
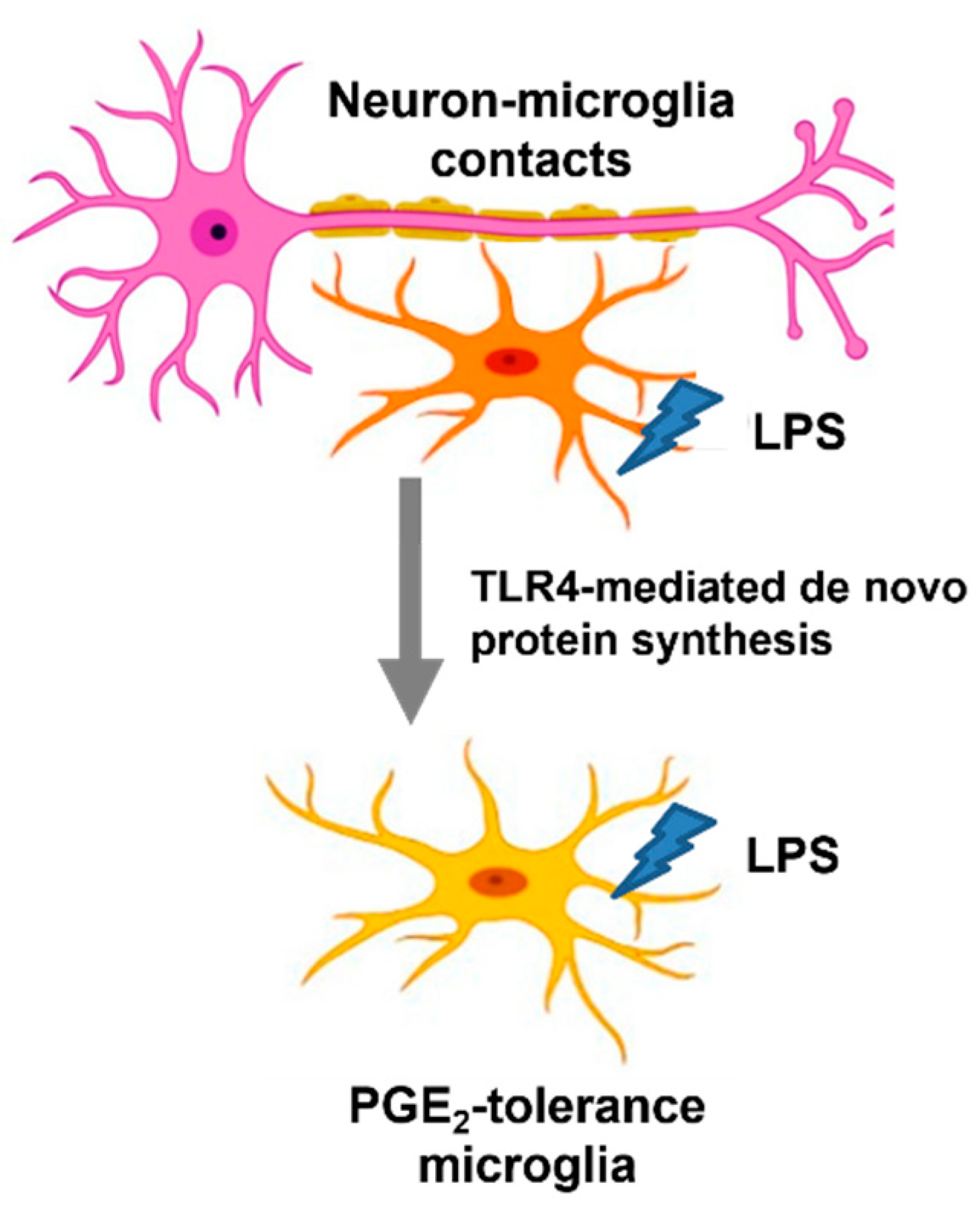
Neuron–Microglia Contacts Govern the PGE2 Tolerance through TLR4-Mediated de Novo Protein Synthesis

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents
2.3. Preparation of Primary Neuron–Glia, Mixed Glia, and Microglia- and Astrocyte-Enriched Cultures
2.4. Cell Treatment
2.5. Quantitative Real Time-PCR
2.6. Measurement of PGE2
2.7. Statistical Analysis
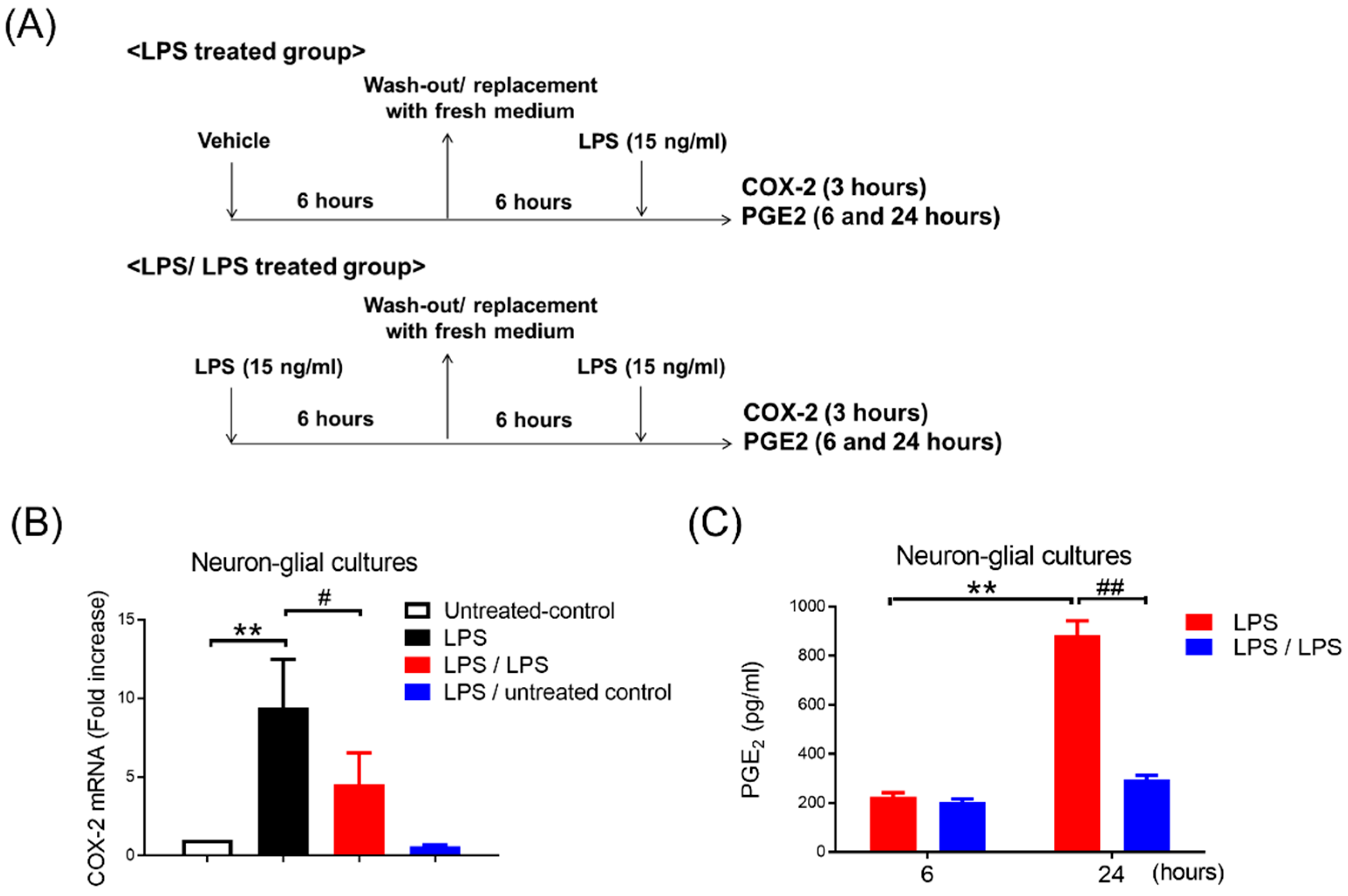
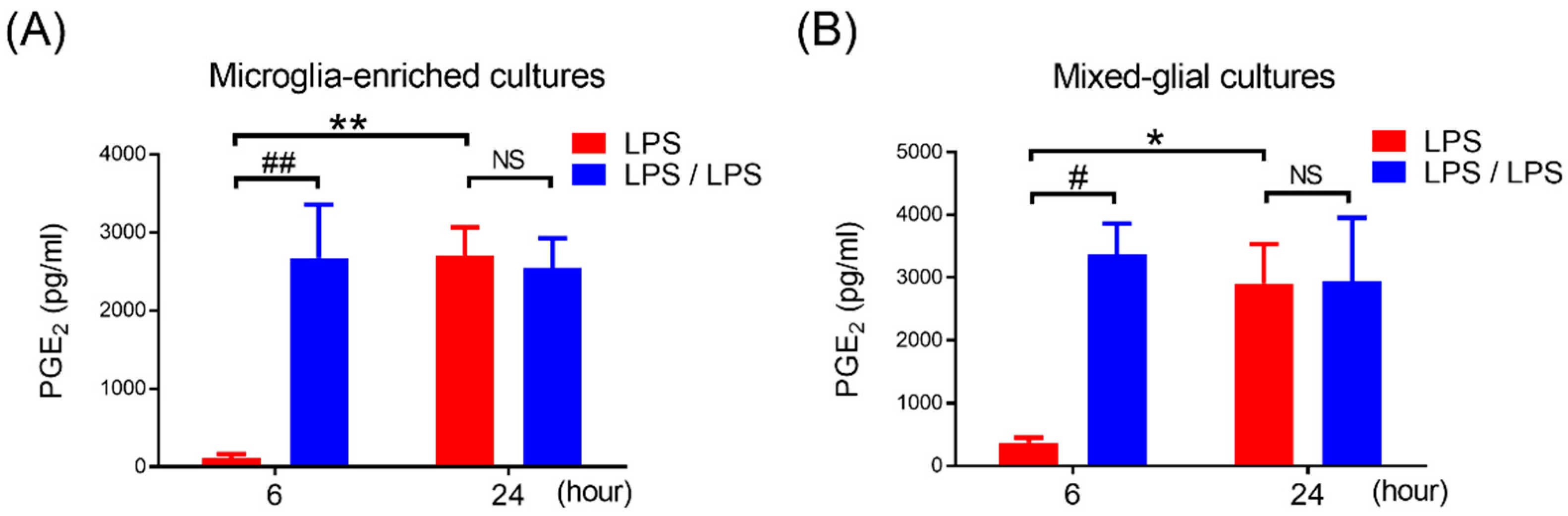
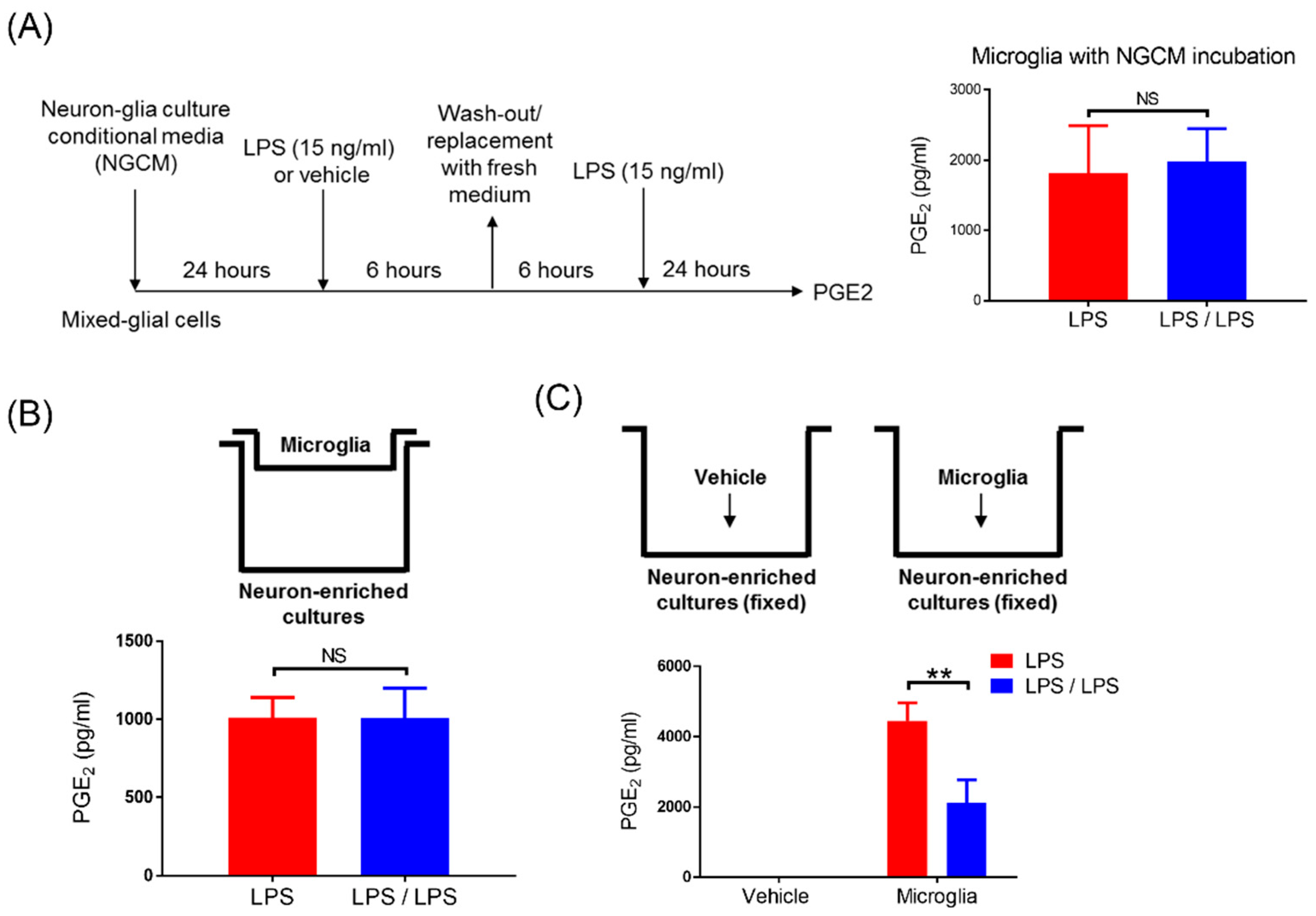
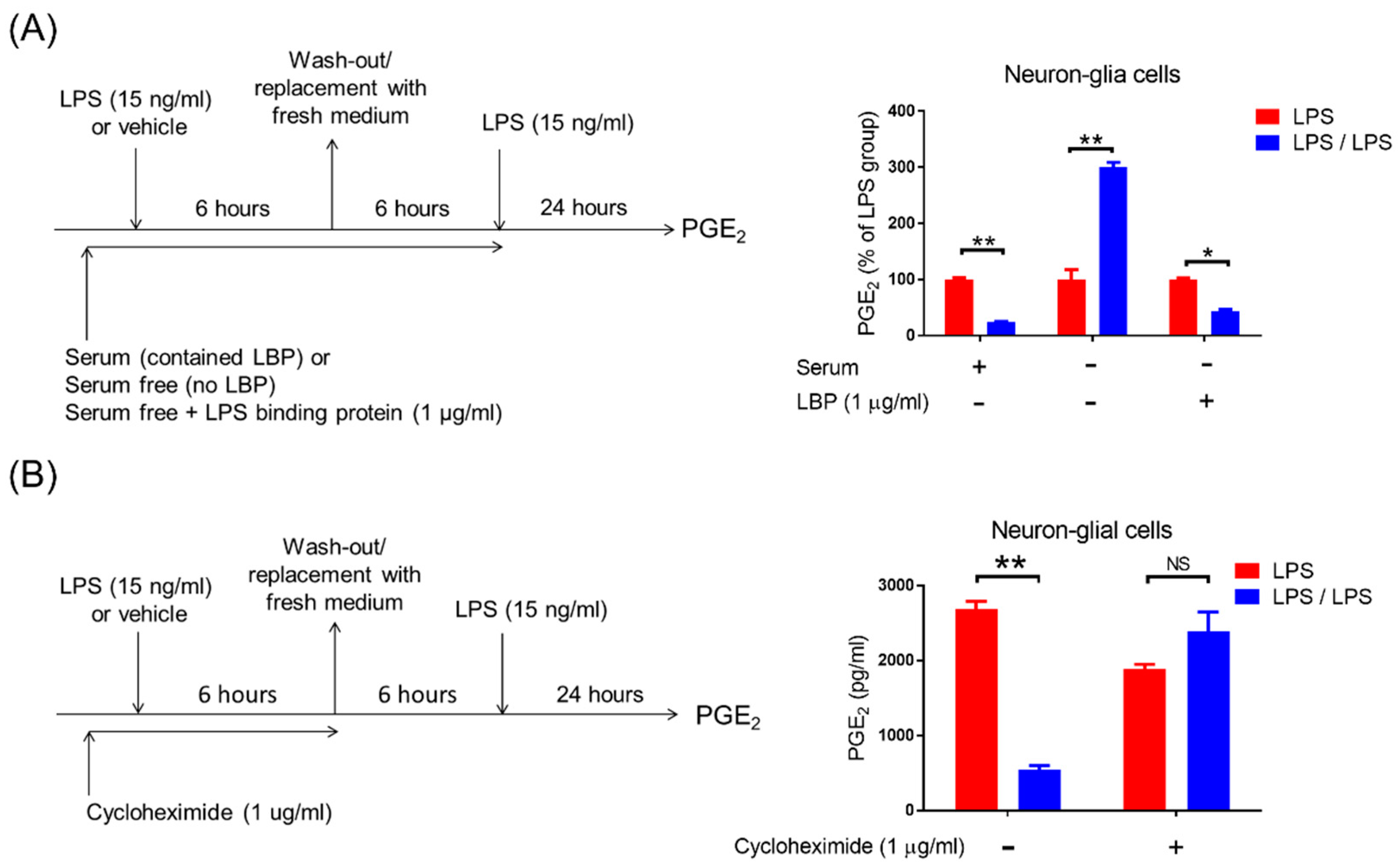
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Chen, S.-D. The changing phenotype of microglia from homeostasis to disease. Transl. Neurodegener. 2012, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Bohlen, C.J.; Friedman, B.A.; Dejanovic, B.; Sheng, M. Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu. Rev. Genet. 2019, 53, 263–288. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharm. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- McDonough, A.; Weinstein, J.R. The role of microglia in ischemic preconditioning. Glia 2020, 68, 455–471. [Google Scholar] [CrossRef]
- Wendeln, A.-C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Neher, J.J.; Cunningham, C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019, 40, 358–374. [Google Scholar] [CrossRef]
