
Non-Targeted Metabolomics Approach Revealed Significant Changes in Metabolic Pathways in Patients with Chronic Traumatic Encephalopathy


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Human Brain Samples
2.3. Sample Preparations
2.4. LC-MS Analysis
2.5. Validation
2.6. Data Processing and Statistical Analysis
2.7. Metabolite Identification and Pathway Analysis
2.8. RNA Sequencing and Analysis
2.9. Immunohistochemistry (IHC)
3. Results
3.1. Metabolic Profiling of Postmortem Brain of the CTE Patients
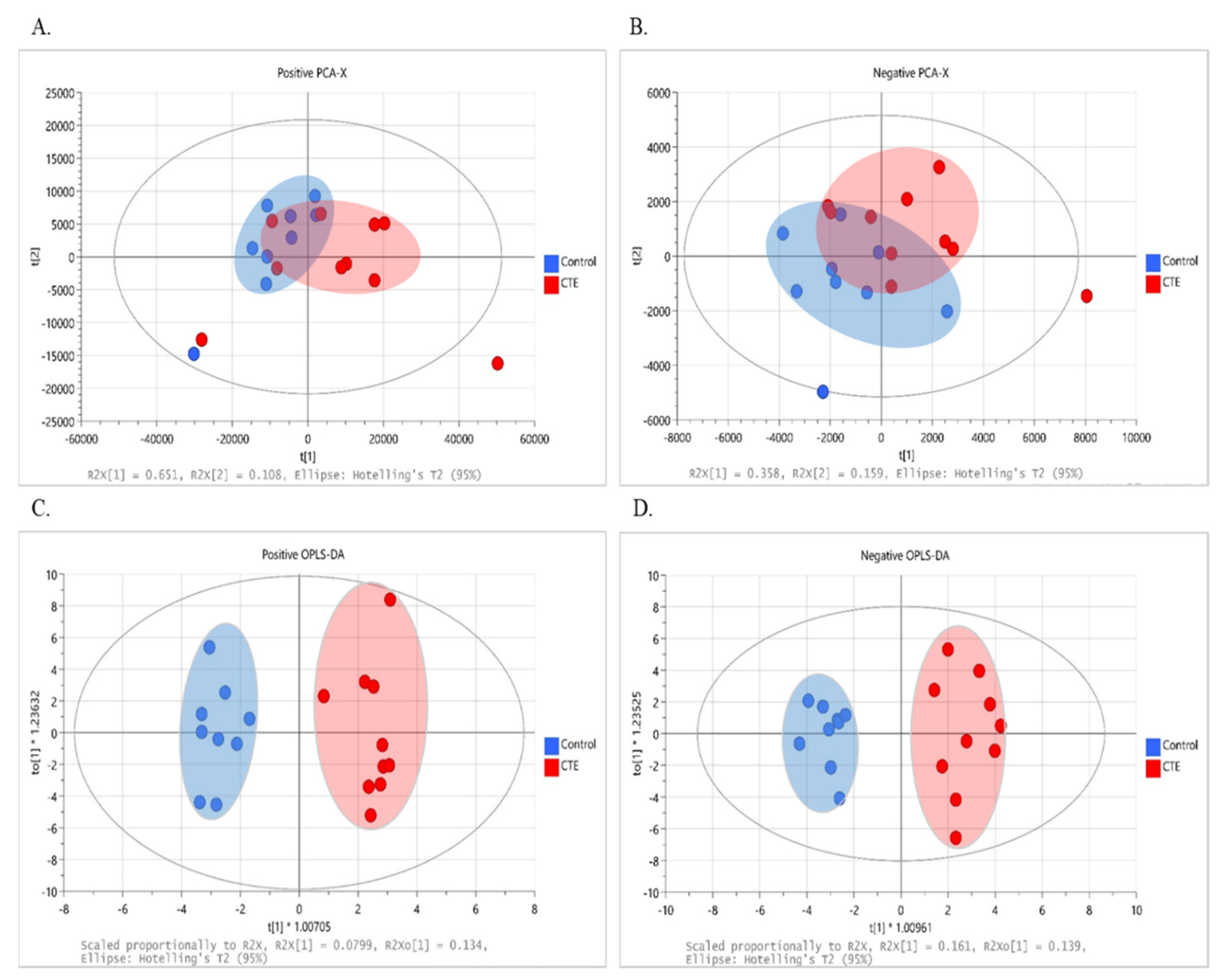
3.2. Multivariate Analysis
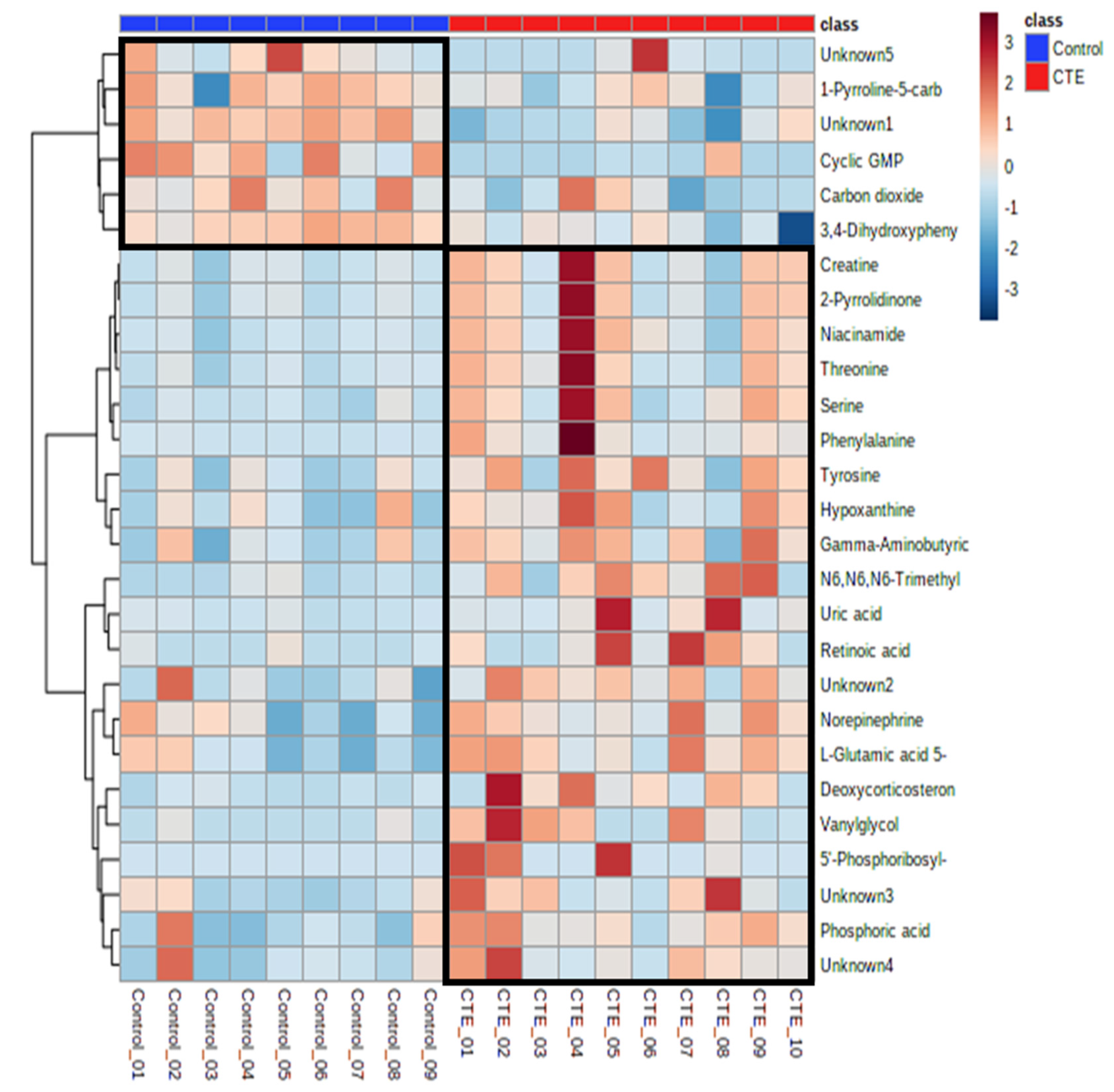
3.3. Identification of Significant Metabolites
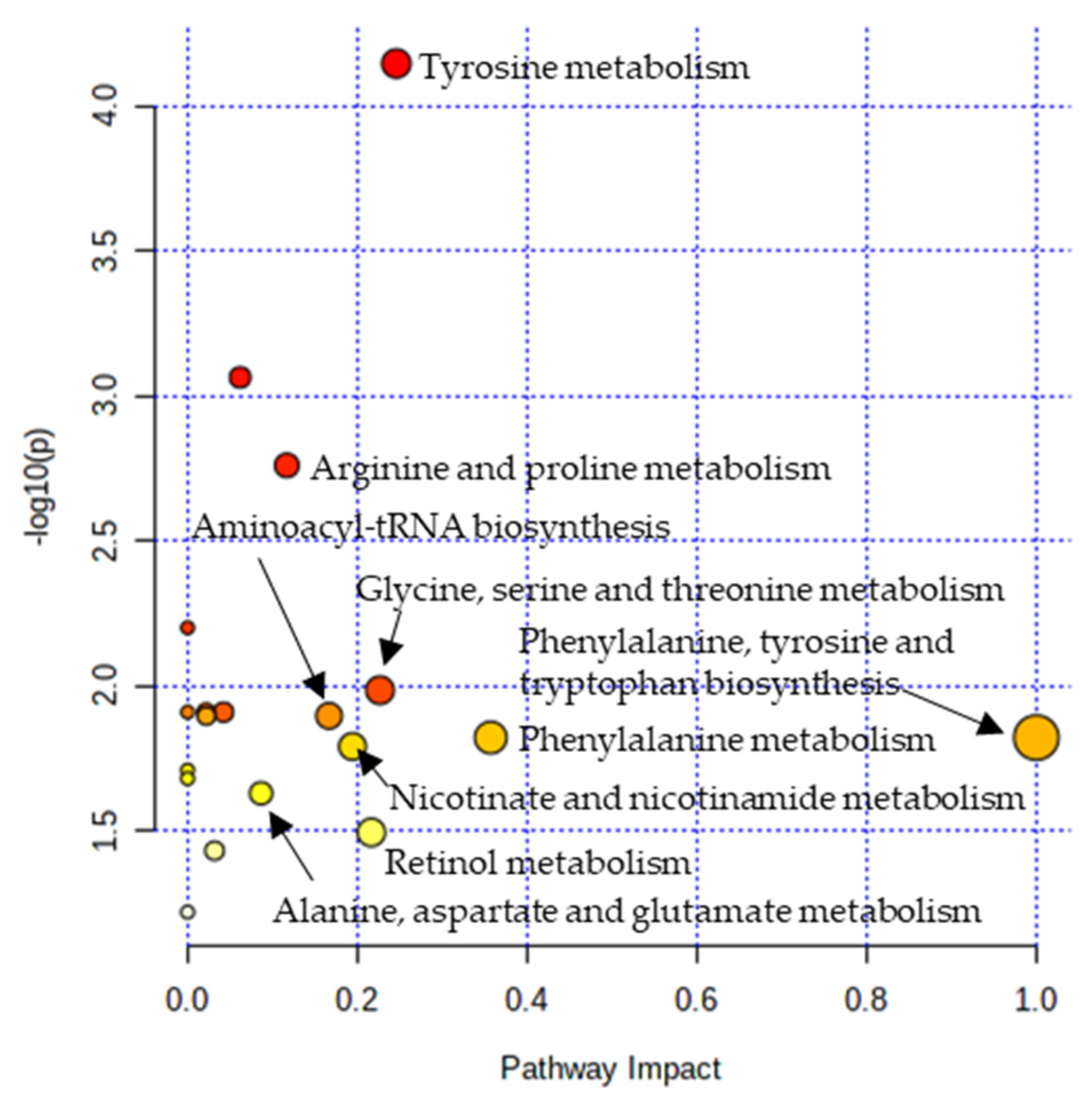
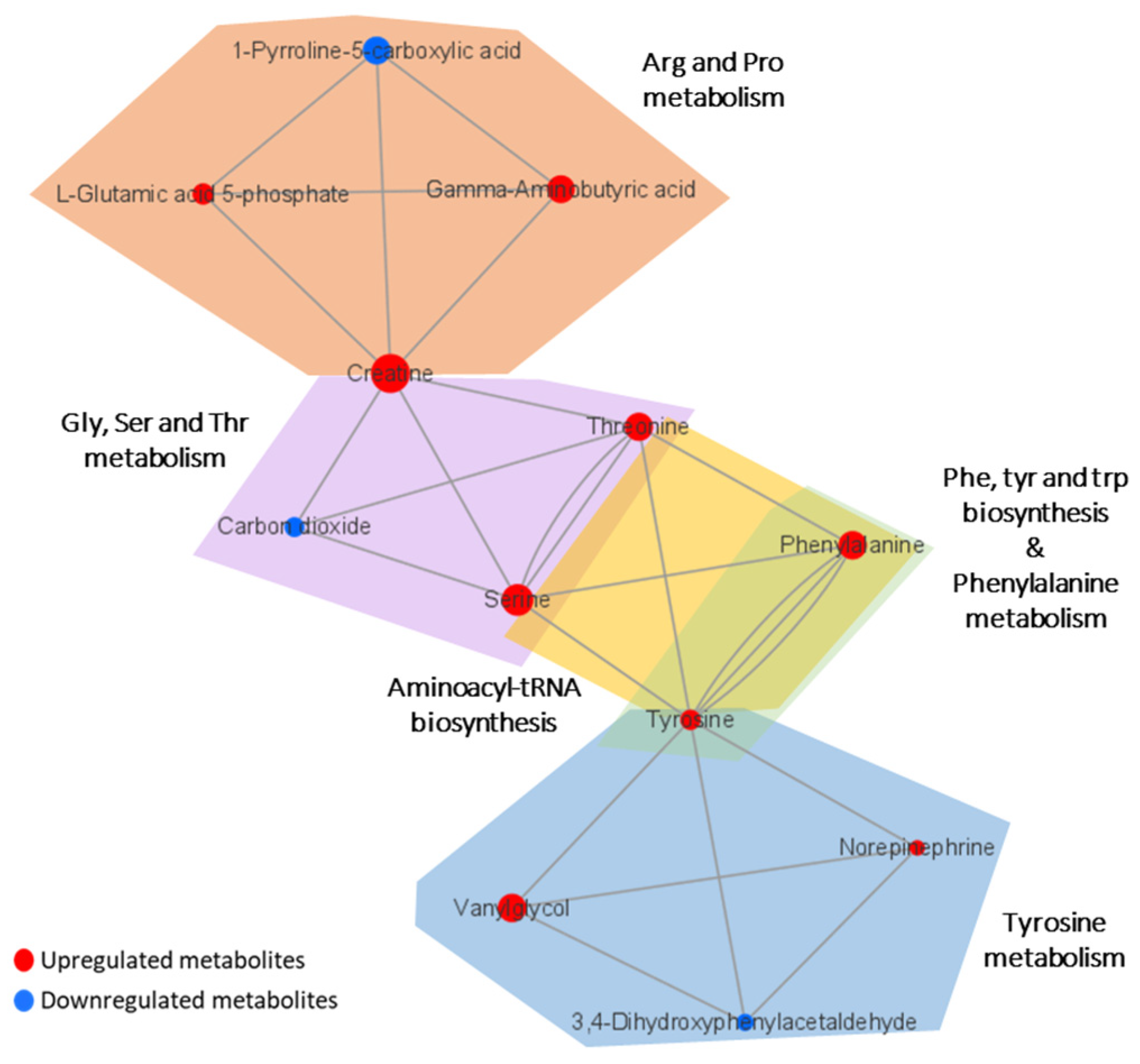
3.4. Metabolic Pathway Analysis
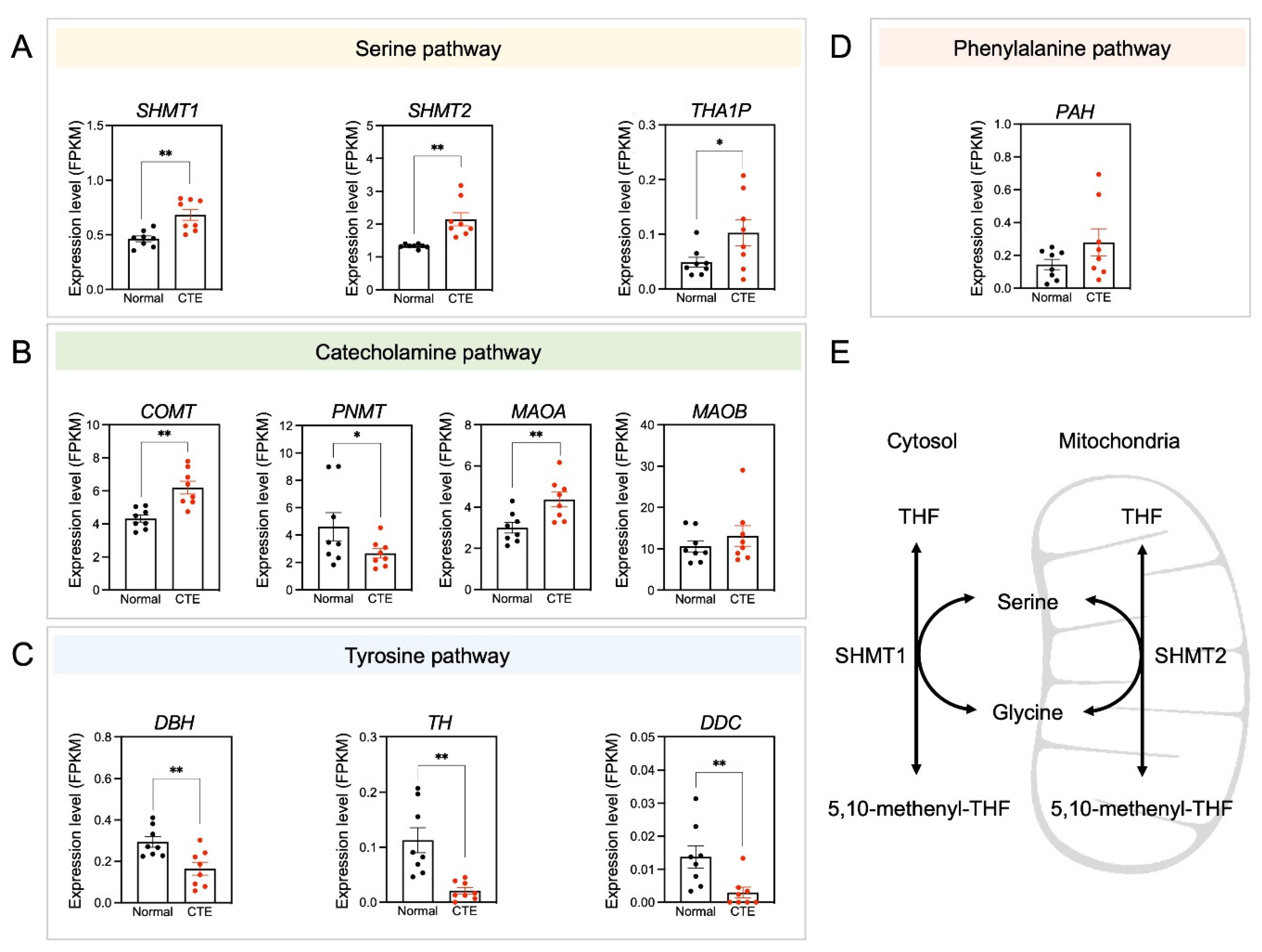
3.5. Alterations of Serine Pathway-Related Gene Expression
3.6. Immunoreactivity of SHMT2 in Astrocytes of CTE Patients
4. Discussion
4.1. Astrocyte Activation
4.2. Phenylalanine Metabolism
4.3. Tyrosine Metabolism
4.4. Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iverson, G.L.; Gardner, A.J.; McCrory, P.; Zafonte, R.; Castellani, R.J. A critical review of chronic traumatic encephalopathy. Neurosci. Biobehav. Rev. 2015, 56, 276–293. [Google Scholar] [CrossRef] [PubMed]
- Tharmaratnam, T.; Iskandar, M.A.; Tabobondung, T.C.; Tobbia, I.; Gopee-Ramanan, P.; Tabobondung, T.A. Chronic Traumatic Encephalopathy in Professional American Football Players: Where Are We Now? Front. Neurol. 2018, 9, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omalu, B.I.; Fitzsimmons, R.P.; Hammers, J.; Bailes, J. Chronic traumatic encephalopathy in a professional American wrestler. J. Forensic Nurs. 2010, 6, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Buckland, M.E.; Sy, J.; Szentmariay, I.; Kullen, A.; Lee, M.; Harding, A.; Halliday, G.; Suter, C.M. Chronic traumatic encephalopathy in two former Australian National Rugby League players. Acta Neuropathol. Commun. 2019, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Stern, R.A.; Nowinski, C.J.; Stein, T.D.; Alvarez, V.E.; Daneshvar, D.H.; Lee, H.S.; Wojtowicz, S.M.; Hall, G.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Omalu, B.; Hammers, J.L.; Bailes, J.; Hamilton, R.L.; Kamboh, M.I.; Webster, G.; Fitzsimmons, R.P. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg. Focus 2011, 31, E3. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.J.; Tannenberg, A.E. Dementia pugilistica in an alcoholic achondroplastic dwarf. Pathology 1996, 28, 102–104. [Google Scholar] [CrossRef]
