
Cochlear Implantation Outcomes in Patients with Auditory Neuropathy Spectrum Disorder of Genetic and Non-Genetic Etiologies: A Multicenter Study

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetic Examinations
2.2. Imaging Studies
2.3. CI Outcome Evaluation
2.4. Statistical Analysis
3. Results
3.1. CI Outcomes in ANSD Patients with Biallelic OTOF Variants
3.2. CI Outcomes in ANSD Patients with Rare Gene Variants
3.3. CI Outcomes in ANSD Patients with CND
3.4. CI Outcomes in ANSD Patients with Indefinite Etiologies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bielecki, I.; Horbulewicz, A.; Wolan, T. Prevalence and risk factors for auditory neuropathy spectrum disorder in a screened newborn population at risk for hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1668–1670. [Google Scholar] [CrossRef] [PubMed]
- Hood, L.J. Auditory Neuropathy/Auditory Synaptopathy. Otolaryngol. Clin. N. Am. 2021, 54, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.; Starr, A. Auditory neuropathy—Neural and synaptic mechanisms. Nat. Rev. Neurol. 2016, 12, 135–149. [Google Scholar] [CrossRef]
- Rance, G. Auditory neuropathy/dys-synchrony and its perceptual consequences. Trends Amplif. 2005, 9, 1–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starr, A.; Picton, T.W.; Sininger, Y.; Hood, L.J.; Berlin, C.I. Auditory neuropathy. Brain 1996, 119, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Hsu, C.J.; Lin, Y.H.; Lin, Y.H.; Yang, S.Y.; Yang, T.H.; Chen, P.L.; Wu, C.C.; Liu, T.C. An integrative approach for pediatric auditory neuropathy spectrum disorders: Revisiting etiologies and exploring the prognostic utility of auditory steady-state response. Sci. Rep. 2020, 10, 9816. [Google Scholar] [CrossRef] [PubMed]
- De Siati, R.D.; Rosenzweig, F.; Gersdorff, G.; Gregoire, A.; Rombaux, P.; Deggouj, N. Auditory Neuropathy Spectrum Disorders: From Diagnosis to Treatment: Literature Review and Case Reports. J. Clin. Med. 2020, 9, 1074. [Google Scholar] [CrossRef]
