuroinflammation [43].
All macrophages, including activated microglia, are functionally heterogeneous and highly plastic. Microglia, as the immunologically active cells of the brain, are the most important immune defense lines of the central nervous system and play an increasingly important role in maintaining normal brain function. Traditionally, microglia are in a resting state (M0 phenotype) under physiological conditions and play an “immune surveillance” role. Under pathological conditions, microglia are rapidly activated, and the activation is accompanied by changes in transcriptional adaptive functions. Neuroinflammatory (M1 polarization) microglia release pro-inflammatory cytokines, chemokines and neurotoxic factors. The neuroprotective (M2 polarization) microglia achieve neuroprotection by promoting tissue repair and regeneration through expressing anti-inflammatory factors [44]. Clinical studies have shown that overactivated M1 phenotype microglia can cause neuronal disability, damage and degeneration; and play an important role in cerebrovascular and neurodegenerative diseases and neurodevelopmental and mental disorders [16,45,46,47]. Microglia secrete proinflammatory factors including Interleukin-1β [26,27,48,49], Interleukin-6 [49,50,51] and TNFα [49,52,53], along with an increased production of ROS [54,55,56], which can cause damage to the surrounding neuronal cells. In AD, M1 phenotype microglia that secrete pro-inflammatory factors maintain the neuroinflammatory microenvironment and continuously aggravate neuronal damage.
The metabolic phenotype determines the polarizing phenotype. Microglia undergo comprehensive phenotypic remodeling to adapt effector functions [43]. To perform this multitude of functions, microglial metabolism must be precisely regulated to ensure adequate energy supply and synthesis of response-specific molecules [57]. Therefore, microglia are metabolically flexible to reprogram the metabolism of microglia to change the neuroinflammatory microenvironment and block the aggravation of neuronal damage. M1-type pro-inflammatory microglia cells showed increased glycolytic metabolism and impaired OXPHOS, while M2-type anti-inflammatory macrophages showed increased OXPHOS (Figure 2). Model M1 utilizes this anabolic metabolism to produce much lower ATP than OXPHOS, necessary for balancing energy production with macromolecular synthesis [45,58,59]. In addition, glycolysis provides food for the pentose-phosphate pathway (PPP). PPP supports the production of amino acids for protein synthesis, ribose for nucleotides, and NADPH needed to produce ROS. In M1 microglia, TCA is interrupted, and the intermediate (acetyl-CoA) induces fatty acids, lipids, and prostaglandin synthesis or is used to induce cytokine production [60]. In the proteomic study on AD of Johnson et al. [61], the module of the protein network linked to glycometabolism emerged as one of the most significantly associated modules with AD pathology and cognitive impairment. Also, the enrichment of microglial proteins in the glucose metabolism module were observed. Among them, glycolysis-related proteins were increased, including lactate dehydrogenase (LDH), pyruvate kinase M(PKM) and glyceraldehyde 3- phosphate dehydrogenase (GAPDH). In addition, Abhishek JHA et al. found that the interruption of TCA cycle resulted in the accumulation of citrate and succinate by coordinating the integration of metabolomics and transcription, and revealed that glutamine metabolism was characterized by M2 polarization [62]. Up-regulation of iNOS and NO production in M1-type macrophages, which nitrosylates proteins in the mitochondrial electron transport chain, leads to suppression of OXPHOS [57]. In most cases, however, the blood-brain barrier prevents the influx of alternative substrates (fatty acids, amino acids, ketones) and therefore the brain needs a continuous supply of glucose. As blood glucose levels decrease, ketones produced by peripheral fatty acid metabolism supplement glucose as an energy source [46]. Fatty acid oxidation (FAO) fuels OXPHOS, and has been shown to affect polarization [63]. Carroll et al. [64] demonstrated in bone marrow-derived macrophages that CoA promotes the localization of TLR4in the lipid raft cell membrane, linking fatty acid synthase to pro-inflammatory macrophage activation.
Polyamine metabolism of microglia can also regulate its energy metabolism. In AD patients, there is typical up-regulation of arginase (ARG), arginine brain deprivation, and a large increase in brain polyamine levels. Arginine generates ornithine by the action of arginase; ornithine generates putrescine by the action of ornithine decarboxylase, and spermidine is generated by spermidine synthase. One of the functions of spermidine is to participate in the post-translational modification of eIF5A to form hydroxybutylamine-modified hypusine, abbreviated as eIF5AH. This modification is necessary for the function of eIF5A. Therefore, the M2-type polarization of macrophages dependent on oxidative phosphorylation is eIF5AH-dependent, which in turn enhances the arginase activity, increases the arginine-converted spermidine, and further enhances its oxidative phosphorylation, and maintains its M2 phenotype. Studies have shown that polyamine metabolism and arginine metabolism of microglia are dependent on the change of polarization phenotype [65,66,67,68,69].
In summary, the harmful proteins tau and Aβ can directly regulate the energy metabolism of microglia, and indirectly affect its energy metabolism through FAO, glutamine metabolism, and polyamine pathway. The polarization of microglia depends on the metabolic pattern of mitochondria, which regulates the polarization of microglia. Therefore, tau and Aβ regulating microglia energy metabolism, and neuron–microglia crosstalk may be new therapeutic approaches for AD.
4. Aβ and Tau as a Regulator of Microglia Metabolism Reprogramming
The relationship between tau and Aβ and microglia metabolism has been confirmed at this stage (Table 1). Microglia can recognize Aβ oligomer and tau as the danger signal. Sung et al. cultured pure primary microglia from neonatal mouse brain tissues. The PMGs were then treated with various forms of Aβ, including monomeric Aβ, oligomeric Aβ and fibrillary Aβ, all forms of Aβ altered the morphology of the microglia, Aβ substantially increased the expression of pro-inflammatory cytokines protein and mRNA [70]. Oligomeric tau was co-located with microglia in animal models as well as in the brains of AD patients using immunofluorescence by Ashley N. Nilson et al. [71]. What is more, people believed Aβ and tau could induce mitochondrial toxicity and metabolic dysfunction. Aβ induces metabolic reprogramming of microglia from OXPHOS to glycolysis, which is dependent on the mTOR-HIF-1α pathway [70]. The mTOR pathway regulates glucose metabolism as part of a mechanism for sensing the energy status of the cell. Phosphorylated mTOR increases the expression of HIF-1α, the master transcriptional regulator of glycolysis [72]. Rosella Abeti et al. found that Aβ affects mitochondrial oxygen consumption and that cells appear to deplete NADH [73]. Joshi AU et al. found iron accumulation in microglia isolated from APP/PS1 mice and in primary IFN+Aβ-treated mouse microglia, with decreased ability to engulf Aβ with increased glycolysis [74]. Consistent with microglia effects, studies have shown that IFNγ+Aβ increases the expression of HK II and PKM2, which catalyze the first and last reactions in glycolysis, while IFNγ+Aβ also increases PFKFB3. PFKFB3 catalyzes the production of fructose 2,6-diphosphate, which is the allosteric activator of phosphofructokinase. PFKFB3 is recognized as a key glycolysis regulator, responsible for the conversion of fructose-6-phosphate to fructose-1,6-diphosphate, a rate-limiting irreversible glycolysis [75,76].
Receptors on microglia that recognize Aβ and tau can also participate in the regulation of metabolic reprogramming. Trigger receptor 2 (TREM2) expressed on myeloid cells is a microglia receptor that recognizes changes in the lipid microenvironment that may occur during Aβ accumulation and neuronal degeneration of AD. The risk of AD is related to TREM2 receptor subtype variants on microglia. Yingjun Zhao et al. proved that the extracellular domain of TREM2 can bind to Aβ [111]. Using genome-wide RNA sequencing and multiphoton microscopy, we further identified metabolically deficient microglia in 5XFAD mice and found that exposure to Aβ triggered acute microglia inflammation with metabolic reprogramming from OXPHOS to glycolysis, following the discovery by Wang Y et al. in a 5XFAD mouse, an AD mouse model, that Aβ affected microglia activation through the receptor TREM2 [70,82]. The study of Tyler K. Ulland et al. showed that TREM2 deficiency impairs mTOR signaling and enhances AMPK activation in microglia. Furthermore, gene expression microarray analyses of sorted microglia from Trem2 KO 5XFAD and 5XFAD mice revealed that TREM2 deficiency was associated with decreased expression of genes encoding translation initiation factors, ribosomal proteins, glucose transporters, glycolytic enzymes, as well as the transcription factor HIF1α that controls glycolysis [77]. TREM2 can also over-activate AKT [78], and then promote glycolysis through mTOR/HIF1α pathway [70]. Similarly, tau can also affect the metabolism of microglia through TREM2 [93,94,95].