- Danielsen, T.; Hauch, C.; Kelly, L.; White, C.L. Chronic Traumatic Encephalopathy (CTE)-Type Neuropathology in a Young Victim of Domestic Abuse. J. Neuropathol. Exp. Neurol. 2021, 80, 624–627. [Google Scholar] [CrossRef]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.S.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef]
- Bieniek, K.F.; Cairns, N.J.; Crary, J.F.; Dickson, D.W.; Folkerth, R.D.; Keene, C.D.; Litvan, I.; Perl, D.P.; Stein, T.D.; Vonsattel, J.P.; et al. The Second NINDS/NIBIB Consensus Meeting to Define Neuropathological Criteria for the Diagnosis of Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2021, 80, 210–219. [Google Scholar] [CrossRef]
- Gavett, B.E.; Stern, R.A.; McKee, A.C. Chronic traumatic encephalopathy: A potential late effect of sport-related concussive and subconcussive head trauma. Clin. Sports Med. 2011, 30, 179–188, xi. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.; Prosapio, J.; Kara, S.; Sonty, S.; Youssef, P.; Nedd, K. Preliminary clinical diagnostic criteria for chronic traumatic encephalopathy: A case report and literature review. Interdiscip. Neurosurg. 2021, 26, 101290. [Google Scholar] [CrossRef]
- Levine, B.; Kovacevic, N.; Nica, E.I.; Cheung, G.; Gao, F.; Schwartz, M.L.; Black, S.E. The Toronto traumatic brain injury study: Injury severity and quantified MRI. Neurology 2008, 70, 771–778. [Google Scholar] [CrossRef]
- Gale, S.D.; Baxter, L.; Roundy, N.; Johnson, S.C. Traumatic brain injury and grey matter concentration: A preliminary voxel based morphometry study. J. Neurol. Neurosurg. Psychiatry 2005, 76, 984–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, R.C.; Hess, C.P.; Brus-Ramer, M.; Possin, K.L.; Cohn-Sheehy, B.I.; Kramer, J.H.; Berger, M.S.; Yaffe, K.; Miller, B.; Rabinovici, G.D. Cavum Septum Pellucidum in Retired American Pro-Football Players. J. Neurotrauma 2016, 33, 157–161. [Google Scholar] [CrossRef]
- Small, G.W.; Kepe, V.; Siddarth, P.; Ercoli, L.M.; Merrill, D.A.; Donoghue, N.; Bookheimer, S.Y.; Martinez, J.; Omalu, B.; Bailes, J.; et al. PET scanning of brain tau in retired national football league players: Preliminary findings. Am. J. Geriatr. Psychiatry 2013, 21, 138–144. [Google Scholar] [CrossRef]
