
Study of Protein Phosphatase 2A (PP2A) Activity in LPS-Induced Tolerance Using Fluorescence-Based and Immunoprecipitation-Aided Methodology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.3. Peritoneal Macrophage Isolation and in Vitro Stimulation
2.4. Phosphatase Assay
2.5. Immunoprecipitation
2.6. Western Blot
2.7. Chromatin Immunoprecipitation Assay (ChIP)
2.8. Statistical Analysis
3. Results
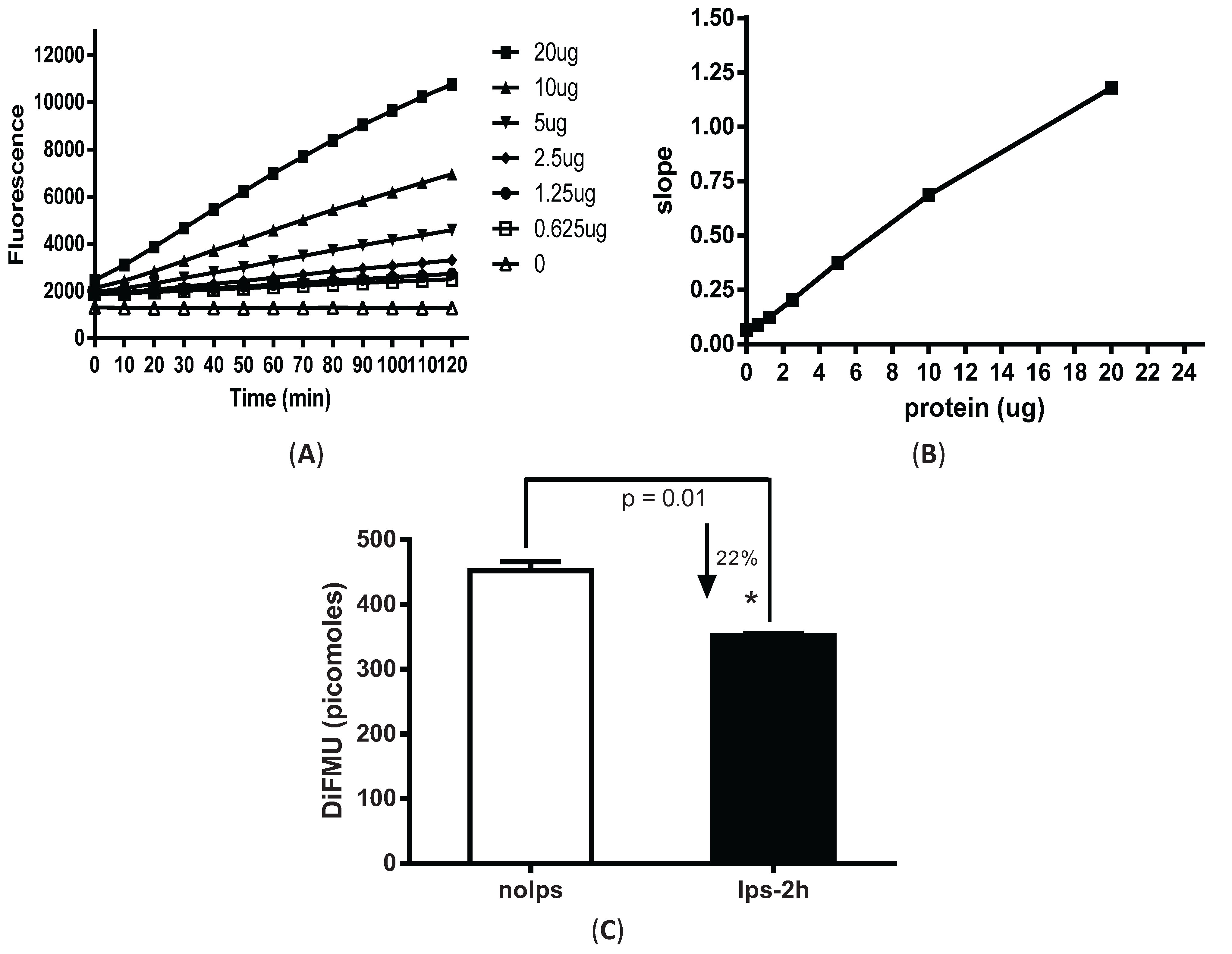