The Toll-like receptor can recognize Aβ and activate microglia [83], and promote metabolic reprogramming by activating the ATP-Citric Acid Lyase (Acly)-driven glucose uptake and cytosolic synthesis of acetyl coenzyme A and oxaloacetic acid [84], or up-regulating PFKFB3 expression [76].Consistent with this, an analysis of resting brain PET by S. Neil Vaishnavi et al. [112] and studies of FDG-PET (for quantitative measurement of local human brain glucose metabolism) in AD patients by Andrei G. Vlassenko et al. [113] showed an increased dependence on aerobic glycolysis in regions associated with Aβ deposition space in the brain of AD patients. Cytochrome c oxidase (COX) and adenine nucleotide transporter (ANT) are targets of chemically synthesized pathological forms of tau, but ANT is a unique mitochondrial target responsible for OXPHOS damage, discovered by Atlante A et al. [114]; Aβ can also enter the mitochondria through the outer membrane transposase (TOM) [115] and cyclophilin D [116] which is a component of the mitochondrial permeability transition pore, and then binds to alcohol dehydrogenase [117], thus increasing Ca2+ influx [117] and TCA cycle disorders (such as abnormal metabolism of α -ketoglutarate [118], affecting the metabolism of microglia [119]. Aβ can also interact with formyl peptide receptors on microglia, and the activation of formyl peptide receptors can then activate the AMPK pathway and inhibit the activation of the mTOR pathway to regulate the metabolism of microglia [79]. The RAGE receptor is related to the recognition of Aβ by microglia. Rashid Deane et al. found that when RAGE receptor was blocked, the pro-inflammatory factors secreted by microglia induced by Aβ decreased [120]. Tau activation of microglia is related to apolipoprotein e gene (APOE) [96,97,98]. APOE may affect many metabolic pathways, such as lipid metabolism [98,100], glucose metabolism [100,101].
Aβ-mediated epigenetic modifications may also be involved in the regulation of microglial metabolism. Lysine acetylation regulates key metabolic enzymes, which thereafter regulate immune processes. Histone deacetylases (HDACs) can act as transcriptional repressors of genes, which regulate glycolytic enzymes and their activity has been linked to inflammatory responses but it is not clear [121]. SITR1 is NAD+ dependent HDACs. Cell level of NAD+ is an important indicator of cell energy state [90]. Grazia Ilaria Caruso et al. found that Aβ can up-regulate the expression of SRIT1 in the nucleus of human microglia cell line HMC3 and activate the enzyme activity of SIRT1 [89]. Lactate is not only a metabolic waste; glycolysis-derived lactate was identified as a substrate for histone lactonization [122] and Rui-Yuan Pan et al. found increased histone lactonization in microglia adjacent to 5XFAD mouse plaques. Moreover, histone lactonization directly stimulated the gene transcription level, and the mRNA level expression of glycolysis-related genes Hif-1α, Pkm2 and Ldha were significantly increased at the same time [91].
In addition, Aβ and tau can indirectly affect metabolism by up-regulating cytokines and pro-inflammatory reactions related to the M1 phenotype through other related signaling pathways, such as IL-1β and IFNγ. Inflammatory cytokine-induced glycolysis program with disruption of the TCA cycle and uncoupling of OXPHOS [80,81,82]. Luxi Wang et al. found that microglia secrete pro-inflammatory cytokines in an inflammatory environment, and thus the expression of GLUT promotes a glucose increase [123]. Furthermore, Casimir Bamberger et al. revealed through Covalent Protein Painting that Aβ was related to inhibition of mitochondrial succinate dehydrogenase, which could be related to racemization of its serine residues [124,125] or to the lysine site of tau [114], which was the basis for the interruption of the TCA cycle in pro-inflammatory microglia, leading to the accumulation of succinic acid [126]. Meihua Jin et al. demonstrated that tau could activate the cGAS/STING pathway in microglia [104], while STING activation could lead to the accumulation of the macrophage metabolite succinate. This stabilizes the transcription factor HIF-1α, leading to a shift to the glycolytic profile [105]. Mammalian targets that inhibit rapamycin complex 1 (mTORC1) can inhibit hexokinase 1-dependent glycolysis and caspase-1 activation, which indicates that inflammasome activation of NLRP3 is involved in macrophage metabolism [127]. Aβ [85,86,87,88] and tau [107,108,109,110] can activate NLRP3 of microglia via converting its metabolism into sugar [107,127]. Bradley L. Heckmann et al. activated RAW264.7 and BMDM at Aβ is closely related to TREM2 and CD63 [128]. CD36 is a type B scavenger receptor that recognizes low density lipoprotein (LDL), oxidized phospholipid and Aβ, and is also considered a FA transporter or uptake promoter [43]. These findings suggest that AD-related stimuli can directly change the metabolic processes of microglia in a variety of ways. However, the precise signaling mechanism involved remains to be fully elucidated.
5. Exosomes as the Mediator of Aβ and Tau Propagation
Neuronal aggregates of tau and Aβ can generally act on adjacent microglia through exosomes or receptors. Exosomes are membrane-bound vesicles released by mammalian cells from the intracellular multivesicular compartments. They play an important role in intercellular communication because exosomes are potential vehicles for transferring cell contents from source cells to recipient cells [129]. In AD, exosomes are related to the release of Aβ oligomer and its extracellular accumulation [130,131,132,133], and the accumulation of hyperphosphorylated tau [134,135,136,137]. In neuronal cytoplasm, APP is endocytosed and cleaved by β-secretase to form APPβ on the early endosome membrane. This early endosome undergoes maturation to form multivesicular bodies (MVB)-containing exosomes, formed by the invagination of early endosome during maturation. Tau protein oligomers are firstly passed through endoplasmic reticulum and Golgi body, and then they become part of secretary vesicles and later exosomes. Under physiological conditions, these Tau- and Aβ-containing exosomes are degraded by lysosomes. But in the case of AD, when there is a problem in lysosomes, then these MVBs containing exosomes are filled with Tau and Aβ, and are released into the extracellular space. From the extracellular space, exosomes are captured by microglia [138].
Many studies have proved that neuron-derived exosomes contain Aβ oligomers and tau, and can be internalized by adjacent cells, activating the immune response or propagation (Table 2). Maitrayee et al. detected exosomes containing Aβ oligomers in the brain of human AD patients and demonstrated that t exosomes could be a vehicle for the spreading of Aβ oligomers [137]. Consistent with this, Aβ and tau were detected in plasma neuron-derived exosomes [133] and exosomes in the cerebrospinal fluid of AD patients [139]. In the AD mouse model, Ahmed et al. isolated Aβ-related exosomes and found that they could be transported to mitochondria [140]. Rajendran et al. demonstrated in neuroblastoma N2a cells that a fraction of Aβ peptides is localized to MVBs and is released in association with exosomes, and Aβ was still seen on the exosomes from the cryoelectron microscopy specimen after several stringent washing which demonstrates that it is not that extracellular soluble Aβ simply ‘‘sticks’’ to the extracellular exosomes, but that Aβ is contained in the exosomes or that Aβ is inserted in the membrane [141]. Saman S et al. detected the presence of tau in isolated exosomes from cerebrospinal fluid of human AD patients and verified in M1C cells (neuroblastoma) that most of their secreted tau occurred through exosome release [142]. Similarly, Fiandaca MS et al. detected tau and Aβ in neurogenic blood exosomes extracts from AD patients [143]. Guix FX et al. contained aggregates of tau in the exosomes of neurons derived from human induced pluripotent stem cells (iPSC) and detected several different forms of tau in plasma-isolated exosomes from AD patients [144]. However, the mechanism by which Aβ and tau are encapsulated in exosomes is not yet clear.