- Chu, C.H.; Chen, S.H.; Wang, Q.; Langenbach, R.; Li, H.; Zeldin, D.; Chen, S.L.; Wang, S.; Gao, H.; Lu, R.B.; et al. PGE2 Inhibits IL-10 Production via EP2-Mediated beta-Arrestin Signaling in Neuroinflammatory Condition. Mol. Neurobiol. 2015, 52, 587–600. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.H.; Wang, S.; Li, C.L.; Chen, S.H.; Hu, C.F.; Chung, Y.L.; Chen, S.L.; Wang, Q.; Lu, R.B.; Gao, H.M.; et al. Neurons and astroglia govern microglial endotoxin tolerance through macrophage colony-stimulating factor receptor-mediated ERK1/2 signals. Brain Behav. Immun. 2016, 55, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Saccaro, L.F.; Schilliger, Z.; Perroud, N.; Piguet, C. Inflammation, Anxiety, and Stress in Attention-Deficit/Hyperactivity Disorder. Biomedicines 2021, 9, 1313. [Google Scholar] [CrossRef] [PubMed]
- Tsay, H.-J.; Liu, H.-K.; Kuo, Y.-H.; Chiu, C.-S.; Liang, C.-C.; Chung, C.-W.; Chen, C.-C.; Chen, Y.-P.; Shiao, Y.-J. EK100 and Antrodin C Improve Brain Amyloid Pathology in APP/PS1 Transgenic Mice by Promoting Microglial and Perivascular Clearance Pathways. Int. J. Mol. Sci. 2021, 22, 10413. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, J.; Schlotterer, A.; Kurowski, L.; Hoffmann, S.; Hammad, S.; Dooley, S.; Buchholz, M.; Hu, J.; Fleming, I.; et al. MicroRNA-124 Alleviates Retinal Vasoregression via Regulating Microglial Polarization. Int. J. Mol. Sci. 2021, 22, 11068. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, K. Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins Other Lipid Mediat. 2010, 91, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagami, T.; Koma, H.; Yamamoto, Y. Pathophysiological Roles of Cyclooxygenases and Prostaglandins in the Central Nervous System. Mol. Neurobiol. 2016, 53, 4754–4771. [Google Scholar] [CrossRef]