- Berlin, C.I.; Hood, L.J.; Morlet, T.; Wilensky, D.; Li, L.; Mattingly, K.R.; Taylor-Jeanfreau, J.; Keats, B.J.; John, P.S.; Montgomery, E.; et al. Multi-site diagnosis and management of 260 patients with auditory neuropathy/dys-synchrony (auditory neuropathy spectrum disorder). Int. J. Audiol. 2010, 49, 30–43. [Google Scholar] [CrossRef]
- Rance, G.; Starr, A. Pathophysiological mechanisms and functional hearing consequences of auditory neuropathy. Brain 2015, 138, 3141–3158. [Google Scholar] [CrossRef]
- Harrison, R.V.; Gordon, K.A.; Papsin, B.C.; Negandhi, J.; James, A.L. Auditory neuropathy spectrum disorder (ANSD) and cochlear implantation. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1980–1987. [Google Scholar] [CrossRef]
- Teagle, H.F.; Roush, P.A.; Woodard, J.S.; Hatch, D.R.; Zdanski, C.J.; Buss, E.; Buchman, C.A. Cochlear implantation in children with auditory neuropathy spectrum disorder. Ear Hear. 2010, 31, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.-y.; Moteki, H.; Miyagawa, M.; Yamasoba, T.; Kashio, A.; Iwasaki, S.; Takahashi, M.; Naito, Y.; Fujiwara, K.; Sugaya, A. Etiology of hearing loss affects auditory skill development and vocabulary development in pediatric cochlear implantation cases. Acta Oto-Laryngol. 2022, 142, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lin, Y.H.; Liu, T.C.; Lin, K.N.; Yang, W.S.; Hsu, C.J.; Chen, P.L.; Wu, C.M. Identifying Children With Poor Cochlear Implantation Outcomes Using Massively Parallel Sequencing. Medicine 2015, 94, e1073. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, A.R.; Han, J.H.; Kim, S.D.; Kim, S.H.; Koo, J.-W.; Oh, S.H.; Choi, B.Y. Outcome of Cochlear Implantation in Prelingually Deafened Children According to Molecular Genetic Etiology. Ear Hear. 2017, 38, e316–e324. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Polineni, S.P.; Davies, C.; Shahal, D.; Mittal, J.; Al-Zaghal, Z.; Sinha, R.; Jindal, U.; Mittal, R. Genotype-Phenotype Correlation for Predicting Cochlear Implant Outcome: Current Challenges and Opportunities. Front. Genet. 2020, 11, 678. [Google Scholar] [CrossRef]