- Geiszler, P.C.; Ugun-Klusek, A.; Lawler, K.; Pardon, M.C.; Yuchun, D.; Bai, L.; Daykin, C.A.; Auer, D.P.; Bedford, L. Dynamic metabolic patterns tracking neurodegeneration and gliosis following 26S proteasome dysfunction in mouse forebrain neurons. Sci. Rep. 2018, 8, 4833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.C.; Stein, T.D.; Kiernan, P.T.; Alvarez, V.E. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015, 25, 350–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O. Metabolomics by Gas Chromatography-Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol. 2016, 114, 21–33. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Kim, Y.H.; Shim, H.S.; Kim, K.H.; Lee, J.; Chung, B.C.; Kowall, N.W.; Ryu, H.; Lee, J. Metabolomic Analysis Identifies Alterations of Amino Acid Metabolome Signatures in the Postmortem Brain of Alzheimer’s Disease. Exp. Neurobiol. 2019, 28, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, X.; Lu, X.; Lin, X.; Xu, G. A data preprocessing strategy for metabolomics to reduce the mask effect in data analysis. Front. Mol. Biosci. 2015, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [Green Version]
- Lever, J.; Krzywinski, M.; Altman, N. Principal component analysis. Nat. Methods 2017, 14, 641–642. [Google Scholar] [CrossRef] [Green Version]
- Worley, B.; Powers, R. Multivariate Analysis in Metabolomics. Curr. Metab. 2013, 1, 92–107. [Google Scholar] [CrossRef]
- Suhre, K.; Schmitt-Kopplin, P. MassTRIX: Mass translator into pathways. Nucleic Acids Res. 2008, 36, W481–W484. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.S.; Lee, S.; Shin, J.Y.; Hwang, Y.J.; Cho, H.; Yoo, S.K.; Kim, Y.; Lim, S.; Kim, Y.K.; Hwang, E.M.; et al. Transcriptome analyses of chronic traumatic encephalopathy show alterations in protein phosphatase expression associated with tauopathy. Exp. Mol. Med. 2017, 49, e333. [Google Scholar] [CrossRef]
- Hyeon, S.J.; Park, J.; Yoo, J.; Kim, S.H.; Hwang, Y.J.; Kim, S.C.; Liu, T.; Shim, H.S.; Kim, Y.; Cho, Y.; et al. Dysfunction of X-linked inhibitor of apoptosis protein (XIAP) triggers neuropathological processes via altered p53 activity in Huntington’s disease. Prog. Neurobiol. 2021, 204, 102110. [Google Scholar] [CrossRef]
- Crowe, A.R.; Yue, W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio. Protoc. 2019, 9, e3465. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, C.; Zheng, M.; Gao, F.; Zhang, J.; Liu, F. Metabolomics Analysis of L-Arginine Induced Gastrointestinal Motility Disorder in Rats Using UPLC-MS After Magnolol Treatment. Front. Pharmacol. 