- Fathali, N.; Ostrowski, R.P.; Lekic, T.; Jadhav, V.; Tong, W.; Tang, J.; Zhang, J.H. Cyclooxygenase-2 inhibition provides lasting protection against neonatal hypoxic-ischemic brain injury. Crit. Care Med. 2010, 38, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Teismann, P. COX-2 in the neurodegenerative process of Parkinson’s disease. Biofactors 2012, 38, 395–397. [Google Scholar] [CrossRef] [Green Version]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in Inflammatory and Degenerative Brain Diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wu, S.; Hickey, R.W.; Rose, M.E.; Chen, J.; Graham, S.H. Neuronal cyclooxygenase-2 activity and prostaglandins PGE2, PGD2, and PGF2 alpha exacerbate hypoxic neuronal injury in neuron-enriched primary culture. Neurochem. Res. 2008, 33, 490–499. [Google Scholar] [CrossRef]
- Hickey, R.W.; Adelson, P.D.; Johnnides, M.J.; Davis, D.S.; Yu, Z.; Rose, M.E.; Chang, Y.F.; Graham, S.H. Cyclooxygenase-2 activity following traumatic brain injury in the developing rat. Pediatr. Res. 2007, 62, 271–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, A.; Chen, D.; Ganesh, T.; Varvel, N.H.; Dingledine, R. The COX-2/prostanoid signaling cascades in seizure disorders. Expert Opin. Ther. Targets 2019, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Hu, Q.; Wang, H.; Yang, H.; Gao, F.; Ren, H.; Chen, D.; Fu, C.; Zheng, L.; Zhen, X.; et al. Induction of COX-2-PGE2 synthesis by activation of the MAPK/ERK pathway contributes to neuronal death triggered by TDP-43-depleted microglia. Cell Death Dis. 2015, 6, e1702. [Google Scholar] [CrossRef] [Green Version]