- Wu, C.C.; Tsai, C.Y.; Lin, Y.H.; Chen, P.Y.; Lin, P.H.; Cheng, Y.F.; Wu, C.M.; Lin, Y.H.; Lee, C.Y.; Erdenechuluun, J.; et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes 2019, 10, 772. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.H.; Wu, P.C.; Tsai, C.Y.; Lin, Y.H.; Lo, M.Y.; Hsu, S.J.; Lin, P.H.; Erdenechuluun, J.; Wu, H.P.; Hsu, C.J.; et al. Hearing Impairment with Monoallelic GJB2 Variants: A GJB2 Cause or Non-GJB2 Cause? J. Mol. Diagn. 2021, 23, 1279–1291. [Google Scholar] [CrossRef]
- Sennaroğlu, L.; Bajin, M.D. Classification and Current Management of Inner Ear Malformations. Balkan Med. J. 2017, 34, 397–411. [Google Scholar] [CrossRef]
- van der Knaap, M.S.; Valk, J. Classification of congenital abnormalities of the CNS. AJNR Am. J. Neuroradiol. 1988, 9, 315–326. [Google Scholar]
- Archbold, S.; Lutman, M.; Marshall, D. Categories of auditory performance. Ann. Otol. Rhinol. Laryngol. Suppl. 1995, 166, 312–314. [Google Scholar]
- Allen, M.C.; Nikolopoulos, T.P.; O’Donoghue, G.M. Speech intelligibility in children after cochlear implantation. Am. J. Otol. 1998, 19, 742–746. [Google Scholar] [PubMed]
- Myers, K.; Nicholson, N. Cochlear Implant Behavioral Outcomes for Children With Auditory Neuropathy Spectrum Disorder: A Mini-Systematic Review. Am. J. Audiol. 2021, 30, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, D.; Chaudhry, A.; Muzaffar, J.; Monksfield, P.; Bance, M. Cochlear Implantation Outcomes in Post Synaptic Auditory Neuropathies: A Systematic Review and Narrative Synthesis. J. Int. Adv. Otol. 2020, 16, 411–431. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, S.; Grati, M.; Cohen-Salmon, M.; El-Amraoui, A.; Mustapha, M.; Salem, N.; El-Zir, E.; Loiselet, J.; Petit, C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat. Genet. 1999, 21, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, N.A.; Drescher, M.J.; Drescher, D.G. Direct interaction of otoferlin with syntaxin 1A, SNAP-25, and the L-type voltage-gated calcium channel Cav1.3. J. Biol. Chem. 2009, 284, 1364–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarelli, R.; del Castillo, I.; Cama, E.; Scimemi, P.; Starr, A. Audibility, speech perception and processing of temporal cues in ribbon synaptic disorders due to OTOF mutations. Hear. Res. 2015, 330, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liu, X. Cochlear Implantation Outcomes in Patients With OTOF Mutations. Front. Neurosci. 2020, 14, 447. [Google Scholar] [CrossRef]