2019, 10, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, H.; Blaszczynski, J.; Billaut, J.C.; Nadal-Desbarats, L.; Pradat, P.F.; Devos, D.; Moreau, C.; Andres, C.R.; Emond, P.; Corcia, P.; et al. Comparative analysis of targeted metabolomics: Dominance-based rough set approach versus orthogonal partial least square-discriminant analysis. J. Biomed. Inform. 2015, 53, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Alosco, M.L.; Mariani, M.L.; Adler, C.H.; Balcer, L.J.; Bernick, C.; Au, R.; Banks, S.J.; Barr, W.B.; Bouix, S.; Cantu, R.C.; et al. Developing methods to detect and diagnose chronic traumatic encephalopathy during life: Rationale, design, and methodology for the DIAGNOSE CTE Research Project. Alzheimers Res. Ther. 2021, 13, 136. [Google Scholar] [CrossRef]
- Stern, R.A.; Daneshvar, D.H.; Baugh, C.M.; Seichepine, D.R.; Montenigro, P.H.; Riley, D.O.; Fritts, N.G.; Stamm, J.M.; Robbins, C.A.; McHale, L.; et al. Clinical presentation of chronic traumatic encephalopathy. Neurology 2013, 81, 1122–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Bailes, J.E.; Huber, J.D.; Rosen, C.L. Linking traumatic brain injury to chronic traumatic encephalopathy: Identification of potential mechanisms leading to neurofibrillary tangle development. J. Neurotrauma 2014, 31, 1129–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Shao, A.; Yao, Y.; Tu, S.; Deng, Y.; Zhang, J. Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 2020, 18, 62. [Google Scholar] [CrossRef] [Green Version]
- Wolosker, H.; Blackshaw, S.; Snyder, S.H. Serine racemase: A glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. USA 1999, 96, 13409–13414. [Google Scholar] [CrossRef] [Green Version]
- Van Horn, M.R.; Sild, M.; Ruthazer, E.S. D-serine as a gliotransmitter and its roles in brain development and disease. Front. Cell Neurosci. 2013, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Perez, E.J.; Tapanes, S.A.; Loris, Z.B.; Balu, D.T.; Sick, T.J.; Coyle, J.T.; Liebl, D.J. Enhanced astrocytic d-serine underlies synaptic damage after traumatic brain injury. J. Clin. Investig. 2017, 127, 3114–3125. [Google Scholar] [CrossRef]
- Wolosker, H.; Dumin, E.; Balan, L.; Foltyn, V.N. D-amino acids in the brain: D-serine in neurotransmission and neurodegeneration. FEBS J. 2008, 275, 3514–3526. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Uno, Y.; Rudolph, U.; Cobb, J.; Liu, J.; Anderson, T.; Levy, D.; Balu, D.T.; Coyle, J.T. Astrocytes in primary cultures express serine racemase, synthesize d-serine and acquire A1 reactive astrocyte features. Biochem. Pharm. 2018, 151, 245–251. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- van den Pol, A.N.; Obrietan, K.; Chen, G. Excitatory actions of GABA after neuronal trauma. J. Neurosci. 1996, 16, 4283–4292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium signaling in neurodegeneration. Mol. Neurodegener. 2009, 4, 20. [Google Scholar] [CrossRef]
