Recruitment of DNA Repair MRN Complex by Intrinsically Disordered Protein Domain Fused to Cas9 Improves Efficiency of CRISPR-Mediated Genome Editing


Abstract
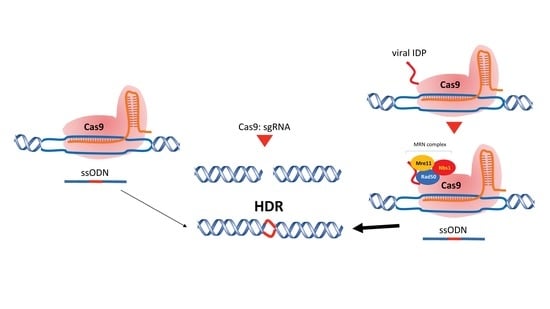
:
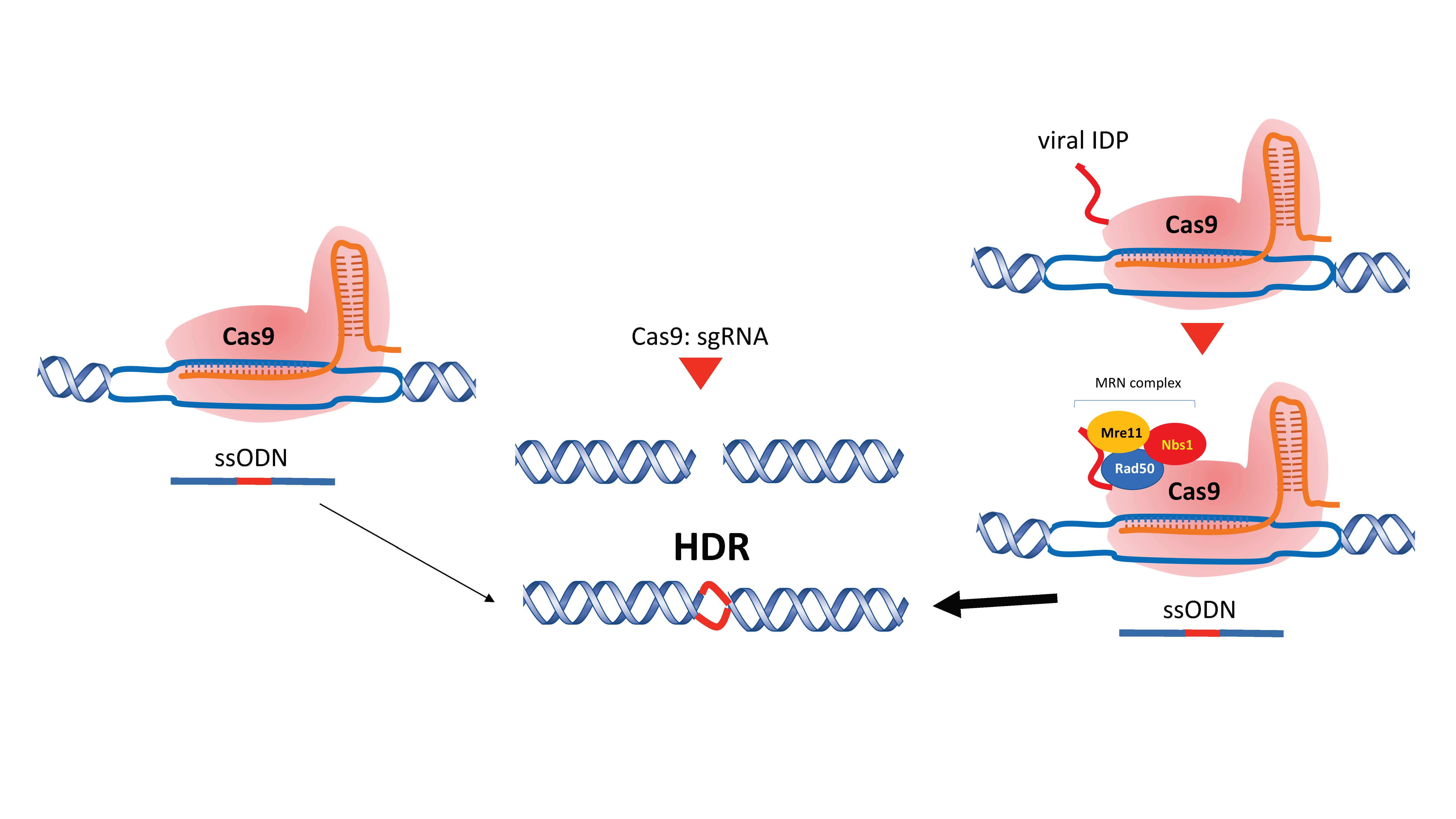{kind=link}
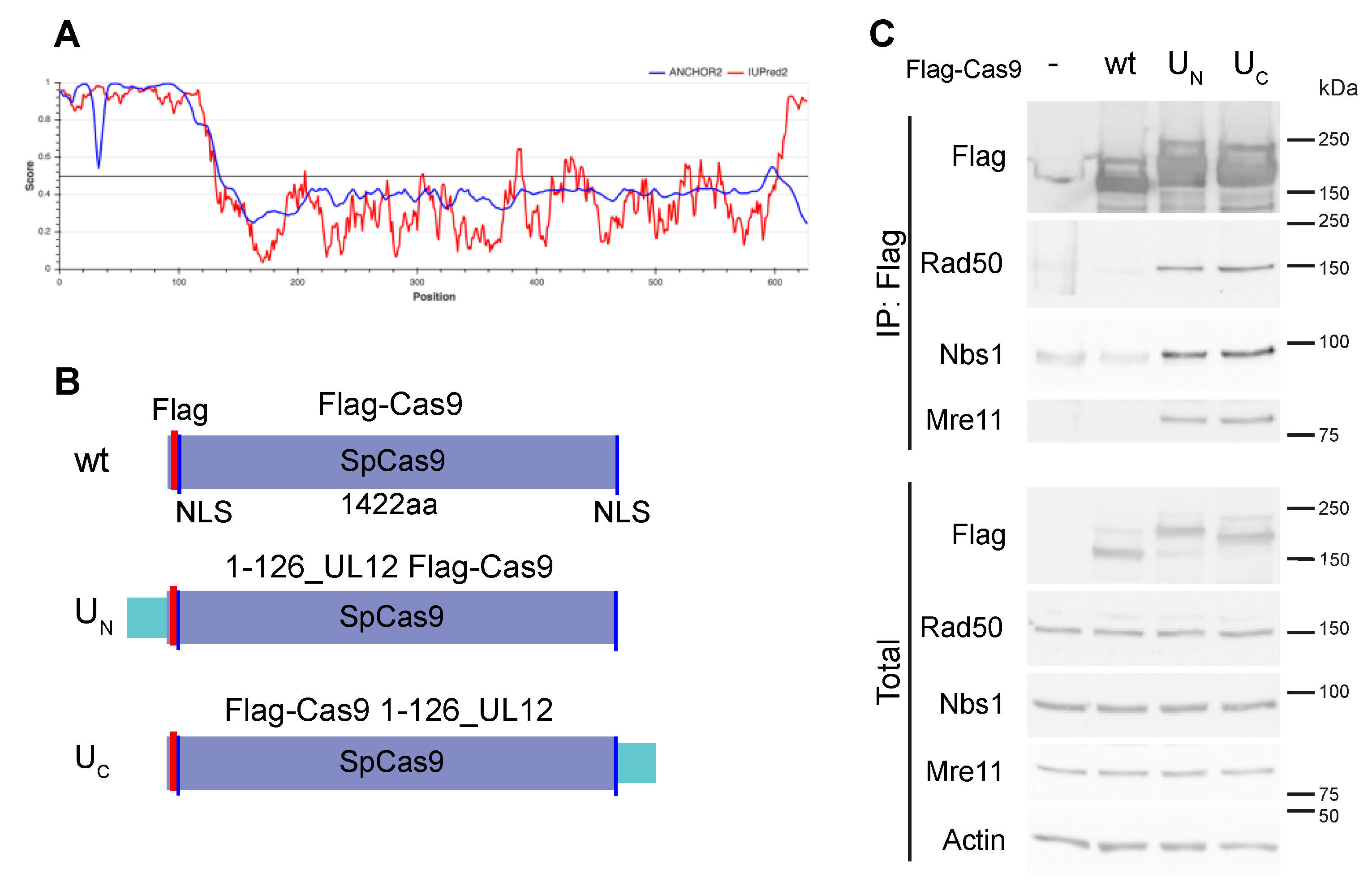{kind=link}
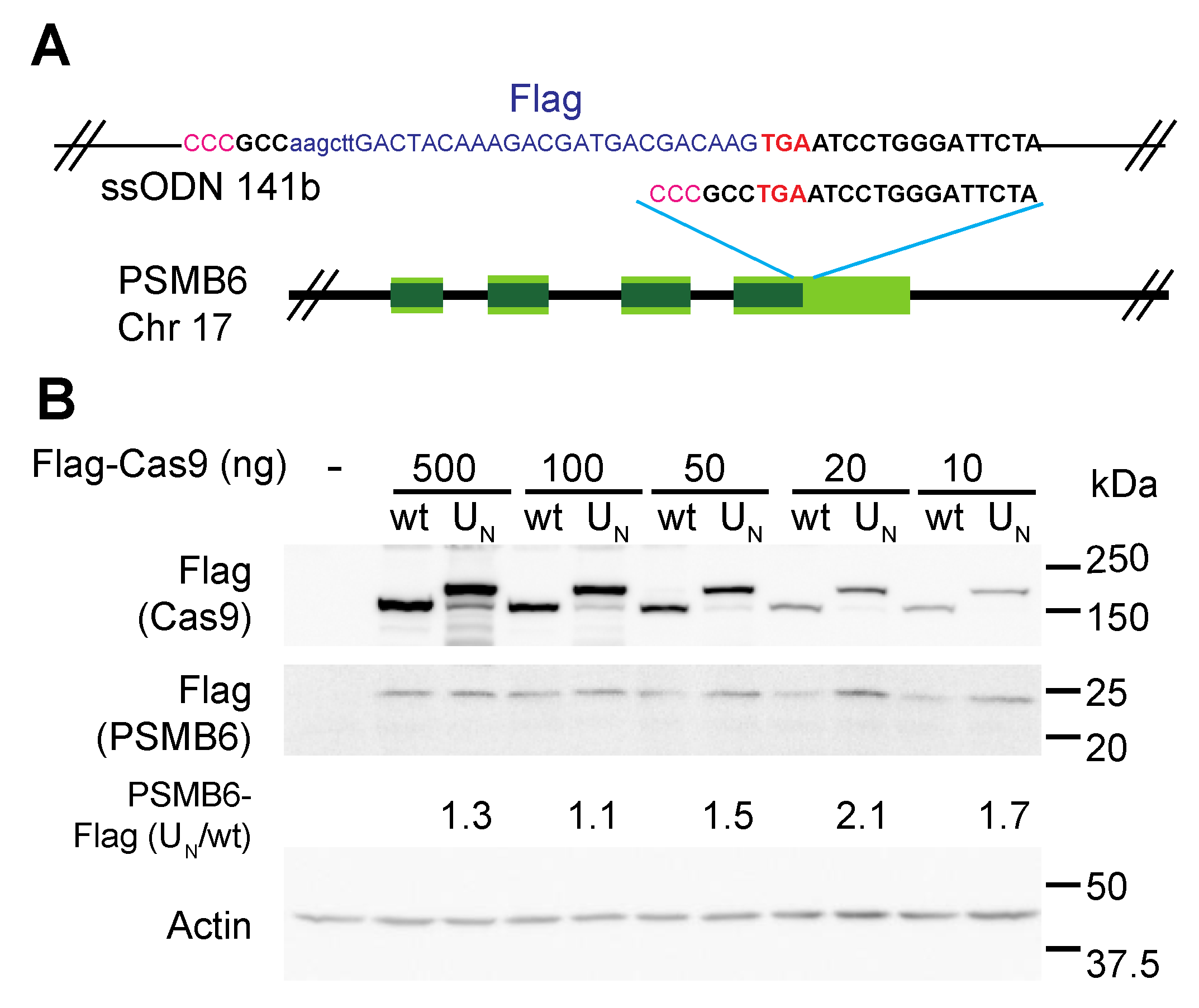{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Plasmids and Transfection
2.3. Immunoblot and Coimmunoprecipitation
2.4. Statistical Analysis
2.5. Flow Cytometry
2.6. Cell Staining
2.7. Recombinant Cas9 Production and Purification
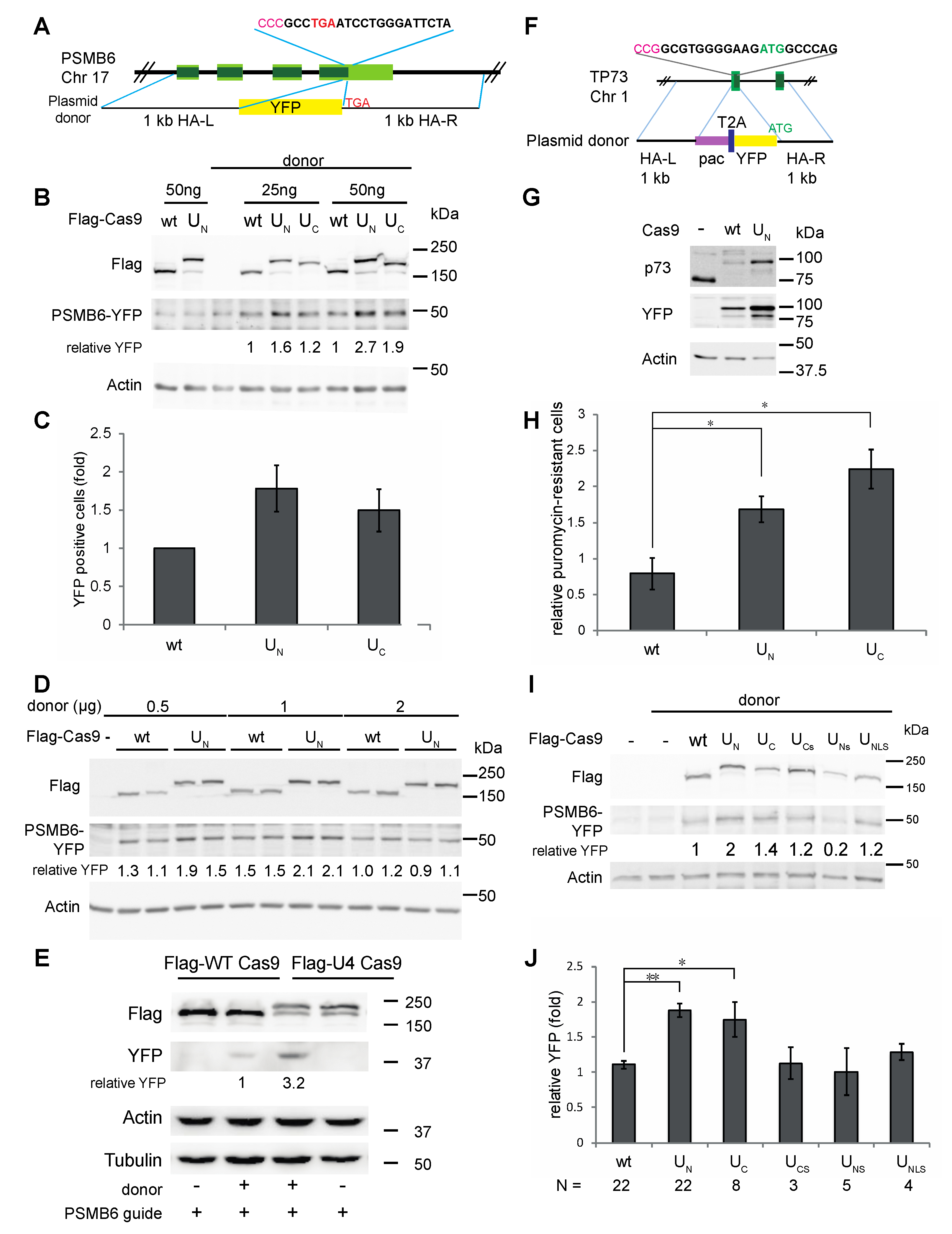
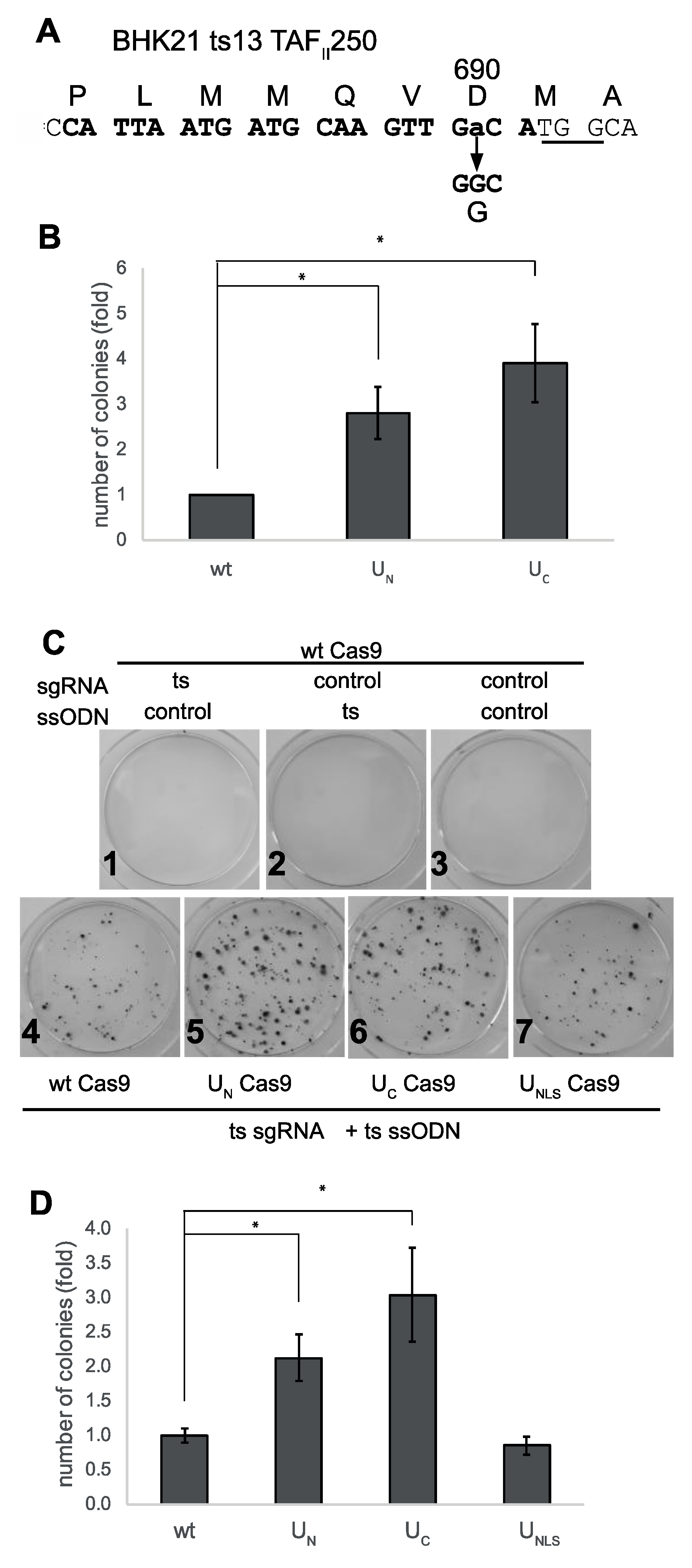
3. Results
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Doudna, J.A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016, 34, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Song, J.; Yang, D.; Xu, J.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. [Google Scholar] [CrossRef]
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.F.; Chin, L. Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep. 2016, 14, 1555–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nihongaki, Y.; Kawano, F.; Nakajima, T.; Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015, 33, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Lomova, A.; Clark, D.N.; Campo-Fernandez, B.; Flores-Bjurstrom, C.; Kaufman, M.L.; Fitz-Gibbon, S.; Wang, X.; Miyahira, E.Y.; Brown, D.; DeWitt, M.A.; et al. Improving Gene Editing Outcomes in Human Hematopoietic Stem and Progenitor Cells by Temporal Control of DNA Repair. Stem Cells 2019, 37, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, M.; Khedher, A.H.Y.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S.; et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.T.; Bashir, S.; Li, X.; Rossius, J.; Chu, V.T.; Rajewsky, K.; Kuhn, R. Enhancement of Precise Gene Editing by the Association of Cas9 With Homologous Recombination Factors. Front. Genet. 2019, 10, 365. [Google Scholar] [CrossRef] [Green Version]
- Reuven, N.B.; Staire, A.E.; Myers, R.S.; Weller, S.K. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol. 2003, 77, 7425–7433. [Google Scholar] [CrossRef]
- Reuven, N.B.; Willcox, S.; Griffith, J.D.; Weller, S.K. Catalysis of strand exchange by the HSV-1 UL12 and ICP8 proteins: Potent ICP8 recombinase activity is revealed upon resection of dsDNA substrate by nuclease. J. Mol. Biol. 2004, 342, 57–71. [Google Scholar] [CrossRef]