- Lee, S.Y.; Han, J.H.; Song, H.K.; Kim, N.J.; Yi, N.; Kyong, J.S.; Choi, B.Y. Central auditory maturation and behavioral outcomes after cochlear implantation in prelingual auditory neuropathy spectrum disorder related to OTOF variants (DFNB9): Lessons from pilot study. PLoS ONE 2021, 16, e0252717. [Google Scholar] [CrossRef]
- Wu, C.C.; Hsu, C.J.; Huang, F.L.; Lin, Y.H.; Lin, Y.H.; Liu, T.C.; Wu, C.M. Timing of cochlear implantation in auditory neuropathy patients with OTOF mutations: Our experience with 10 patients. Clin. Otolaryngol. Off. J. ENT-UK Off. J. Neth. Soc. Oto-Rhino-Laryngol. Cervico-Facial Surg. 2018, 43, 352–357. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Liu, T.C.; Wang, S.H.; Hsu, C.J.; Wu, C.M. Genetic characteristics in children with cochlear implants and the corresponding auditory performance. Laryngoscope 2011, 121, 1287–1293. [Google Scholar] [CrossRef]
- Strom, T.M.; Hörtnagel, K.; Hofmann, S.; Gekeler, F.; Scharfe, C.; Rabl, W.; Gerbitz, K.D.; Meitinger, T. Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy and Deafness (DIDMOAD) Caused by Mutations in a Novel Gene (Wolframin) Coding for a Predicted Transmembrane Protein. Hum. Mol. Genet. 1998, 7, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Rendtorff, N.D.; Lodahl, M.; Boulahbel, H.; Johansen, I.R.; Pandya, A.; Welch, K.O.; Norris, V.W.; Arnos, K.S.; Bitner-Glindzicz, M.; Emery, S.B.; et al. Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. Am. J. Med. Genet. A 2011, 155, 1298–1313. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, K.; Nakamura, N.; Ghadami, M.; Matsumoto, N.; Kishino, T.; Ohta, T.; Niikawa, N.; Yoshiura, K. Confirmation of genetic homogeneity of nonsyndromic low-frequency sensorineural hearing loss by linkage analysis and a DFNA6/14 mutation in a Japanese family. J. Hum. Genet. 