- Weber, J.T. Altered calcium signaling following traumatic brain injury. Front. Pharm. 2012, 3, 60. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.L.; Guan, P.P.; Liang, Y.Y.; Huang, X.S.; Wang, P. Calcium Ions Stimulate the Hyperphosphorylation of Tau by Activating Microsomal Prostaglandin E Synthase 1. Front. Aging Neurosci. 2019, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- ten Hoedt, A.E.; de Sonneville, L.M.; Francois, B.; ter Horst, N.M.; Janssen, M.C.; Rubio-Gozalbo, M.E.; Wijburg, F.A.; Hollak, C.E.; Bosch, A.M. High phenylalanine levels directly affect mood and sustained attention in adults with phenylketonuria: A randomised, double-blind, placebo-controlled, crossover trial. J. Inherit. Metab. Dis. 2011, 34, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Sharman, R.; Sullivan, K.; Young, R.M.; McGill, J. Depressive symptoms in adolescents with early and continuously treated phenylketonuria: Associations with phenylalanine and tyrosine levels. Gene 2012, 504, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.D.; Montenigro, P.H.; Alvarez, V.E.; Xia, W.; Crary, J.F.; Tripodis, Y.; Daneshvar, D.H.; Mez, J.; Solomon, T.; Meng, G.; et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. 2015, 130, 21–34. [Google Scholar] [CrossRef]
- Anand, B.G.; Dubey, K.; Shekhawat, D.S.; Kar, K. Intrinsic property of phenylalanine to trigger protein aggregation and hemolysis has a direct relevance to phenylketonuria. Sci. Rep. 2017, 7, 11146. [Google Scholar] [CrossRef]
- Lin, A.P.; Ramadan, S.; Stern, R.A.; Box, H.C.; Nowinski, C.J.; Ross, B.D.; Mountford, C.E. Changes in the neurochemistry of athletes with repetitive brain trauma: Preliminary results using localized correlated spectroscopy. Alzheimers Res. 2015, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernstrom, J.D.; Fernstrom, M.H. Tyrosine, phenylalanine, and catecholamine synthesis and function in the brain. J. Nutr. 2007, 137, 1539S–1547S. [Google Scholar] [CrossRef] [PubMed]