3.1. Fluorescence-Based Measurement of Phosphatase Activity Changes in LPS-Stimulated Peritoneal Macrophages
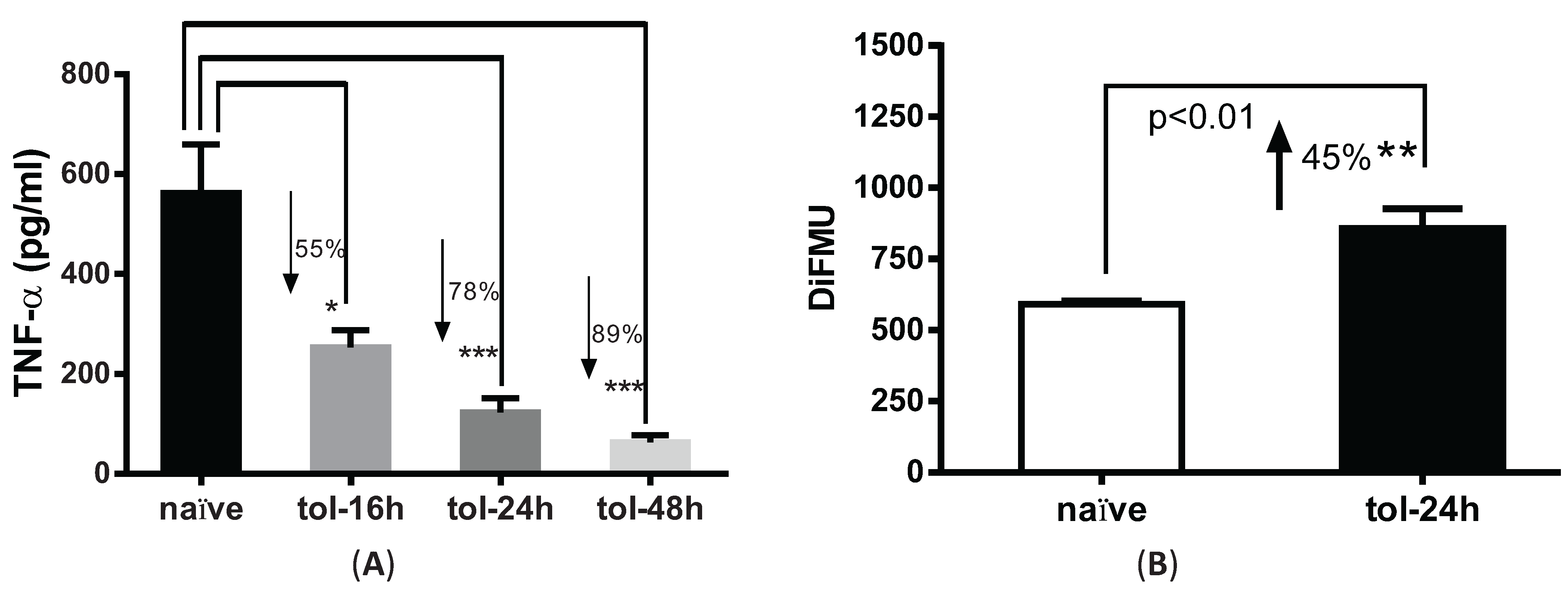
3.2. LPS Pre-Treatment Induced Tolerization and Upregulation of