In addition, Aβ and tau are not only found in exosomes, but also internalized by adjacent cells. Charisse Winston et al. confirmed whether exosomes spread the pathological forms of tau in vivo [144]. Wang Y et al. found that exosomes containing tau in the conditioned medium from organotypic hippocampal slices seeded tau accumulation in adjacent microglia [148]. Microglia depletion was found to inhibit the propagation of tau in a P301S tau transgenic mouse model by Asai H et al. [149]. The synergy of Aβ recognition, internalization and clearance, and cell activation may involve microglia receptors, such as scavenger receptors (SR-AI/II), CD36, RAGE, Fc receptors, TLR (Toll-like receptor), and complement receptors. Exosomes boosted the uptake of Aβ by primary cultured microglia in vitro, discovered by Michael B Dinkins et al. in AD 5XFAD mouse model [152]. Crehan et al. treated primary rat microglia with Aβ after blocking complement receptor 1 and found reduced microglia activation [153]. Ahmed Elsherbini et al. found in the AD 5XFAD mouse model that exosome-mediated Aβ acted on mitochondria, binding to and aggregation of mitochondrial outer membrane protein 1(VDAC1) [140]. Indra Sethy Coraci et al. verified the expression of CD36 in microglia in the brain of AD patients and confirmed the binding of Aβ to CD36 in the Bowes-CD36 cell line, which was transfected with human CD36 mammalian expression vector and produced a cell line expressing CD36 [154]. Microglia surface receptors are involved in the internalization of Aβ and tau in exosomes, consistent with their involvement in reprogramming of microglia metabolism.
6. Conclusions
In conclusion, the inflammatory microenvironment mediated by microglia is crucial for the onset and progression of AD. The aggregates, in combination with Aβ and tau accumulation, can cause neuronal dysfunction and glial activation, followed by neuroinflammation in AD. Aβ and Tau are produced in neurons and released into the extracellular space, where they can be degraded or cleared by microglia, or enter microglia via receptors on their surface to promote glycolysis by regulating mitochondrial function, aggravating the neuroinflammatory microenvironment and neuron injury. Therefore, it has previously been observed that it is urgent to explore the means for the treatment of neurodegenerative diseases by reprogramming microglia metabolism (Figure 3).
The regulation of Aβ and tau on microglia metabolism is mainly focused on the effect on mitochondrial function. Mitochondrial metabolism, which includes the tricarboxylic acid cycle, OXPHOS, FAO, nucleotide synthesis, and amino acid metabolism, is at the core of the cellular metabolic network. Both Aβ and tau mutations can cause mitochondrial dysfunction, and Aβ has been identified as a crucial role in the formation of free radicals, oxidative damage, and mitochondrial dysfunction [155]. The development of Aβ plaques has been proven to cause severe damage to mitochondria in studies. The ETC function of mitochondria can be exacerbated by the combined impacts of Aβ and ROS, a break-down of the electron transfer cycle by preventing cytochrome oxidase from producing reactive oxygen species (ROS) [156,157,158]. Lener et al. utilized the respiratory inhibitors rotenone and antimycin to create mitochondrial damage and increase the quantity of ROS in neural cells, and discovered that the level of A was also rising. In the brain of AD patients, the activities of various mitochondrial localization enzymes decreased, and in most neurons, the number of intact mitochondria decreased [159]. The changes in some mitochondrial enzyme activities found in AD are related to the pathology of Aβ [160]. Recent studies have found that Tau oligomers alter memory consolidation, reduce the levels of synaptophysin and neuron-specific protein 11 associated with synaptic vesicles, and also reduce the level of Complex I. Brain samples from pR5 transgenic mice overexpressing tau protein mutant P301 showed that mitochondrial complex enzyme activity decreased, mitochondrial depolarization, respiratory damage and ROS level increased [161]. It is suggested that Tau protein or Tau fragment can directly or indirectly affect the activity or expression level of mitochondrial respiratory enzymes, and make mitochondrial function disorder.
In addition, in the inflammatory environment in which neurons are located, not only the microglia play a role, but also the crosstalk between microglia and astrocytes plays an important role. Microglia generally respond faster than astrocytes to pathological stimuli, inducing astrocyte activation and determining the fate of astrocytes [162]. Microglia under the condition of neuritis release ATP and stimulate astrocytes to release glutamic acid. Glutamic acid is a well-recognized modulator of neuronal excitability, which can act, for example, through metabolic glutamate receptors, indicating that microglia play an important role in the regulation of astrocyte-mediated excitatory neurotransmission [163]. Activated microglia negatively affected the activity and expression of the glutamic acid-aspartic acid transporter in cultured astrocytes [164]. On the other hand, inflammatory mediators released by activated microglia increase the uptake of extracellular glutamate, which is dependent on glutamate transporter 1, in astrocytes [165]. Increased extracellular glutamate uptake in the inflammatory response is associated with increased expression of GLUT1. Aβ and tau interact strictly with microglia and neurons in the microenvironment of neuroinflammation, and microglia metabolism reprogramming is the central point for regulating neuroinflammation. Reversing microglia phenotype by targeting microglia metabolism may provide a new strategy for the treatment of AD.
Author Contributions
Conceptualization, X.Y. and J.S.; writing—original draft preparation, Y.Z. and B.L.; visualization, L.X. and S.Y.; writing—review and editing, J.F. and J.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (82102733 and 81772794); Jilin Provincial Research Foundation for the Development of Science and Technology Projects (20200703009ZP, 20190201164JC and 20191008011TC); Jilin Provincial Health Technology Innovation Project (2021JC034 and 2018SCZ021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Abbott, A. Could drugs prevent Alzheimer’s? These trials aim to find out. Nature 2022, 603, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; de Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vecsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cyto-kines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchy-ma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef]
- Villegas-Llerena, C.; Phillips, A.; Garcia-Reitboeck, P.; Hardy, J.; Pocock, J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 2016, 36, 74–81. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Merighi, S.; Borea, P.A.; Varani, K.; Vincenzi, F.; Travagli, A.; Nigro, M.; Pasquini, S.; Suresh, R.R.; Kim, S.W.; Volkow, N.D.; et al. Pathophysiological Role and Medicinal Chemistry of A2A Adenosine Receptor Antagonists in Alzheimer’s Disease. Molecules 2022, 27, 2680. [Google Scholar] [CrossRef]
- Lillo, A.; Raich, I.; Lillo, J.; Perez-Olives, C.; Navarro, G.; Franco, R. Expression of the Adenosine A2A-A3 Receptor Heteromer in Different Brain Regions and Marked Upregula-tion in the Microglia of the Transgenic APPSw, Ind Alzheimer’s Disease Model. Biomedicines 2022, 10, 214. [Google Scholar] [CrossRef]
- Pocock, J.M.; Kettenmann, H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007, 30, 527–535. [Google Scholar] [CrossRef]
- López-López, A.; Gelpi, E.; Lopategui, D.M.; Vidal-Taboada, J.M. Association of the CX3CR1-V249I Variant with Neurofibrillary Pathology Progression in Late-Onset Alzheimer’s Disease. Mol. Neurobiol. 2017, 55, 2340–2349. [Google Scholar] [CrossRef]
- Marinelli, S.; Basilico, B.; Marrone, M.C.; Ragozzino, D. Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Semin. Cell Dev. Biol. 2019, 94, 138–151. [Google Scholar] [CrossRef]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2015, 136, 10–17. [Google Scholar] [CrossRef]