- Schumacher, A.J.; Mohni, K.N.; Kan, Y.; Hendrickson, E.A.; Stark, J.M.; Weller, S.K. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog. 2012, 8, e1002862. [Google Scholar] [CrossRef]
- Balasubramanian, N.; Bai, P.; Buchek, G.; Korza, G.; Weller, S.K. Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J. Virol. 2010, 84, 12504–12514. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018. [Google Scholar] [CrossRef]
- Talavera, A.; Basilico, C. Temperature sensitive mutants of BHK cells affected in cell cycle progression. J. Cell. Physiol. 1977, 92, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.B.; Antoku, S.; Weller, S.K. The UL12.5 gene product of herpes simplex virus type 1 exhibits nuclease and strand exchange activities but does not localize to the nucleus. J. Virol. 2004, 78, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Kremers, G.J.; Goedhart, J.; van Munster, E.B.; Gadella, T.W., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry 2006, 45, 6570–6580. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, S.; Jitrapakdee, S.; Wallace, J.C. Development of a bicistronic vector driven by the human polypeptide chain elongation factor 1alpha promoter for creation of stable mammalian cell lines that express very high levels of recombinant proteins. Biochem. Biophys. Res. Commun. 1998, 252, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Guzman, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Akhmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Jacobovitch, Y.; Dantes, A.; Bernheim, R.; Peleg, Y. Applications of the Restriction Free (RF) cloning procedure for molecular manipulations and protein expression. J. Struct. Biol. 2010, 172, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Hemsley, A.; Arnheim, N.; Toney, M.D.; Cortopassi, G.; Galas, D.J. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 1989, 17, 6545–6551. [Google Scholar] [CrossRef] [Green Version]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Turoverov, K.K.; Kuznetsova, I.M.; Fonin, A.V.; Darling, A.L.; Zaslavsky, B.Y.; Uversky, V.N. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem. Sci. 2019, 44, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, J.C.; Weller, S.K.; Weber, P.C. The product of the UL12.5 gene of herpes simplex virus type 1 is a capsid-associated nuclease. J. Virol. 1997, 71, 3039–3047. [Google Scholar] [PubMed]