- Hoozemans, J.J.; Veerhuis, R.; Janssen, I.; van Elk, E.J.; Rozemuller, A.J.; Eikelenboom, P. The role of cyclo-oxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: Implications for Alzheimer’s disease. Brain Res. 2002, 951, 218–226. [Google Scholar] [CrossRef]
- Johansson, J.U.; Woodling, N.S.; Shi, J.; Andreasson, K.I. Inflammatory Cyclooxygenase Activity and PGE2 Signaling in Models of Alzheimer’s Disease. Curr. Immunol. Rev. 2015, 11, 125–131. [Google Scholar] [CrossRef]
- Bartels, A.L.; Leenders, K.L. Cyclooxygenase and neuroinflammation in Parkinson’s disease neurodegeneration. Curr. Neuropharmacol. 2010, 8, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Wang, Q.; Shi, J.; Lokteva, L.; Breyer, R.M.; Montine, T.J.; Andreasson, K. The prostaglandin E2EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Du, L.; Hong, J.S. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J. Pharmacol. Exp. Ther. 2000, 293, 607–617. [Google Scholar]
- Qin, L.; Li, G.; Qian, X.; Liu, Y.; Wu, X.; Liu, B.; Hong, J.-S.; Block, M.L. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia 2005, 52, 78–84. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Nahid, A.; Satoh, M.; Chan, E.K. MicroRNA in TLR signaling and endotoxin tolerance. Cell. Mol. Immunol. 