- Borst, K.; Schwabenland, M.; Prinz, M. Microglia metabolism in health and disease. Neurochem. Int. 2018, 130, 104331. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflamma-tory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Jiang, D.; Gong, F.; Ge, X.; Lv, C.; Huang, C.; Feng, S.; Zhou, Z.; Rong, Y.; Wang, J.; Ji, C.; et al. Neuron-derived exosomes-transmitted miR-124-3p protect traumatically injured spinal cord by suppressing the activation of neurotoxic microglia and astrocytes. J. Nanobiotechnol. 2020, 18, 105. [Google Scholar] [CrossRef]
- Men, Y.; Yelick, J.; Jin, S.; Tian, Y.; Chiang, M.S.R.; Higashimori, H.; Brown, E.; Jarvis, R.; Yang, Y. Exosome reporter mice reveal the involvement of exosomes in mediating neuron to astroglia communication in the CNS. Nat. Commun. 2019, 10, 4136. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Han, Z.; Hu, T.; Zhang, S.; Ge, X.; Huang, S.; Wang, L.; Yu, J.; Li, W.; Wang, Y.; et al. Neuron-derived exosomes with high miR-21-5p expression promoted polarization of M1 microglia in culture. Brain Behav. Immun. 2019, 83, 270–282. [Google Scholar] [CrossRef]
- Brites, D.; Fernandes, A. Neuroinflammation and Depression: Microglia Activation, Extracellular Microvesicles and mi-croRNA Dysregulation. Front. Cell. Neurosci. 2015, 9, 476. [Google Scholar] [CrossRef] [Green Version]
- Turola, E.; Furlan, R.; Bianco, F.; Matteoli, M.; Verderio, C. Microglial microvesicle secretion and intercellular signaling. Front. Physiol. 2012, 3, 149. [Google Scholar] [CrossRef] [Green Version]
- Potolicchio, I.; Carven, G.J.; Xu, X.; Stipp, C.; Riese, R.J.; Stern, L.J.; Santambrogio, L. Proteomic analysis of microglia-derived exosomes: Metabolic role of the aminopeptidase CD13 in neuro-peptide catabolism. J. Immunol. 2005, 175, 2237–2243. [Google Scholar] [CrossRef] [Green Version]
- Paolicelli, R.C.; Bergamini, G.; Rajendran, L. Cell-to-cell Communication by Extracellular Vesicles: Focus on Microglia. Neuroscience 2019, 405, 148–157. [Google Scholar] [CrossRef]
- Hamley, I.W. The amyloid beta peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Xie, L.; Lai, Y.; Lei, F.; Liu, S.; Liu, R.; Wang, T. Exploring the association between interleukin-1beta and its interacting proteins in Alzheimer’s disease. Mol. Med. Rep. 2015, 11, 3219–3228. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wu, J. Amyloid-beta: A double agent in Alzheimer’s disease? Biomed. Pharm. 2021, 139, 111575. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; Gonzalez, H.M.; Leger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar]
- Ulbright, T.M.; Roth, L.M. Recent developments in the pathology of germ cell tumors. Semin. Diagn. Pathol. 1987, 4, 304–319. [Google Scholar]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The Many Faces of Tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, W.; Hanger, D.P.; Miller, C.C.J.; Lovestone, S. The Importance of Tau Phosphorylation for Neurodegenerative Diseases. Front. Neurol. 2013, 4, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Small, S.A.; Duff, K. Linking Abeta and tau in late-onset Alzheimer’s disease: A dual pathway hypothesis. Neuron 2008, 60, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Hagemeyer, N.; Kierdorf, K.; Frenzel, K.; Xue, J.; Ringelhan, M.; Abdullah, Z.; Godin, I.; Wieghofer, P.; Jordão, M.J.C.; Ulas, T.; et al. Transcriptome-based profiling of yolk sac-derived macrophages reveals a role for Irf8 in macrophage maturation. EMBO J. 2016, 35, 1730–1744. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Erny, D.; Hagemeyer, N. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 2017, 18, 385–392. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Chausse, B.; Kakimoto, P.A.; Kann, O. Microglia and lipids: How metabolism controls brain innate immunity. Semin. Cell Dev. Biol. 2020, 112, 137–144. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Hallowell, R.W.; Collins, S.L.; Craig, J.M.; Zhang, Y.; Oh, M.; Illei, P.B.; Chan-Li, Y.; Vigeland, C.; Mitzner, W.; Scott, A.L.; et al. mTORC2 signalling regulates M2 macrophage differentiation in response to helminth infection and adaptive thermogenesis. Nat. Commun. 2017, 8, 14208. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2017, 66, 1200–1212. [Google Scholar] [CrossRef]
- Yang, S.; Qin, C.; Hu, Z.-W.; Zhou, L.-Q.; Yu, H.-H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.-J.; Tian, D.-S. Microglia reprogram metabolic profiles for phenotype and function changes in central nervous system. Neurobiol. Dis. 2021, 152, 105290. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, A.B.; Hennessy, E.; Murray, C.L.; Nazmi, A.; Delaney, H.J.; Healy, D.; Fagan, S.G.; Rooney, M.; Stewart, E.; Lewis, A.; et al. Acute systemic inflammation exacerbates neuroinflammation in Alzheimer’s disease: IL-1beta drives amplified responses in primed astrocytes and neuronal network dysfunction. Alzheim. Dement. 2021, 17, 1735–1755. [Google Scholar] [CrossRef]
- Babic Leko, M.; Perkovic, M.N.; Klepac, N.; Strac, D.S.; Borovecki, F.; Pivac, N.; Hof, P.R.; Simic, G. IL-1beta, IL-6, IL-10, and TNFalpha Single Nucleotide Polymorphisms in Human Influence the Suscep-tibility to Alzheimer’s Disease Pathology. J. Alzheim. Dis. 2020, 75, 1029–1047. [Google Scholar] [CrossRef]
- Baril, A.-A.; Beiser, A.S.; Redline, S.; McGrath, E.R.; Gottlieb, D.J.; Aparicio, H.; Seshadri, S.; Himali, J.J.; Pase, M.P. Interleukin-6 Interacts with Sleep Apnea Severity when Predicting Incident Alzheimer’s Disease Dementia. J. Alzheim. Dis. 2021, 79, 1451–1457. [Google Scholar] [CrossRef]
- Lyra, E.S.N.M.; Goncalves, R.A.; Pascoal, T.A.; Lima-Filho, R.A.S.; Resende, E.P.F.; Vieira, E.L.M.; Teixeira, A.L.; de Souza, L.C.; Peny, J.A.; Fortuna, J.T.S.; et al. Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic altera-tions in Alzheimer’s disease. Transl. Psychiatry 2021, 11, 251. [Google Scholar] [CrossRef]
- Xu, C.; Wu, J.; Wu, Y.; Ren, Z.; Yao, Y.; Chen, G.; Fang, E.F.; Noh, J.H.; Liu, Y.U.; Wei, L.; et al. TNF-alpha-dependent neuronal necroptosis regulated in Alzheimer’s disease by coordination of RIPK1-p62 complex with autophagic UVRAG. Theranostics 2021, 11, 9452–9469. [Google Scholar] [CrossRef]
- Jayaraman, A.; Htike, T.T.; James, R.; Picon, C.; Reynolds, R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocam-pus. Acta Neuropathol. Commun. 2021, 9, 159. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheim. Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Moulton, M.J.; Barish, S.; Ralhan, I.; Chang, J.; Goodman, L.D.; Harland, J.G.; Marcogliese, P.C.; Johansson, J.O.; Ioannou, M.S.; Bellen, H.J. Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer’s disease–associated genes. Proc. Natl. Acad. Sci. USA 2021, 118, e2112095118. [Google Scholar] [CrossRef]
- Taupin, P. A dual activity of ROS and oxidative stress on adult neurogenesis and Alzheimer’s disease. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 16–21. [Google Scholar] [CrossRef]
- Wang, A.; Luan, H.H.; Medzhitov, R. An evolutionary perspective on immunometabolism. Science 2019, 363, eaar3932. [Google Scholar] [CrossRef]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2015, 213, 15–23. [Google Scholar] [CrossRef]
- Weiss, P.A.; Hofmann, H.M.; Kainer, F.; Haas, J.G. Fetal outcome in gestational diabetes with elevated amniotic fluid insulin levels: Dietary versus insulin treatment. Diabetes Res. Clin. Pract. 1988, 5, 1–7. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.G.; Zaslona, Z.; Galvan-Pena, S.; Koppe, E.L.; Sevin, D.C.; Angiari, S.; Triantafilou, M.; Triantafilou, K.; Modis, L.K.; O’Neill, L.A. An unexpected link between fatty acid synthase and cholesterol synthesis in proinflammatory macro-phage activation. J. Biol. Chem. 2018, 293, 5509–5521. [Google Scholar] [CrossRef] [Green Version]
- Puleston, D.J.; Buck, M.D.; Geltink, R.I.K.; Kyle, R.L.; Caputa, G.; O’Sullivan, D.; Cameron, A.M.; Castoldi, A.; Musa, Y.; Kabat, A.M.; et al. Polyamines and eIF5A Hypusination Modulate Mitochondrial Respiration and Macrophage Activation. Cell Metab. 2019, 30, 352–363.e8. [Google Scholar] [CrossRef] [Green Version]
- Polis, B.; Srikanth, K.D.; Elliott, E.; Gil-Henn, H.; Samson, A.O. L-Norvaline Reverses Cognitive Decline and Synaptic Loss in a Murine Model of Alzheimer’s Disease. Neurotherapeutics 2018, 15, 1036–1054. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, S.M.; Atanasova, M.; Dimitrov, I.; Doytchinova, I.A. Cellular polyamines condense hyperphosphorylated Tau, triggering Alzheimer’s disease. Sci. Rep. 2020, 10, 10098. [Google Scholar] [CrossRef]