Phosphatase Activity in Peritoneal Macrophages
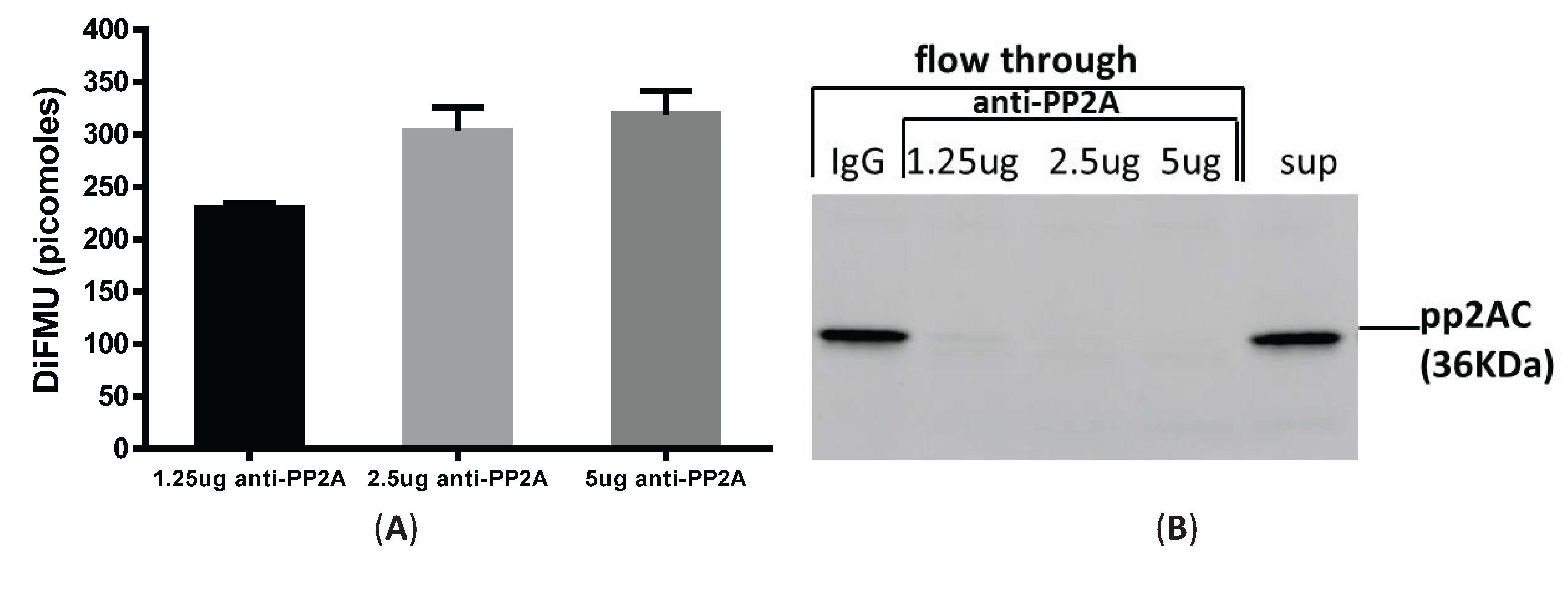
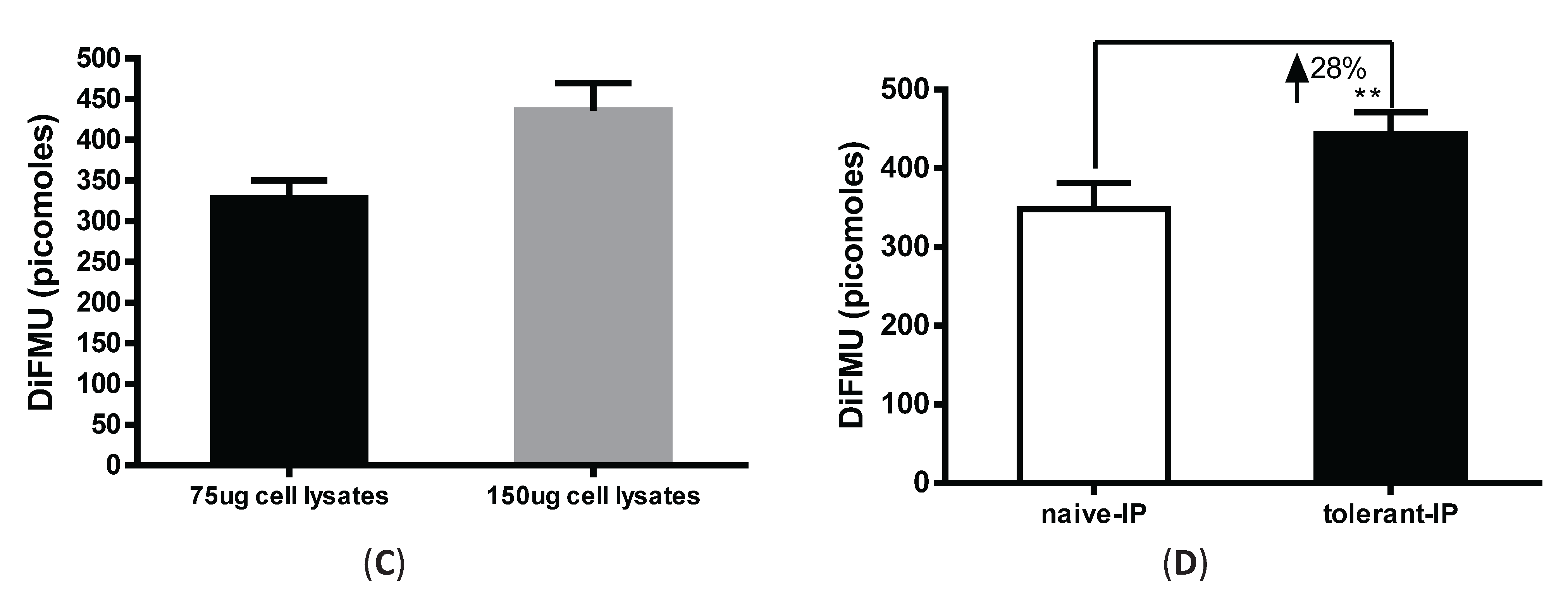
3.3. Immunoprecipitation-Aided Fluorescence Method to Examine PP2A Activity in Tolerized Macrophages