2011, 8, 388–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Kim, H.M. Dynamic lipopolysaccharide transfer cascade to TLR4/MD2 complex via LBP and CD14. BMB Rep. 2017, 50, 55–57. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.K.; Kim, S.J.; Rah, S.H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.O.; Park, B.S.; Yoon, T.Y.; et al. Reconstruction of LPS Transfer Cascade Reveals Structural Determinants within LBP, CD14, and TLR4-MD2 for Efficient LPS Recognition and Transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.E.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Crowley, G.; Hong, S. Understanding microglial diversity and implications for neuronal function in health and disease. Dev. Neurobiol. 2021, 81, 507–523. [Google Scholar] [CrossRef]
- Deczkowska, A.; Amit, I.; Schwartz, M. Microglial immune checkpoint mechanisms. Nat. Neurosci. 2018, 21, 779–786. [Google Scholar] [CrossRef]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia–Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Posfai, B.; Cserep, C.; Orsolits, B.; Denes, A. New Insights into Microglia-Neuron Interactions: A Neuron’s Perspective. Neuroscience 2019, 405, 103–117. [Google Scholar] [CrossRef] [Green Version]
- Gomes, F.; Spohr, T.; Martinez, R.; Neto, V.M. Cross-talk between neurons and glia: Highlights on soluble factors. Braz. J. Med Biol. Res. 2001, 34, 611–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Leak, R.K.; Hu, X. Neurotransmitter receptors on microglia. Stroke Vasc. Neurol. 