- Sandusky-Beltran, L.A.; Kovalenko, A.; Placides, D.S.; Ratnasamy, K.; Ma, C.; Hunt, J.B.; Liang, H.; Calahatian, J.I.T.; Michalski, C.; Fahnestock, M.; et al. Aberrant AZIN2 and polyamine metabolism precipitates tau neuropathology. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Hunt, J.B.; Nash, K.R.; Placides, D.; Moran, P.; Selenica, M.-L.B.; Abuqalbeen, F.; Ratnasamy, K.; Slouha, N.; Rodriguez-Ospina, S.; Savlia, M.; et al. Sustained Arginase 1 Expression Modulates Pathological Tau Deposits in a Mouse Model of Tauopathy. J. Neurosci. 2015, 35, 14842–14860. [Google Scholar] [CrossRef] [Green Version]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef]
- Nilson, A.N.; English, K.C.; Gerson, J.E.; Whittle, T.B.; Crain, C.N.; Xue, J.; Sengupta, U.; Castillo-Carranza, D.L.; Zhang, W.; Gupta, P.; et al. Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neuro-degenerative Diseases. J. Alzheim. Dis. 2017, 55, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α–mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [Green Version]
- Abeti, R.; Abramov, A.Y.; Duchen, M.R. Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 2011, 134, 1658–1672. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, A.; Mela, V.; Harty, C.; Minogue, A.M.; Costello, D.A.; Kerskens, C.; Lynch, M.A. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and com-promised function in APP/PS1 mice. Brain Pathol. 2019, 29, 606–621. [Google Scholar] [CrossRef] [Green Version]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Araiz, A.; Finucane, O.M.; Keogh, S.; Lynch, M.A. Anti-TLR2 antibody triggers oxidative phosphorylation in microglia and increases phagocytosis of beta-amyloid. J. Neuroinflammation 2018, 15, 247. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.-C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Sayed, F.A.; Kodama, L.; Fan, L.; Carling, G.K.; Udeochu, J.C.; Le, D.; Li, Q.; Zhou, L.; Wong, M.Y.; Horowitz, R.; et al. AD-linked R47H-TREM2 mutation induces disease-enhancing microglial states via AKT hyperactivation. Sci. Transl. Med. 2021, 13, eabe3947. [Google Scholar] [CrossRef]
- Cui, Y.-H.; Le, Y.; Gong, W.; Proost, P.; Van Damme, J.; Murphy, W.J.; Wang, J.M. Bacterial Lipopolysaccharide Selectively Up-Regulates the Function of the Chemotactic Peptide Receptor Formyl Peptide Receptor 2 in Murine Microglial Cells. J. Immunol. 2002, 168, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Holland, R.; McIntosh, A.; Finucane, O.; Mela, V.; Rubio-Araiz, A.; Timmons, G.; McCarthy, S.; Gun’Ko, Y.; Lynch, M. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav. Immun. 2018, 68, 183–196. [Google Scholar] [CrossRef]
- Voloboueva, L.A.; Emery, J.F.; Sun, X.; Giffard, R.G. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mito-chondrial glucose-regulated protein 75/mortalin. FEBS Lett. 2013, 587, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.; Kolbe, C.-C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 2019, 51, 997–1011.e7. [Google Scholar] [CrossRef]
- Van Zeller, M.; Dias, D.; Sebastiao, A.M.; Valente, C.A. NLRP3 Inflammasome: A Starring Role in Amyloid-beta- and Tau-Driven Pathological Events in Alz-heimer’s Disease. J. Alzheim. Dis. 2021, 83, 939–961. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef] [Green Version]
- Akhter, R.; Shao, Y.; Formica, S.; Khrestian, M.; Bekris, L.M.; Akhter, R. TREM2 alters the phagocytic, apoptotic and inflammatory response to Abeta42 in HMC3 cells. Mol. Immunol. 2021, 131, 171–179. [Google Scholar] [CrossRef]
- Caruso, G.I.; Spampinato, S.F.; Costantino, G.; Merlo, S.; Sortino, M.A. SIRT1-Dependent Upregulation of BDNF in Human Microglia Challenged with Abeta: An Early but Transient Response Rescued by Melatonin. Biomedicines 2021, 9, 466. [Google Scholar] [CrossRef] [PubMed]
- Li, X. SIRT1 and energy metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, R.Y.; He, L.; Zhang, J.; Liu, X.; Liao, Y.; Gao, J.; Liao, Y.; Yan, Y.; Li, Q.; Zhou, X.; et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzhei-mer’s disease. Cell Metab. 2022, 34, 634–648.e6. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Teubner, B.J.W.; Tummers, B.; Boada-Romero, E.; Harris, L.; Yang, M.; Guy, C.S.; Zakharenko, S.S.; Green, D.R. LC3-Associated Endocytosis Facilitates beta-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 2019, 178, 536–551.e14. [Google Scholar] [CrossRef] [PubMed]
- Supiev, T.K.; Solov’Ev, M.M.; Galiapin, A.S.; Izenbaev, N.B. The use of charcoal sorbents as the drainage materials in the treatment of suppurative diseases of the mandibulofacial region in children. Grek. Bull. Surg. 1988, 140, 72–74. [Google Scholar]
- Yang, J.; Fu, Z.; Zhang, X.; Xiong, M.; Meng, L.; Zhang, Z. TREM2 ectodomain and its soluble form in Alzheimer’s disease. J. Neuroinflammation 2020, 17, 204. [Google Scholar] [CrossRef]
- Bolós, M.; Llorens-Martín, M.; Jurado-Arjona, J.; Hernández, F.; Rábano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimer Dis. 2015, 50, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Serrano, J.R.; Hoyle, R.; Holtzman, D.M. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Wang, C.; Xiong, M.; Gratuze, M.; Bao, X.; Shi, Y.; Andhey, P.S.; Manis, M.; Schroeder, C.; Yin, Z.; Madore, C.; et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and de-creases synaptic phagocytosis by microglia. Neuron 2021, 109, 1657–1674.e7. [Google Scholar] [CrossRef]
- Shi, Y.; Initiative, A.D.N.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Zhao, J.; Davis, M.D.; Martens, Y.A.; Shinohara, M.; Graff-Radford, N.R.; Younkin, S.G.; Wszolek, Z.K.; Kanekiyo, T.; Bu, G. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 2017, 26, 2690–2700. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, X.; Zhao, L. Human ApoE Isoforms Differentially Modulate Brain Glucose and Ketone Body Metabolism: Implications for Alzheimer’s Disease Risk Reduction and Early Intervention. J. Neurosci. 2018, 38, 6665–6681. [Google Scholar] [CrossRef]
- Drzezga, A.; Riemenschneider, M.; Strassner, B.; Grimmer, T.; Peller, M.; Knoll, A.; Wagenpfeil, S.; Minoshima, S.; Schwaiger, M.; Kurz, A. Cerebral glucose metabolism in patients with AD and different APOE genotypes. Neurology 2005, 64, 102–107. [Google Scholar] [CrossRef]
- Wang, F.; Qi, X.M.; Wertz, R.; Mortensen, M.; Hagen, C.; Evans, J.; Sheinin, Y.; James, M.; Liu, P.; Tsai, S.; et al. p38gamma MAPK Is Essential for Aerobic Glycolysis and Pancreatic Tumorigenesis. Cancer Res. 2020, 80, 3251–3264. [Google Scholar] [CrossRef]
- Perea, J.R.; Avila, J.; Bolos, M. Dephosphorylated rather than hyperphosphorylated Tau triggers a pro-inflammatory pro-file in microglia through the p38 MAPK pathway. Exp. Neurol. 2018, 310, 14–21. [Google Scholar] [CrossRef]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Gomes, M.T.R.; Guimarães, E.S.; Marinho, F.V.; Macedo, I.; Aguiar, E.R.G.R.; Barber, G.N.; Moraes-Vieira, P.M.M.; Alves-Filho, J.C.; Oliveira, S.C. STING regulates metabolic reprogramming in macrophages via HIF-1α during Brucella infection. PLoS Pathog. 2021, 17, e1009597. [Google Scholar] [CrossRef]
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflam-masome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [Green Version]
- Panda, C.; Voelz, C.; Habib, P.; Mevissen, C.; Pufe, T.; Beyer, C.; Gupta, S.; Slowik, A. Aggregated Tau-PHF6 (VQIVYK) Potentiates NLRP3 Inflammasome Expression and Autophagy in Human Microglial Cells. Cells 2021, 10, 1652. [Google Scholar] [CrossRef]
- Jiang, S.; Maphis, N.M.; Binder, J.; Chisholm, D.; Weston, L.; Duran, W.; Peterson, C.; Zimmerman, A.; Mandell, M.A.; Jett, S.D.; et al. Proteopathic tau primes and activates interleukin-1beta via myeloid-cell-specific MyD88- and NLRP3-ASC-inflammasome pathway. Cell Rep. 2021, 36, 109720. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [Green Version]
- Vaishnavi, S.N.; Vlassenko, A.G.; Rundle, M.M.; Snyder, A.Z.; Mintun, M.A.; Raichle, M.E. Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. USA 2010, 107, 17757–17762. [Google Scholar] [CrossRef] [Green Version]
- Vlassenko, A.G.; Vaishnavi, S.N.; Couture, L.; Sacco, D.; Shannon, B.J.; Mach, R.H.; Morris, J.C.; Raichle, M.E.; Mintun, M.A. Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta) deposition. Proc. Natl. Acad. Sci. USA 2010, 107, 17763–17767. [Google Scholar] [CrossRef] [Green Version]