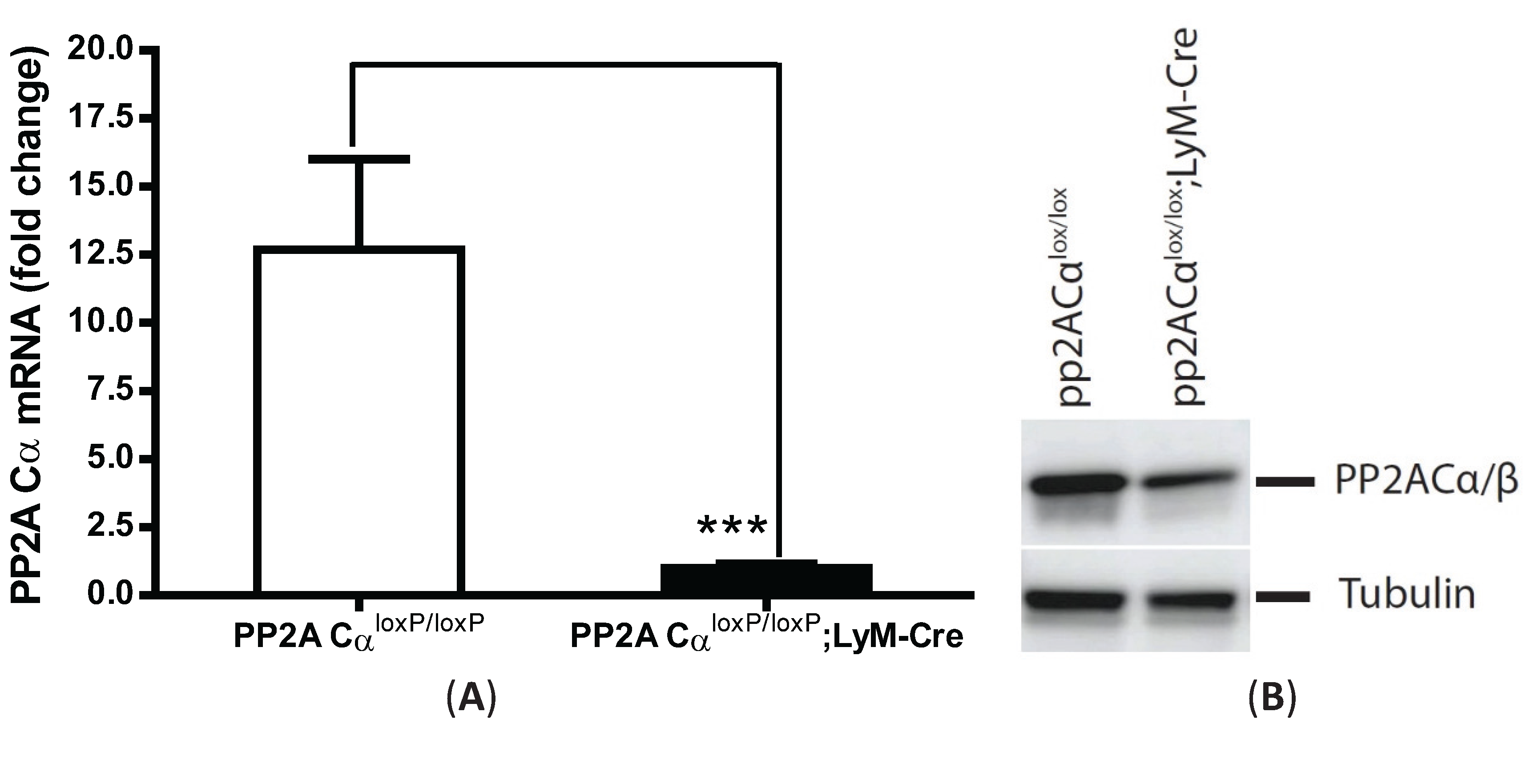
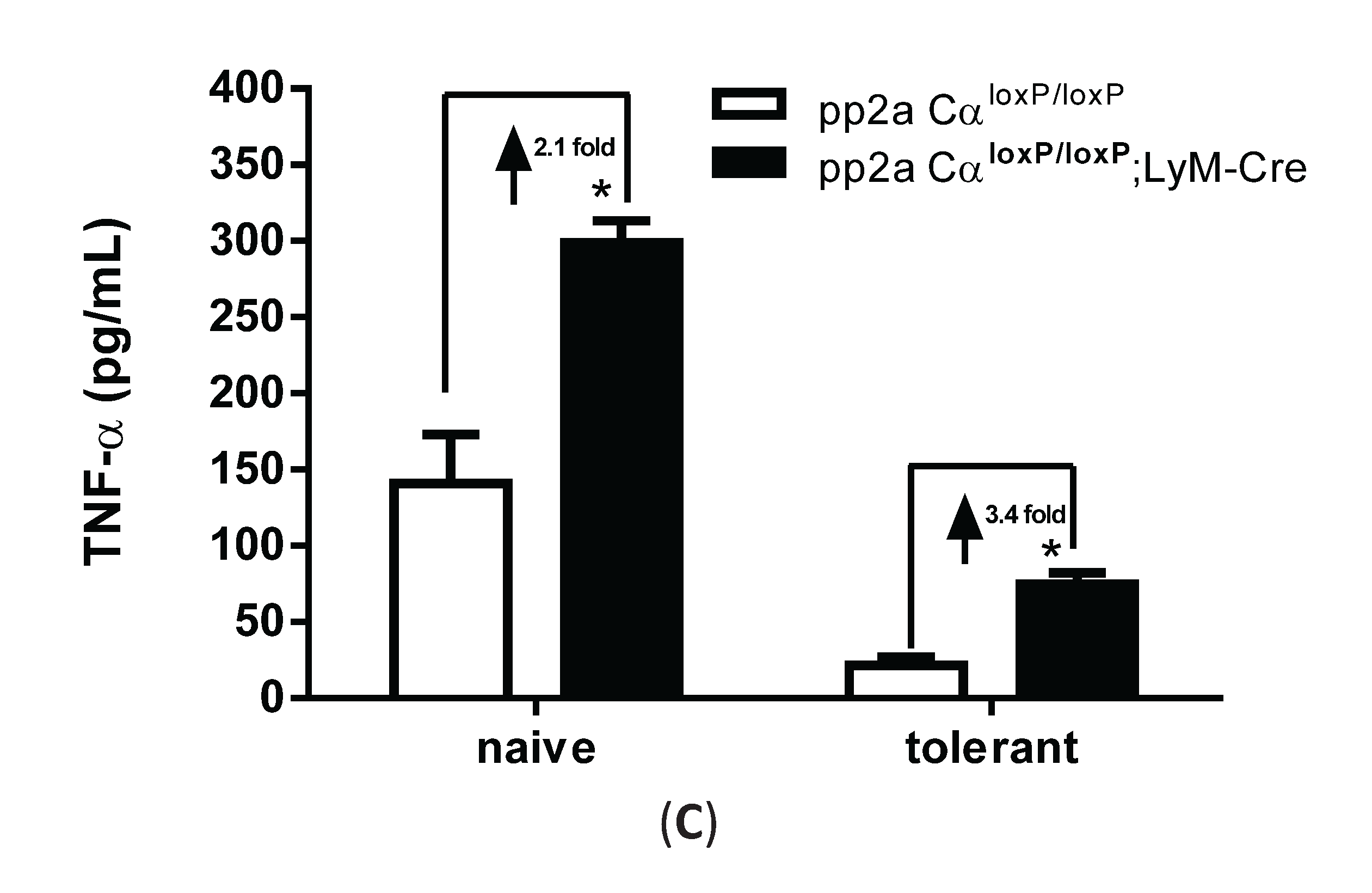
3.4. PP2ACα-Conditional Knockout Macrophages Released More TNF-α upon LPS Stimulation in both Naïve and Tolerized Cells