2016, 1, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Veremeyko, T.; Yung, A.W.; Dukhinova, M.; Strekalova, T.; Ponomarev, E.D. The Role of Neuronal Factors in the Epigenetic Reprogramming of Microglia in the Normal and Diseased Central Nervous System. Front. Cell. Neurosci. 2019, 13, 453. [Google Scholar] [CrossRef] [Green Version]
- Mead, E.L.; Mosley, A.; Eaton, S.; Dobson, L.; Heales, S.J.; Pocock, J.M. Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. J. Neurochem. 2012, 121, 287–301. [Google Scholar] [CrossRef]
- Lyons, A.; Downer, E.; Crotty, S.; Nolan, Y.; Mills, K.; Lynch, M.A. CD200 Ligand Receptor Interaction Modulates Microglial Activation In Vivo and In Vitro: A Role for IL-4. J. Neurosci. 2007, 27, 8309–8313. [Google Scholar] [CrossRef] [PubMed]
- Lehrman, E.K.; Wilton, D.K.; Litvina, E.Y.; Welsh, C.A.; Chang, S.T.; Frouin, A.; Walker, A.J.; Heller, M.D.; Umemori, H.; Chen, C.; et al. CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 2018, 100, 120–134.e6. [Google Scholar] [CrossRef] [Green Version]
- Mott, R.T.; Ait-Ghezala, G.; Town, T.; Mori, T.; Vendrame, M.; Zeng, J.; Ehrhart, J.; Mullan, M.; Tan, J. Neuronal expression of CD22: Novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia 2004, 46, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Pluvinage, J.V.; Haney, M.S.; Smith, B.A.H.; Sun, J.; Iram, T.; Bonanno, L.; Li, L.; Lee, D.P.; Morgens, D.W.; Yang, A.C.; et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 2019, 568, 187–192. [Google Scholar] [CrossRef] [PubMed]