2002, 47, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Young, T.L.; Ives, E.; Lynch, E.; Person, R.; Snook, S.; MacLaren, L.; Cater, T.; Griffin, A.; Fernandez, B.; Lee, M.K.; et al. Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum. Mol. Genet. 2001, 10, 2509–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasakura-Kimura, N.; Masuda, M.; Mutai, H.; Masuda, S.; Morimoto, N.; Ogahara, N.; Misawa, H.; Sakamoto, H.; Saito, K.; Matsunaga, T. WFS1 and GJB2 mutations in patients with bilateral low-frequency sensorineural hearing loss. Laryngoscope 2017, 127, E324–E329. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.A.; Saito, M.; Makepeace, C.; Permutt, M.A.; Schlesinger, P.; Mueckler, M. Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J. Biol. Chem. 2003, 278, 52755–52762. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Hosoya, M.; Oishi, N.; Okano, H.; Fujioka, M.; Ogawa, K. Expression pattern of wolframin, the WFS1 (Wolfram syndrome-1 gene) product, in common marmoset (Callithrix jacchus) cochlea. Neuroreport 2016, 27, 833–836. [Google Scholar] [CrossRef]
- Nishio, S.Y.; Takumi, Y.; Usami, S.I. Laser-capture micro dissection combined with next-generation sequencing analysis of cell type-specific deafness gene expression in the mouse cochlea. Hear Res. 2017, 348, 87–97. [Google Scholar] [CrossRef]
- Guan, J.; Wang, H.; Lan, L.; Wu, Y.; Chen, G.; Zhao, C.; Wang, D.; Wang, Q. Recurrent de novo WFS1 pathogenic variants in Chinese sporadic patients with nonsyndromic sensorineural hearing loss. Mol. Genet. Genom. Med. 2020, 8, e1367. [Google Scholar] [CrossRef]
- Hogewind, B.F.; Pennings, R.J.; Hol, F.A.; Kunst, H.P.; Hoefsloot, E.H.; Cruysberg, J.R.; Cremers, C.W. Autosomal dominant optic neuropathy and sensorineual hearing loss associated with a novel mutation of WFS1. Mol. Vis. 2010, 16, 26–35. [Google Scholar]