- Impellizzeri, D.; Campolo, M.; Bruschetta, G.; Crupi, R.; Cordaro, M.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Traumatic Brain Injury Leads to Development of Parkinson’s Disease Related Pathology in Mice. Front. Neurosci. 2016, 10, 458. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshini, M.; Kamal, M.A.; Greig, N.H.; Reale, M.; Abuzenadah, A.M.; Chaudhary, A.G.; Damanhouri, G.A. Alzheimer’s disease and type 2 diabetes: Exploring the association to obesity and tyrosine hydroxylase. CNS Neurol. Disord. Drug Targets 2012, 11, 482–489. [Google Scholar] [CrossRef]
- Chen, Y.; Lian, Y.; Ma, Y.; Wu, C.; Zheng, Y.; Xie, N. The expression and significance of tyrosine hydroxylase in the brain tissue of Parkinsons disease rats. Exp. Med. 2017, 14, 4813–4816. [Google Scholar] [CrossRef]
- van de Rest, O.; Bloemendaal, M.; de Heus, R.; Aarts, E. Dose-Dependent Effects of Oral Tyrosine Administration on Plasma Tyrosine Levels and Cognition in Aging. Nutrients 2017, 9, 1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepma, M.; Deinum, J.; Asplund, C.L.; Rombouts, S.A.; Tamsma, J.T.; Tjeerdema, N.; Spape, M.M.; Garland, E.M.; Robertson, D.; Lenders, J.W.; et al. Neurocognitive function in dopamine-beta-hydroxylase deficiency. Neuropsychopharmacology 2011, 36, 1608–1619. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, K.; Tabuchi, K.; Ojika, M.; Zucca, F.A.; Zecca, L.; Ito, S. Norepinephrine and its metabolites are involved in the synthesis of neuromelanin derived from the locus coeruleus. J. Neurochem. 2015, 135, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Li, S.; Feng, H.; Chen, Y. Iron Metabolism Disorders for Cognitive Dysfunction After Mild Traumatic Brain Injury. Front. Neurosci. 2021, 15, 587197. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Sapuan, A.; Dineen, R.A.; Auer, D.P. Life span pigmentation changes of the substantia nigra detected by neuromelanin-sensitive MRI. Mov. Disord. 2018, 33, 1792–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zecca, L.; Zucca, F.A.; Wilms, H.; Sulzer, D. Neuromelanin of the substantia nigra: A neuronal black hole with protective and toxic characteristics. Trends Neurosci. 2003, 26, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, H.I.L.; Riphagen, J.M.; Ramakers, I.; Verhey, F.R.J. Alzheimer’s disease pathology: Pathways between central norepinephrine activity, memory, and neuropsychiatric symptoms. Mol. Psychiatry 2021, 26, 897–906. [Google Scholar] [CrossRef]
- Raskind, M.A.; Peskind, E.R.; Halter, J.B.; Jimerson, D.C. Norepinephrine and MHPG levels in CSF and plasma in Alzheimer’s disease. Arch. Gen. Psychiatry 1984, 41, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Pillet, L.E.; Taccola, C.; Cotoni, J.; Thiriez, H.; Andre, K.; Verpillot, R. Correction: Correlation between cognition and plasma noradrenaline level in Alzheimer’s disease: A potential new blood marker of disease evolution. Transl. Psychiatry 2020, 10, 409. [Google Scholar] [CrossRef]