- von Saucken, V.E.; Jay, T.R.; Landreth, G.E. The effect of amyloid on microglia-neuron interactions before plaque onset occurs independently of TREM2 in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2020, 145, 105072. [Google Scholar] [CrossRef]
- Bohannon, J.K.; Hernandez, A.; Enkhbaatar, P.; Adams, W.L.; Sherwood, E.R. The immunobiology of toll-like receptor 4 agonists: From endotoxin tolerance to immunoadjuvants. Shock 2013, 40, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, A.; Bohannon, J.K.; Luan, L.; Fensterheim, B.A.; Guo, Y.; Patil, N.K.; McAdams, C.; Wang, J.; Sherwood, E.R. The role of MyD88- and TRIF-dependent signaling in monophosphoryl lipid A-induced expansion and recruitment of innate immunocytes. J. Leukoc. Biol. 2016, 100, 1311–1322. [Google Scholar] [CrossRef]
- Nahid, M.A.; Pauley, K.M.; Satoh, M.; Chan, E.K. miR-146a is critical for endotoxin-induced tolerance: Implication in Innate Immunity. J. Biol. Chem. 2009, 284, 34590–34599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajmone-Cat, M.A.; Mancini, M.; De Simone, R.; Cilli, P.; Minghetti, L. Microglial polarization and plasticity: Evidence from organotypic hippocampal slice cultures. Glia 2013, 61, 1698–1711. [Google Scholar] [CrossRef] [PubMed]
- Ajmone-Cat, M.A.; Nicolini, A.; Minghetti, L. Prolonged exposure of microglia to lipopolysaccharide modifies the intracellular signaling pathways and selectively promotes prostaglandin E2 synthesis. J. Neurochem. 