- Atlante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 2008, 1777, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Pozueta, J.; Lefort, R.; Shelanski, M.L. Synaptic changes in Alzheimer’s disease and its models. Neuroscience 2013, 251, 51–65. [Google Scholar] [CrossRef]
- Singer, T.P.; Ramsay, R.R.; McKeown, K.; Trevor, A.; Castagnoli, N.E., Jr. Mechanism of the neurotoxicity of 1-methyl-4-phenylpyridinium (MPP+), the toxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 17–23. [Google Scholar] [CrossRef]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [Green Version]
- Shakespear, M.R.; Iyer, A.; Cheng, C.Y.; Das Gupta, K.; Singhal, A.; Fairlie, D.P.; Sweet, M.J. Lysine Deacetylases and Regulated Glycolysis in Macrophages. Trends Immunol. 2018, 39, 473–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pavlou, S.; Du, X.; Bhuckory, M.; Xu, H.; Chen, M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol. Neurodegener. 2019, 14, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, I.; Yamada, N.; Sakuraba, Y.; Kamenosono, M.; Tutumi, S. Suppression of Mitochondrial Succinate Dehydrogenase, a Primary Target of β-Amyloid, and Its Derivative Racemized at Ser Residue. J. Neurochem. 2002, 65, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, C.; Pankow, S.; Martinez-Bartolome, S.; Ma, M.; Diedrich, J.; Rissman, R.A.; Yates, J.R., 3rd. Protein Footprinting via Covalent Protein Painting Reveals Structural Changes of the Proteome in Alz-heimer’s Disease. J. Proteome Res. 2021, 20, 2762–2771. [Google Scholar] [CrossRef] [PubMed]
- Fairley, L.H.; Wong, J.H.; Barron, A.M. Mitochondrial Regulation of Microglial Immunometabolism in Alzheimer’s Dis-ease. Front. Immunol. 2021, 12, 624538. [Google Scholar] [CrossRef] [PubMed]
- Shippy, D.C.; Ulland, T.K. Microglial Immunometabolism in Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 563446. [Google Scholar] [CrossRef]
- Gerlach, D.; Köhler, W.; Knöll, H.; Moravek, L.; Weeks, C.R.; Ferretti, J.J. Purification and characterization of streptococcus pyogenes erythrogenic toxin type a produced by a cloned gene in streptococcus sanguis. Ser. A Med. Microbiol. Infect. Dis. Virol. Parasitol. 1987, 266, 347–358. [Google Scholar] [CrossRef]
- Pascual, M.; Ibáñez, F.; Guerri, C. Exosomes as mediators of neuron-glia communication in neuroinflammation. Neural Regen. Res. 2020, 15, 796–801. [Google Scholar] [CrossRef]
- Shaheen, H.; Singh, S.; Melnik, R. A Neuron-Glial Model of Exosomal Release in the Onset and Progression of Alzhei-mer’s Disease. Front. Comput. Neurosci. 2021, 15, 653097. [Google Scholar] [CrossRef]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Bi-omarker Potential in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef]
- Xiao, T.; Zhang, W.; Jiao, B.; Pan, C.-Z.; Liu, X.; Shen, L. The role of exosomes in the pathogenesis of Alzheimer’ disease. Transl. Neurodegener. 2017, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Wang, S.-K.; Zhang, Y.; Rostami, A.; Kenkare, A.; Casella, G.; Yuan, Z.-Q.; Li, X. Role of extracellular vesicles in neurodegenerative diseases. Prog. Neurobiol. 2021, 201, 102022. [Google Scholar] [CrossRef]
- Quek, C.; Hill, A.F. The role of extracellular vesicles in neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2017, 483, 1178–1186. [Google Scholar] [CrossRef]
- Csernansky, J.G.; Tacke, U.; Rusen, D.; Hollister, L.E. The Effect of Benzodiazepines on Tardive Dyskinesia Symptoms. J. Clin. Psychopharmacol. 1988, 8, 154–155. [Google Scholar] [CrossRef]
- Sinha, M.S.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Verma, H.; Dhiman, M.; Tell, G.; Gigli, G.L.; Janes, F.; Mantha, A.K. Brain Exosomes: Friend or Foe in Alzheimer’s Disease? Mol. Neurobiol. 2021, 58, 6610–6624. [Google Scholar] [CrossRef]
- Jia, L.; Qiu, Q.; Zhang, H.; Chu, L.; Du, Y.; Zhang, J.; Zhou, C.; Liang, F.; Shi, S.; Wang, S.; et al. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimer Dement. 2019, 15, 1071–1080. [Google Scholar] [CrossRef]
- Elsherbini, A.; Qin, H.; Zhu, Z.; Tripathi, P.; Crivelli, S.M.; Bieberich, E. In vivo evidence of exosome-mediated Abeta neurotoxicity. Acta Neuropathol. Commun. 2020, 8, 100. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [Green Version]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [Green Version]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [Green Version]
- Guix, F.X.; Corbett, G.T.; Cha, D.J.; Mustapic, M.; Liu, W.; Mengel, D.; Chen, Z.; Aikawa, E.; Young-Pearse, T.; Kapogiannis, D.; et al. Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2018, 19, 663. [Google Scholar] [CrossRef] [Green Version]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gelé, P.; Drobeck, H.; Bégard, S.; Galas, M.-C.; Delacourte, A.; Beauvillain, J.-C.; Buée, L.; et al. Alkalizing Drugs Induce Accumulation of Amyloid Precursor Protein By-products in Luminal Vesicles of Multivesicular Bodies. J. Biol. Chem. 2007, 282, 18197–18205. [Google Scholar] [CrossRef] [Green Version]
- Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A.F. Inhibition of gamma-secretase causes increased secretion of amyloid precursor protein C-terminal frag-ments in association with exosomes. FASEB J. 2008, 22, 1469–1478. [Google Scholar] [CrossRef]
- Perez-Gonzalez, R.; Gauthier, S.A.; Kumar, A.; Levy, E. The Exosome Secretory Pathway Transports Amyloid Precursor Protein Carboxyl-terminal Fragments from the Cell into the Brain Extracellular Space. J. Biol. Chem. 2012, 287, 43108–43115. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Kruger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Lee, S.; Kim, W.; Li, Z.; Hall, G.F. Accumulation of Vesicle-Associated Human Tau in Distal Dendrites Drives Degeneration and Tau Secretion in anIn SituCellular Tauopathy Model. Int. J. Alzheimer Dis. 2012, 2012, 172837. [Google Scholar] [CrossRef]
- Crotti, A.; Sait, H.R.; McAvoy, K.M.; Estrada, K.; Ergun, A.; Szak, S.; Marsh, G.; Jandreski, L.; Peterson, M.; Reynolds, T.L.; et al. BIN1 favors the spreading of Tau via extracellular vesicles. Sci. Rep. 2019, 9, 9477. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crehan, H.; Hardy, J.; Pocock, J. Blockage of CR1 prevents activation of rodent microglia. Neurobiol. Dis. 2013, 54, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Coraci, I.S.; Husemann, J.; Berman, J.W.; Hulette, C.; Dufour, J.H.; Campanella, G.K.; Luster, A.D.; Silverstein, S.C.; El-Khoury, J.B. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can medi-ate production of reactive oxygen species in response to beta-amyloid fibrils. Am. J. Pathol. 2002, 160, 101–112. [Google Scholar] [CrossRef]
- Sinha, M.; Bhowmick, P.; Banerjee, A.; Chakrabarti, S. Antioxidant role of amyloid beta protein in cell-free and biological systems: Implication for the pathogenesis of Alzheimer disease. Free Radic. Biol. Med. 2013, 56, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheim. Dis. 2010, 20 (Suppl. S2), S265–S279. [Google Scholar] [CrossRef] [Green Version]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial Dysfunction: Different Routes to Alzheimer’s Disease Therapy. Oxidative Med. Cell. Longev. 2014, 2014, 780179. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, H.M.; Swerdlow, R.H. Relationships Between Mitochondria and Neuroinflammation: Implications for Alzhei-mer’s Disease. Curr. Top Med. Chem. 2016, 16, 849–857. [Google Scholar] [CrossRef]
- Huang, Z.; Yan, Q.; Wang, Y.; Zou, Q.; Li, J.; Liu, Z.; Cai, Z. Role of Mitochondrial Dysfunction in the Pathology of Amyloid-beta. J. Alzheim. Dis. 2020, 78, 505–514. [Google Scholar] [CrossRef]
- David, D.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Dröse, S.; Brandt, U.; Müller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Jo, M.; Kim, J.-H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2018, 25, 227–240. [Google Scholar] [CrossRef]
- Pascual, O.; Ben Achour, S.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2011, 109, E197–E205. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Takeuchi, H.; Doi, Y.; Kawanokuchi, J.; Sonobe, Y.; Jin, S.; Yawata, I.; Li, H.; Yasuoka, S.; Mizuno, T.; et al. Excitatory amino acid transporter expression by astrocytes is neuroprotective against microglial excitotoxicity. Brain Res. 2008, 1210, 11–19. [Google Scholar] [CrossRef]
- Tilleux, S.; Goursaud, S.; Hermans, E. Selective up-regulation of GLT-1 in cultured astrocytes exposed to soluble media-tors released by activated microglia. Neurochem. Int. 2009, 55, 35–40. [Google Scholar] [CrossRef]
Figure 1.