- Matthews, K.L.; Chen, C.P.; Esiri, M.M.; Keene, J.; Minger, S.L.; Francis, P.T. Noradrenergic changes, aggressive behavior, and cognition in patients with dementia. Biol. Psychiatry 2002, 51, 407–416. [Google Scholar] [CrossRef]
- Mehta, R.; Giri, S.; Mallick, B.N. REM sleep loss-induced elevated noradrenaline could predispose an individual to psychosomatic disorders: A review focused on proposal for prediction, prevention, and personalized treatment. EPMA J. 2020, 11, 529–549. [Google Scholar] [CrossRef]
- Plotegher, N.; Berti, G.; Ferrari, E.; Tessari, I.; Zanetti, M.; Lunelli, L.; Greggio, E.; Bisaglia, M.; Veronesi, M.; Girotto, S.; et al. DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Sci. Rep. 2017, 7, 40699. [Google Scholar] [CrossRef]
- Panneton, W.M.; Kumar, V.B.; Gan, Q.; Burke, W.J.; Galvin, J.E. The neurotoxicity of DOPAL: Behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS ONE 2010, 5, e15251. [Google Scholar] [CrossRef]
- Burke, W.J.; Li, S.W.; Williams, E.A.; Nonneman, R.; Zahm, D.S. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: Implications for Parkinson’s disease pathogenesis. Brain Res. 2003, 989, 205–213. [Google Scholar] [CrossRef]
- Tartaglia, M.C.; Hazrati, L.N.; Davis, K.D.; Green, R.E.; Wennberg, R.; Mikulis, D.; Ezerins, L.J.; Keightley, M.; Tator, C. Chronic traumatic encephalopathy and other neurodegenerative proteinopathies. Front. Hum. Neurosci. 2014, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Rt | MW | Ionization Method | VIP | p-Value | Fold Change | Fold Change Direction | Name |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.06 | 118.93 | Negative | 1.71 | 0.0003 | 5.17 | Down | Unknown 1 |
| 2 | 1.25 | 123.04 | Positive | 1.69 | 0.0030 | 0.92 | Up | Niacinamide |
| 3 | 9.26 | 204.10 | Positive | 1.63 | 0.0279 | 1.20 | Up | Tyrosine |
| 4 | 1.17 | 120.07 | Positive | 1.62 | 0.0172 | 2.44 | Up | Threonine |
| 5 | 1.27 | 132.10 | Positive | 1.58 | 0.0350 | 4.13 | Up | Creatine |
| 6 | 1.00 | 104.11 | Positive | 1.56 | 0.0435 | 2.37 | Up | Gamma-Aminobutyric acid |
| 7 | 1.27 | 86.10 | Positive | 1.55 | 0.0435 | 2.57 | Up | 2-Pyrrolidinone |
| 8 | 0.95 | 225.99 | Negative | 1.45 | 0.0101 | 1.35 | Up | l-Glutamic acid 5-phosphate |