- Payne, M.; Yang, Z.; Katz, B.J.; Warner, J.E.; Weight, C.J.; Zhao, Y.; Pearson, E.D.; Treft, R.L.; Hillman, T.; Kennedy, R.J.; et al. Dominant optic atrophy, sensorineural hearing loss, ptosis, and ophthalmoplegia: A syndrome caused by a missense mutation in OPA1. Am. J. Ophthalmol. 2004, 138, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bette, S.; Zimmermann, U.; Wissinger, B.; Knipper, M. OPA1, the disease gene for optic atrophy type Kjer, is expressed in the inner ear. Histochem. Cell Biol. 2007, 128, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Maeda-Katahira, A.; Nakamura, N.; Hayashi, T.; Katagiri, S.; Shimizu, S.; Ohde, H.; Matsunaga, T.; Kaga, K.; Nakano, T.; Kameya, S.; et al. Autosomal dominant optic atrophy with OPA1 gene mutations accompanied by auditory neuropathy and other systemic complications in a Japanese cohort. Mol. Vis. 2019, 25, 559–573. [Google Scholar] [PubMed]
- Santarelli, R.; Rossi, R.; Scimemi, P.; Cama, E.; Valentino, M.L.; La Morgia, C.; Caporali, L.; Liguori, R.; Magnavita, V.; Monteleone, A. OPA1-related auditory neuropathy: Site of lesion and outcome of cochlear implantation. Brain 2015, 138, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Ham, M.; Han, J.; Osann, K.; Smith, M.; Kimonis, V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion 2019, 46, 262–269. [Google Scholar] [CrossRef]
- Walton, J.; Gibson, W.P.; Sanli, H.; Prelog, K. Predicting cochlear implant outcomes in children with auditory neuropathy. Otol. Neurotol. 2008, 29, 302–309. [Google Scholar] [CrossRef]
- Yousef, M.; Mesallam, T.A.; Garadat, S.N.; Almasaad, A.; Alzhrani, F.; Alsanosi, A.; Hagr, A. Audiologic Outcome of Cochlear Implantation in Children With Cochlear Nerve Deficiency. Otol. Neurotol. 2021, 42, 38–46. [Google Scholar] [CrossRef]
- Wei, X.; Li, Y.; Chen, B.; Gong, Y.; Fu, Q.J.; Liu, T.; Cui, D.; Su, Q.; Shi, Y. Predicting Auditory Outcomes From Radiological Imaging in Cochlear Implant Patients With Cochlear Nerve Deficiency. Otol. Neurotol. 2017, 38, 685–693. [Google Scholar] [CrossRef]
- Peng, K.A.; Kuan, E.C.; Hagan, S.; Wilkinson, E.P.; Miller, M.E. Cochlear Nerve Aplasia and Hypoplasia: Predictors of Cochlear Implant Success. Otolaryngol. Head Neck Surg. 2017, 157, 392–400. [Google Scholar] [CrossRef]