Pathological Aβ and tau formation in Alzheimer’s disease. (A) Cleavage sites for γ-secretase. (B) In the non-amyloidogenic pathway, APP gets chopped up by α-secretase and γ-secretase, which is called the nonamyloidogenic pathway, resulting in extracellular products called sAPPα and secreted fragment called P3, which are soluble, and leaves the membrane-bound APP intracellular domain (AICD). (C) In the amyloidogenic pathway, APP is first cleaved by β-secretase instead followed by the processing by γ-secretase, producing the secreted extracellular products called sAPPβ and Aβ40/Aβ42, and the same membrane-bound AICD. (D) Microtubules are strong cylindrical polymers that provide structural support to neurons. Tau is the major microtubule-associated protein in neurons and it stabilizes microtubule architecture. Under pathological conditions, tau becomes hyperphosphorylated and detaches from microtubules. Phosphorylated tau then aggregates to form paired helical filaments (PHFs) and NFTs.
Figure 1.
Pathological Aβ and tau formation in Alzheimer’s disease. (A) Cleavage sites for γ-secretase. (B) In the non-amyloidogenic pathway, APP gets chopped up by α-secretase and γ-secretase, which is called the nonamyloidogenic pathway, resulting in extracellular products called sAPPα and secreted fragment called P3, which are soluble, and leaves the membrane-bound APP intracellular domain (AICD). (C) In the amyloidogenic pathway, APP is first cleaved by β-secretase instead followed by the processing by γ-secretase, producing the secreted extracellular products called sAPPβ and Aβ40/Aβ42, and the same membrane-bound AICD. (D) Microtubules are strong cylindrical polymers that provide structural support to neurons. Tau is the major microtubule-associated protein in neurons and it stabilizes microtubule architecture. Under pathological conditions, tau becomes hyperphosphorylated and detaches from microtubules. Phosphorylated tau then aggregates to form paired helical filaments (PHFs) and NFTs.

Figure 2.
The role of microglia in neuroinflammation. (A) In a dichotomy model, the M1-type microglia typically express pro-inflammatory cytokines, chemokines and neurotoxic factors, while the M2-type microglia generally produce anti-inflammatory, neuroprotective and wound-healing factors. (B) In the healthy brain, microglia are metabolically flexible and can use glucose to support homeostatic “immune surveillance” functions. (C) Under pathological conditions, such as AD, microglia display a metabolic reprogramming featured by broken TCA cycle. HIF1α, hypoxia inducible factor-1α; PKM, Pyruvate kinase isozyme typeM2; PFKFB3, phosphofructokinase-2/fructose-2,6-bisphosphatase 3; OXPHOS, Oxidative phosphorylation; LDH, lactate dehydrogenase. Brown rectangle: glucose transporter; yellow cycle: TCA cycle; purple rectangle: oxidative phosphorylation; orange rectangle: increased glycolysis.
Figure 2.
The role of microglia in neuroinflammation. (A) In a dichotomy model, the M1-type microglia typically express pro-inflammatory cytokines, chemokines and neurotoxic factors, while the M2-type microglia generally produce anti-inflammatory, neuroprotective and wound-healing factors. (B) In the healthy brain, microglia are metabolically flexible and can use glucose to support homeostatic “immune surveillance” functions. (C) Under pathological conditions, such as AD, microglia display a metabolic reprogramming featured by broken TCA cycle. HIF1α, hypoxia inducible factor-1α; PKM, Pyruvate kinase isozyme typeM2; PFKFB3, phosphofructokinase-2/fructose-2,6-bisphosphatase 3; OXPHOS, Oxidative phosphorylation; LDH, lactate dehydrogenase. Brown rectangle: glucose transporter; yellow cycle: TCA cycle; purple rectangle: oxidative phosphorylation; orange rectangle: increased glycolysis.

Figure 3.
Tau and Aβ proteins, which are, accumulated inside the cytoplasm of the neuron gets encapsulated inside the exosomes in MVBs. These Tau- and Aβ-containing exosomes are degraded by lysosomes, but in AD when there is a problem in lysosomes; then these MVBs containing exosomes are filled with Tau and Aβ, and are released into the extracellular space. From the extracellular space, exosomes are captured by microglia. In addition, the accumulated Aβ oligomer and tau oligomer may act on microglia adjacent to neurons with free forms, and cause metabolic reprogramming of microglia through receptors or related signal pathways, thus maintaining the neuroinflammatory microenvironment. Purple cycle: intracellular Aβ; purple rectangle: APP; light purple wavy rectangle: Aβ oligomers; orange rectangle: tau; green rectangle: TREM2; brown triangle: APOE; yellow rectangle: RAGE.
Figure 3.
Tau and Aβ proteins, which are, accumulated inside the cytoplasm of the neuron gets encapsulated inside the exosomes in MVBs. These Tau- and Aβ-containing exosomes are degraded by lysosomes, but in AD when there is a problem in lysosomes; then these MVBs containing exosomes are filled with Tau and Aβ, and are released into the extracellular space. From the extracellular space, exosomes are captured by microglia. In addition, the accumulated Aβ oligomer and tau oligomer may act on microglia adjacent to neurons with free forms, and cause metabolic reprogramming of microglia through receptors or related signal pathways, thus maintaining the neuroinflammatory microenvironment. Purple cycle: intracellular Aβ; purple rectangle: APP; light purple wavy rectangle: Aβ oligomers; orange rectangle: tau; green rectangle: TREM2; brown triangle: APOE; yellow rectangle: RAGE.