- Henderson, J.O.; Ball-Goodrich, L.J.; Parris, D.S. Structure-function analysis of the herpes simplex virus type 1 UL12 gene: Correlation of deoxyribonuclease activity in vitro with replication function. Virology 1998, 243, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.N.; Weller, S.K. The exonuclease activity of HSV-1 UL12 is required for in vivo function. Virology 1998, 244, 442–457. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Adler, J.; Myers, N.; Biran, A.; Reuven, N.; Shaul, Y. Oncogenic addiction to high 26S proteasome level. Cell Death Dis. 2018, 9, 773. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.A.; Saffran, H.A.; Duguay, B.A.; Smiley, J.R. Herpes simplex virus UL12.5 targets mitochondria through a mitochondrial localization sequence proximal to the N terminus. J. Virol. 2009, 83, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Sekiguchi, T.; Noguchi, E.; Sunamoto, H.; Ohba, T.; Nishimoto, T. The CCG1/TAFII250 gene is mutated in thermosensitive G1 mutants of the BHK21 cell line derived from golden hamster. Gene 1994, 141, 267–270. [Google Scholar]
- Grunewald, J.; Zhou, R.; Garcia, S.P.; Iyer, S.; Lareau, C.A.; Aryee, M.J.; Joung, J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 2019. [Google Scholar] [CrossRef]
- Myers, N.; Olender, T.; Savidor, A.; Levin, Y.; Reuven, N.; Shaul, Y. The Disordered Landscape of the 20S Proteasome Substrates Reveals Tight Association with Phase Separated Granules. Proteomics 2018, 18, e1800076. [Google Scholar] [CrossRef] [PubMed]
- Cumberworth, A.; Lamour, G.; Babu, M.M.; Gsponer, J. Promiscuity as a functional trait: Intrinsically disordered regions as central players of interactomes. Biochem. J. 2013, 454, 361–369. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Blocquel, D.; Habchi, J.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. Structural disorder in viral proteins. Chem. Rev. 2014, 114, 6880–6911. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.I.; Kim, E.T.; Meyerson, N.R.; Pancholi, N.J.; Mohni, K.N.; Enard, D.; Petrov, D.A.; Weller, S.K.; Weitzman, M.D.; Sawyer, S.L. An Intrinsically Disordered Region of the DNA Repair Protein Nbs1 Is a Species-Specific Barrier to Herpes Simplex Virus 1 in Primates. Cell Host Microbe 2016, 20, 178–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.; Weller, S.K. HSV-I and the cellular DNA damage response. Futur. Virol. 