| 9 | 9.87 | 187.10 | Negative | 1.44 | 0.0220 | 7.01 | Up | N6,N6,N6-Trimethyl-l-lysine |
| 10 | 1.09 | 343.99 | Negative | 1.43 | 0.0030 | 0.15 | Down | Cyclic GMP |
| 11 | 0.94 | 104.03 | Negative | 1.41 | 0.0101 | 3.00 | Up | Serine |
| 12 | 12.07 | 329.23 | Negative | 1.36 | 0.0076 | 1.47 | Up | Deoxycorticosterone |
| 13 | 1.22 | 353.03 | Positive | 1.34 | 0.0452 | 2.89 | Up | 5′-Phosphoribosyl-N-formylglycinamide |
| 14 | 1.16 | 219.08 | Negative | 1.34 | 0.0217 | 2.48 | Up | Vanylglycol |
| 15 | 9.80 | 679.51 | Positive | 1.33 | 0.0279 | 1.32 | Up | Unknown 2 |
| 16 | 1.04 | 136.05 | Positive | 1.33 | 0.0022 | 2.38 | Down | 1-Pyrroline-5-carboxylic acid |
| 17 | 13.90 | 335.22 | Negative | 1.24 | 0.0425 | 0.72 | Up | Retinoic acid |
| 18 | 1.35 | 187.00 | Negative | 1.23 | 0.0002 | 0.69 | Down | 3,4-Dihydroxyphenylacetaldehyde |
| 19 | 0.94 | 96.92 | Negative | 1.23 | 0.0279 | 2311.30 | Up | Phosphoric acid |
| 20 | 1.08 | 135.03 | Negative | 1.21 | 0.0435 | 8.82 | Up | Hypoxanthine |
| 21 | 1.55 | 164.07 | Negative | 1.17 | 0.0006 | 2.47 | Up | Phenylalanine |
| 22 | 1.25 | 78.96 | Negative | 1.15 | 0.0279 | 1.12 | Down | Carbon dioxide |
| 23 | 1.00 | 204.00 | Negative | 1.14 | 0.0350 | 0.43 | Up | Norepinephrine |
| 24 | 0.97 | 134.89 | Negative | 1.13 | 0.0172 | 0.67 | Up | Unknown 3 |
| 25 | 0.99 | 167.02 | Negative | 1.13 | 0.0030 | 1.27 | Up | Uric acid |
| 26 | 0.92 | 160.91 | Negative | 1.07 | 0.0350 | 1.79 | Up | Unknown 4 |
| 27 | 12.57 | 316.25 | Positive | 1.06 | 0.0181 | 1.84 | Down | Unknown 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Kim, S.; Kim, Y.H.; Park, U.; Lee, J.; McKee, A.C.; Kim, K.H.; Ryu, H.; Lee, J. Non-Targeted Metabolomics Approach Revealed Significant Changes in Metabolic Pathways in Patients with Chronic Traumatic Encephalopathy. Biomedicines 2022, 10, 1718. https://doi.org/10.3390/biomedicines10071718
Lee J, Kim S, Kim YH, Park U, Lee J, McKee AC, Kim KH, Ryu H, Lee J. Non-Targeted Metabolomics Approach Revealed Significant Changes in Metabolic Pathways in Patients with Chronic Traumatic Encephalopathy. Biomedicines. 2022; 10(7):1718. https://doi.org/10.3390/biomedicines10071718
Chicago/Turabian StyleLee, Jinkyung, Suhyun Kim, Yoon Hwan Kim, Uiyeol Park, Junghee Lee, Ann C. McKee, Kyoung Heon Kim, Hoon Ryu, and Jeongae Lee. 2022. "Non-Targeted Metabolomics Approach Revealed Significant Changes in Metabolic Pathways in Patients with Chronic Traumatic Encephalopathy" Biomedicines 10, no. 7: 1718. https://doi.org/10.3390/biomedicines10071718