- Han, J.J.; Suh, M.W.; Park, M.K.; Koo, J.W.; Lee, J.H.; Oh, S.H. A Predictive Model for Cochlear Implant Outcome in Children with Cochlear Nerve Deficiency. Sci. Rep. 2019, 9, 1154. [Google Scholar] [CrossRef] [PubMed]
- Archbold, S.; Lutman, M.E.; Nikolopoulos, T. Categories of auditory performance: Inter-user reliability. Br. J. Audiol. 1998, 32, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Nikolopoulos, T.P.; Dyar, D.; O’Donoghue, G.M. Reliability of a rating scale for measuring speech intelligibility after pediatric cochlear implantation. Otol. Neurotol. 2001, 22, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Govaerts, P.J.; De Beukelaer, C.; Daemers, K.; De Ceulaer, G.; Yperman, M.; Somers, T.; Schatteman, I.; Offeciers, F.E. Outcome of cochlear implantation at different ages from 0 to 6 years. Otol. Neurotol. 2002, 23, 885–890. [Google Scholar] [CrossRef]
- Ramos-Macías, Á.; Borkoski-Barreiro, S.; Falcón-González, J.C.; Plasencia, D.P. Results in cochlear implanted children before 5 years of age. a long term follow up. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 2183–2189. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Cases (N) |
|---|---|
| OTOF | |
| OTOF c.[1498C>T];[5098G>C] | 1 |
| OTOF c.[2521G>A];[5098G>C] | 2 |
| OTOF c.[3704_3719del];[5098G>C] | 2 |
| OTOF c.[3864G>A];[5098G>C] | 1 |
| OTOF c.[4030C>T];[5098G>C] | 1 |
| OTOF c.[4961-1G>A];[5098G>C] | 1 |
| OTOF c.[5000C>A];[5098G>C] | 1 |
| OTOF c.[5098G>C];[5098G>C] | 7 |
| OTOF c.[5098G>C];[5203C>T] | 1 |
| OTOF c.[5098G>C];[5566C>T] | 1 |
| Other rare genes | |
| WFS1 c.[2051C>T];[2051=] | 1 |
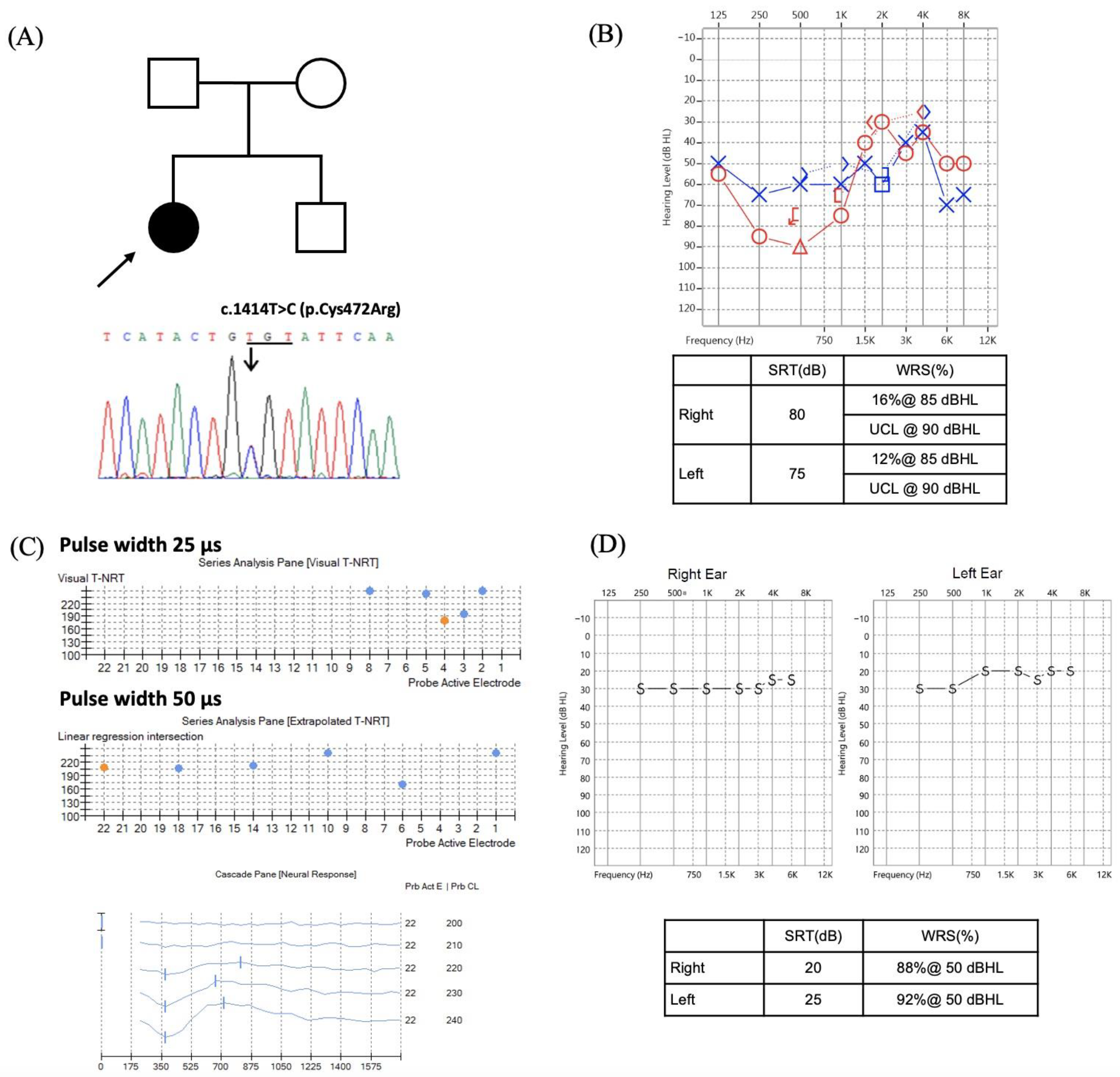
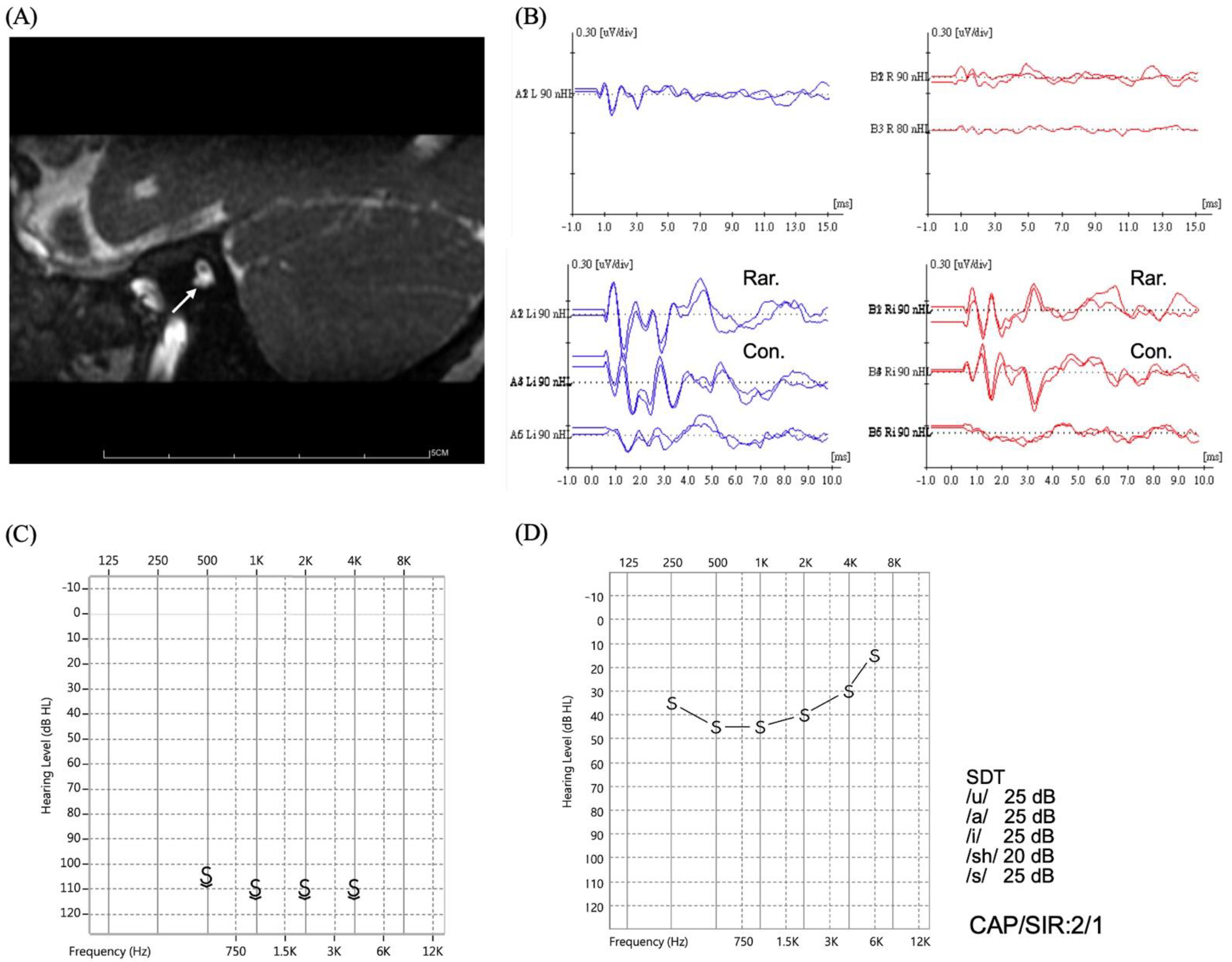
| OPA1 c.[1414T>C];[1414=] | 1 |
| OTOF Variants N = 18 | Rare Gene Variants a N = 2 | CND N = 10 | Indefinite N = 6 | Total N = 36 | p Value | |
|---|---|---|---|---|---|---|
| Sex (M:F) | 13:5 | 0:2 | 5:5 | 6:0 | 24:12 | 0.034 b |
| Age at implantation of the first ear (y), mean ± SD [range] | 2.8 ± 1.3 [1–5.5] | 9.0 ± 9.1 [2.6–15.4] | 3.1 ± 1.7 [1.3–6.2] | 6.6 ± 10.4 [1.7–27.7] | 3.9 ± 4.9 [1–27.7] | 0.734 c |