2015, 10, 383–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.C.; Hsu, M.N.; Chang, C.W.; Lin, M.W.; Hwu, J.R.; Tu, Y.; Hu, Y.C. Synthetic switch to minimize CRISPR off-target effects by self-restricting Cas9 transcription and translation. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reuven, N.; Adler, J.; Broennimann, K.; Myers, N.; Shaul, Y. Recruitment of DNA Repair MRN Complex by Intrinsically Disordered Protein Domain Fused to Cas9 Improves Efficiency of CRISPR-Mediated Genome Editing. Biomolecules 2019, 9, 584. https://doi.org/10.3390/biom9100584
Reuven N, Adler J, Broennimann K, Myers N, Shaul Y. Recruitment of DNA Repair MRN Complex by Intrinsically Disordered Protein Domain Fused to Cas9 Improves Efficiency of CRISPR-Mediated Genome Editing. Biomolecules. 2019; 9(10):584. https://doi.org/10.3390/biom9100584
Chicago/Turabian StyleReuven, Nina, Julia Adler, Karin Broennimann, Nadav Myers, and Yosef Shaul. 2019. "Recruitment of DNA Repair MRN Complex by Intrinsically Disordered Protein Domain Fused to Cas9 Improves Efficiency of CRISPR-Mediated Genome Editing" Biomolecules 9, no. 10: 584. https://doi.org/10.3390/biom9100584