2003, 87, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Jope, R.S. Glycogen synthase kinase-3 regulates inflammatory tolerance in astrocytes. Neuroscience 2010, 169, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Schaafsma, W.; Zhang, X.; van Zomeren, K.C.; Jacobs, S.; Georgieva, P.B.; Wolf, S.A.; Kettenmann, H.; Janova, H.; Saiepour, N.; Hanisch, U.K.; et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav. Immun. 2015, 48, 205–221. [Google Scholar] [CrossRef]
- Chistyakov, D.V.; Astakhova, A.A.; Azbukina, N.V.; Goriainov, S.V.; Chistyakov, V.V.; Sergeeva, M.G. Cellular Model of Endotoxin Tolerance in Astrocytes: Role of Interleukin 10 and Oxylipins. Cells 2019, 8, 1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, H.-C.; Lee, K.-F.; Chen, S.-L.; Chiu, S.-C.; Lee, L.-Y.; Chen, W.-P.; Chen, C.-C.; Chu, C.-H. Neuron–Microglia Contacts Govern the PGE2 Tolerance through TLR4-Mediated de Novo Protein Synthesis. Biomedicines 2022, 10, 419. https://doi.org/10.3390/biomedicines10020419
Kuo H-C, Lee K-F, Chen S-L, Chiu S-C, Lee L-Y, Chen W-P, Chen C-C, Chu C-H. Neuron–Microglia Contacts Govern the PGE2 Tolerance through TLR4-Mediated de Novo Protein Synthesis. Biomedicines. 2022; 10(2):419. https://doi.org/10.3390/biomedicines10020419
Chicago/Turabian StyleKuo, Hsing-Chun, Kam-Fai Lee, Shiou-Lan Chen, Shu-Chen Chiu, Li-Ya Lee, Wan-Ping Chen, Chin-Chu Chen, and Chun-Hsien Chu. 2022. "Neuron–Microglia Contacts Govern the PGE2 Tolerance through TLR4-Mediated de Novo Protein Synthesis" Biomedicines 10, no. 2: 419. https://doi.org/10.3390/biomedicines10020419