| Preoperative hearing thresholds (dBHL), mean ± SD [range] | 88.2 ± 14.5 [57.5–106.3] | 66.3 ± 20.6 [53.8–90] | 100.9 ± 12.1 [80–120] | 97.3 ± 8.7 [90–110] | 91.2 ± 15.9 [53.8–120] | 0.022 c |
| Bilateral implantation (N) | 9 | 1 | 2 | 2 | 14 | 0.457 b |
| Follow up (y), mean ± SD [range] | 4.3 ± 2.4 [0.8–7.6] | 3.2 ± 0.9 [2.6–3.8] | 3.5 ± 4 [1–13.8] | 4.3 ± 4.3 [0.6–10.1] | 4.0 ± 3.1 [0.6–13.8] | 0.707 c |
| OTOF Variants N = 18 | Rare Gene Variants a N = 2 | CND N = 10 | Indefinite N = 6 | Total N = 36 | p Value | |
|---|---|---|---|---|---|---|
| CI-aided hearing thresholds (dBHL), mean ± SD [range] | 25.6 ± 4.1 [20–35] | 20.8 ± 1.9 [18.8–22.5] | 38.9 ± 9.3 [25–55] | 28.3 ± 0.7 [27.5–28.8] | 28.3 ± 7.8 [18.8–55] | <0.001 b |
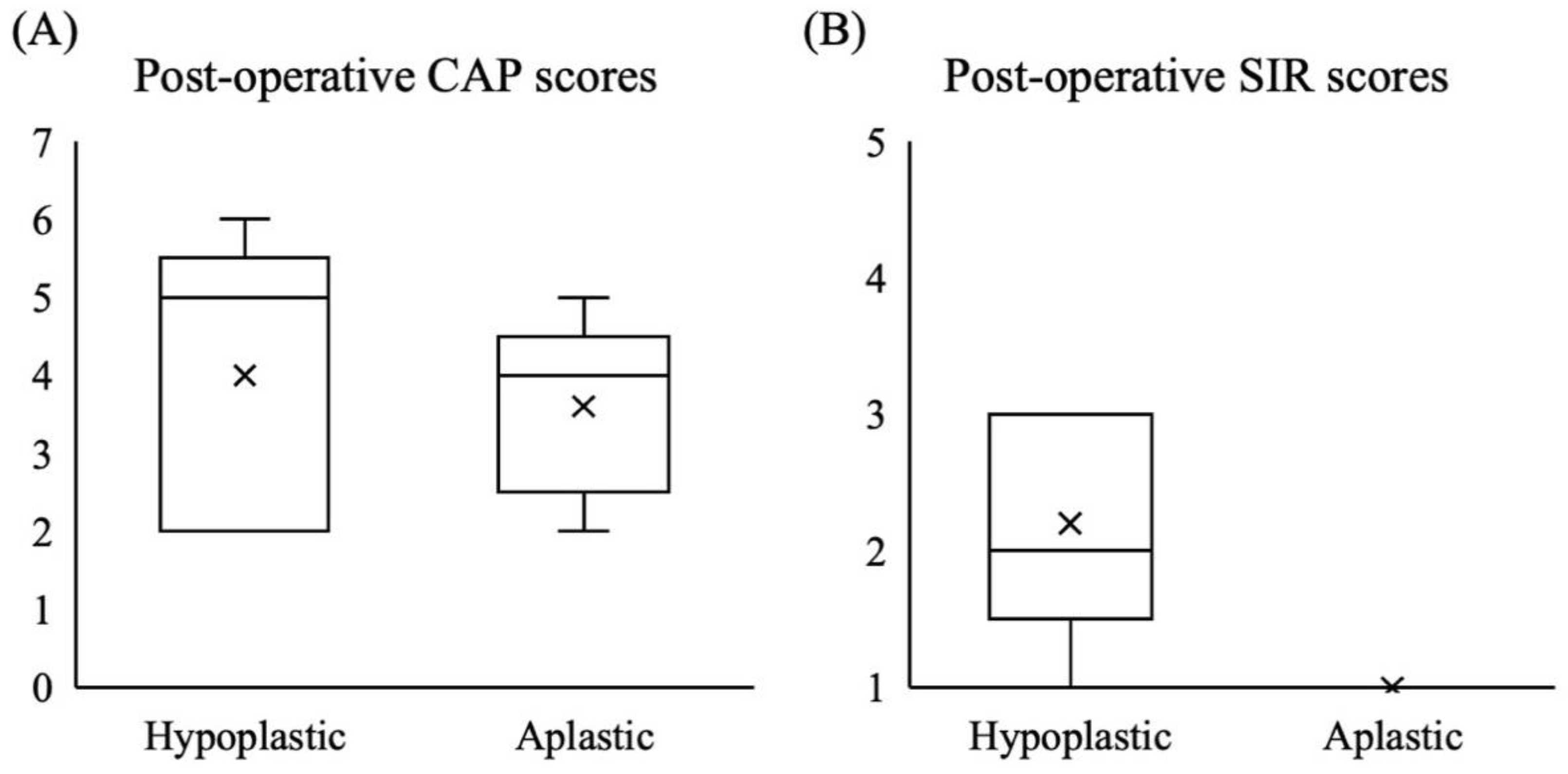
| CAP score, median [range] | 7 [5–7] | 7 [6,7] | 4 [2–6] | 5 [2–7] | 6 [2–7] | 0.021 b |
| SIR score, median [range] | 5 [2–5] | 5 [5] | 1 [1–3] | 4 [1–5] | 4 [1–5] | 0.006 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, P.-H.; Wu, H.-P.; Wu, C.-M.; Chiang, Y.-T.; Hsu, J.S.; Tsai, C.-Y.; Wang, H.; Tseng, L.-H.; Chen, P.-Y.; Yang, T.-H.; et al. Cochlear Implantation Outcomes in Patients with Auditory Neuropathy Spectrum Disorder of Genetic and Non-Genetic Etiologies: A Multicenter Study. Biomedicines 2022, 10, 1523. https://doi.org/10.3390/biomedicines10071523
Lin P-H, Wu H-P, Wu C-M, Chiang Y-T, Hsu JS, Tsai C-Y, Wang H, Tseng L-H, Chen P-Y, Yang T-H, et al. Cochlear Implantation Outcomes in Patients with Auditory Neuropathy Spectrum Disorder of Genetic and Non-Genetic Etiologies: A Multicenter Study. Biomedicines. 2022; 10(7):1523. https://doi.org/10.3390/biomedicines10071523
Chicago/Turabian StyleLin, Pei-Hsuan, Hung-Pin Wu, Che-Ming Wu, Yu-Ting Chiang, Jacob Shujui Hsu, Cheng-Yu Tsai, Han Wang, Li-Hui Tseng, Pey-Yu Chen, Ting-Hua Yang, and et al. 2022. "Cochlear Implantation Outcomes in Patients with Auditory Neuropathy Spectrum Disorder of Genetic and Non-Genetic Etiologies: A Multicenter Study" Biomedicines 10, no. 7: 1523. https://doi.org/10.3390/biomedicines10071523