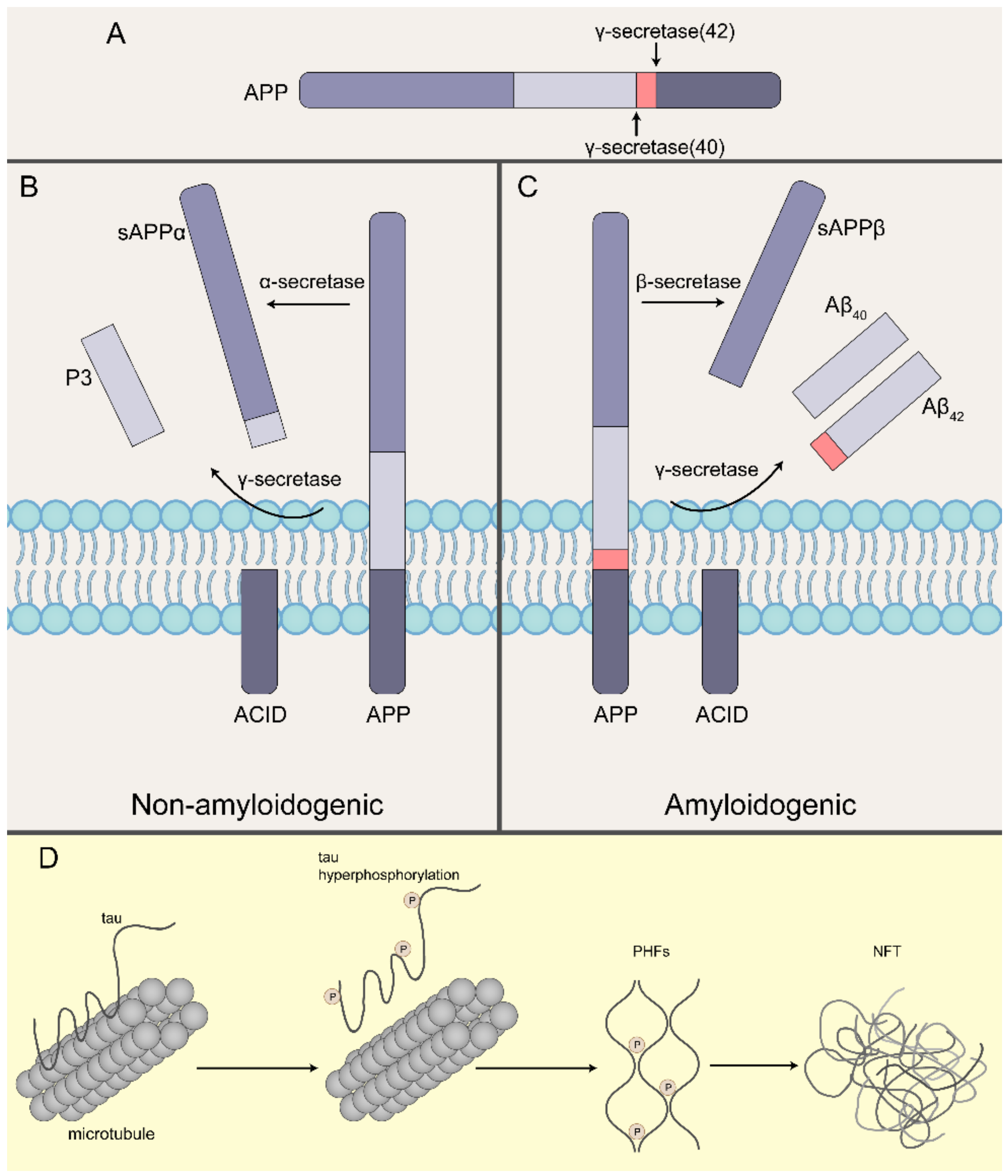{kind=link}
{kind=link}
{kind=link}
Table 1.
Effect of Aβ oligomers and tau oligomers on microglia metabolism.
| Receptor | Signaling Pathway | Metabolism | References | |
|---|---|---|---|---|
| Aβ | mTOR/HIF1α | up-regulation of glycolysis-related protein transcription | [70] | |
| TREM2 | Akt/mTOR/HIF1α | up-regulation of glycolysis-related protein transcription | [77,78] | |
| Formyl peptide receptor | AMPK/mTOR | up-regulation of glycolysis-related protein transcription S | [79] | |
| RAGE Receptor | Inflammatory cytokines | Interruption of TCA cycle and uncoupling of OXPHOS | [80,81,82] | |
| Toll-like receptor | Up-regulation of the expression of PFKFB3; activation of ATP- citrate lyase; down-regulation of Arg1 | [76,83,84] | ||
| NLRP3 | Increased glycolysis | [76,85,86,87,88] | ||
| SRIT1 | Increased glycolysis | [89,90] | ||
| Histone lactylation | Up-regulation of transcriptional levels of glycolysis related genes HIF1α, PKM2 and LDHA | [77,91] | ||
| CD36 | FA transporter or uptake promoter | [43,92] | ||
| Tau | TREM2 | Akt/mTOR/HIF1α | up-regulation of glycolysis-related protein transcription | [93,94,95] |
| APOE | Lipid metabolism; glucose metabolism | [96,97,98,99,100,101] | ||
| p38 AMPK | Up-regulation of the expression of PFKFB3 | [102,103] | ||
| cGAS/STING | accumulation of the metabolite succinate | [104,105] | ||
| NLRP3 | Increased glycolysis | [106,107,108,109,110] |
Abbreviations: mTOR, mammalian target of rapamycin; HIF1α, hypoxia inducible factor-1α; TREM2, Triggering Receptor Expressed On Myeloid Cells 2; Akt, Serine/Threonine Kinase; AMPK, Adenosine 5‘-monophosphate (AMP)-activated protein kinase; RAGE, the Receptor of Advanced Glycation EndproductsTCA, tricarboxylic acid; OXPHOS, Oxidative phosphorylation; PFKFB3, phosphofructokinase-2/fructose-2,6-bisphosphatase 3; Arg1, Arginase 1; NLRP3, NOD-like receptor thermal protein domain associated protein 3; SRIT1, Sirtuin 1; PKM2, Pyruvate kinase isozyme typeM2; LDHA, lactate dehydrogenase A; APOE, Apolipoprotein E; FA, fatty acid; cGAS, Cyclic GMP-AMP Synthase; STING, Stimulator Of Interferon Response cGAMP Interactor.
Table 2.
AD-associated exosomes contain Aβ and tau.
| Protein | Location | Species | The Uptake of Adjacent Cells | References |
|---|---|---|---|---|
| Aβ | SH-SY5Y cells and rat primary neurons | APP, AICD, C-terminal fragments (CTF) | [145] | |
| N2a cells | Aβ | [141] | ||
| CHO-APP695 cells | CTF, APP, Aβ | [146] | ||
| APP transgenic mice, brain from AD patients | CTF, APP, Aβ | [147] | ||
| brain samples of temporal neocortex from AD subjects, SH-SY5Y cell | Aβ oligomers | Peripheral neurons | [137] | |
| Tau | M1C cells and CSF samples from patients with AD | Phosphotau Species Associated with Neurodegeneration/dimerized or trimerized tau species | [142] | |
| N2a cells, rat primary neurons and CSF of patients with AD | Hypophosphorylated tau | Peripheral neurons | [148] | |
| transgenic mice with rapid tau propagation | Peripheral microglia | [149] | ||
| lamprey CNS | hyperphosphorylated tau | [150] | ||
| PS19 mice, CSF of patients with AD | p-Tau | Peripheral microglia | [151] | |
| Aβ and tau | plasma or serum from AD patients | P-S396-tau, P-T181-tau, and Aβ1-42 | [133,143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhao, Y.; Liu, B.; Wang, J.; Xu, L.; Yu, S.; Fu, J.; Yan, X.; Su, J. Aβ and Tau Regulate Microglia Metabolism via Exosomes in Alzheimer’s Disease. Biomedicines 2022, 10, 1800. https://doi.org/10.3390/biomedicines10081800
AMA Style
Zhao Y, Liu B, Wang J, Xu L, Yu S, Fu J, Yan X, Su J. Aβ and Tau Regulate Microglia Metabolism via Exosomes in Alzheimer’s Disease. Biomedicines. 2022; 10(8):1800. https://doi.org/10.3390/biomedicines10081800
Chicago/Turabian StyleZhao, Yuanxin, Buhan Liu, Jian Wang, Long Xu, Sihang Yu, Jiaying Fu, Xiaoyu Yan, and Jing Su. 2022. "Aβ and Tau Regulate Microglia Metabolism via Exosomes in Alzheimer’s Disease" Biomedicines 10, no. 8: 1800. https://doi.org/10.3390/biomedicines10081800
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.