3.5. LPS Tolerance Did Not Change PP2A Activities in PP2ACαl°x/l°x;lyM-Cre Conditional Knockout Macrophages





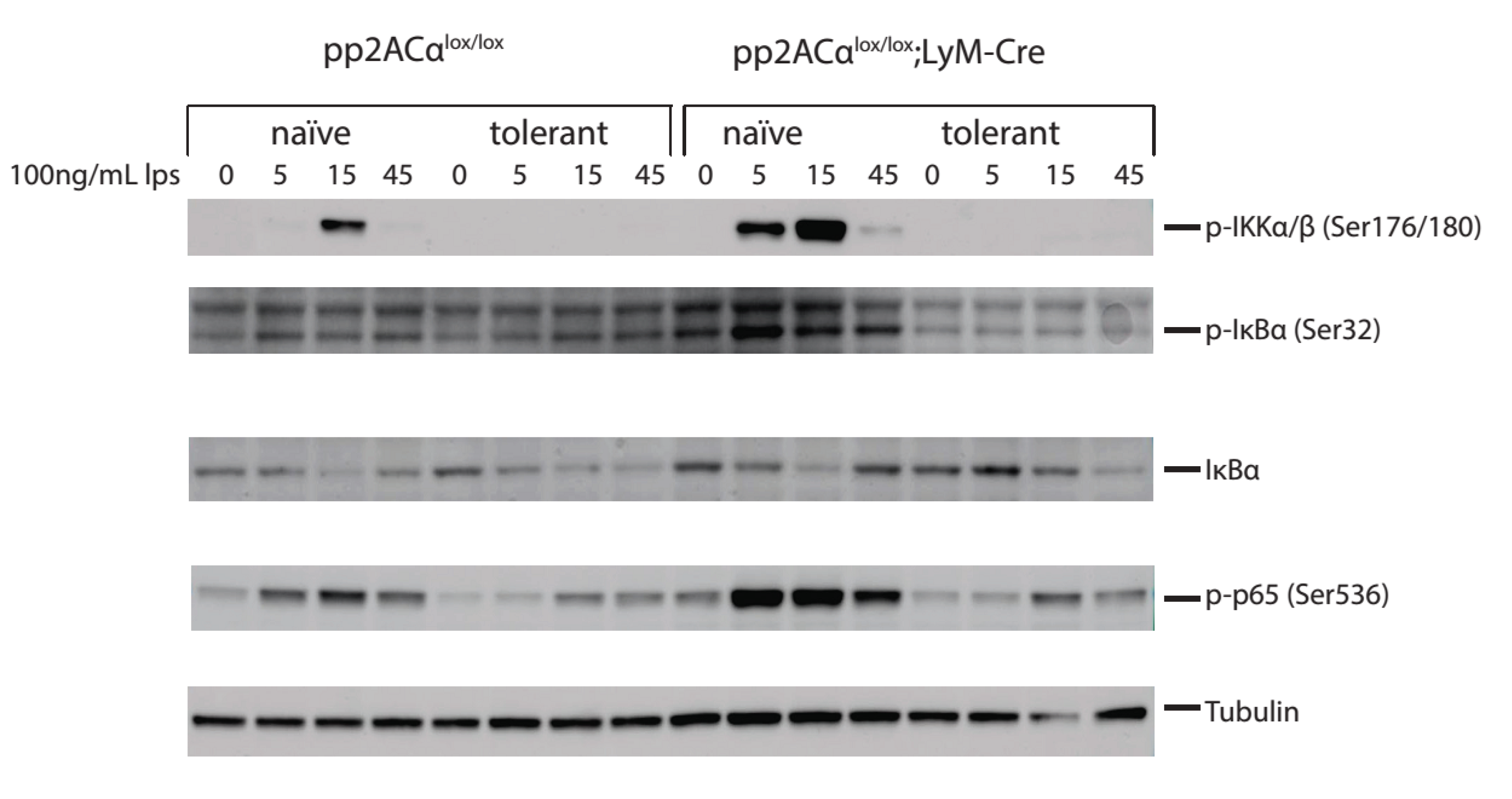
3.6. PP2ACα Deletion Did Not Induce Phosphorylation-Mediated Degradation of IκBα

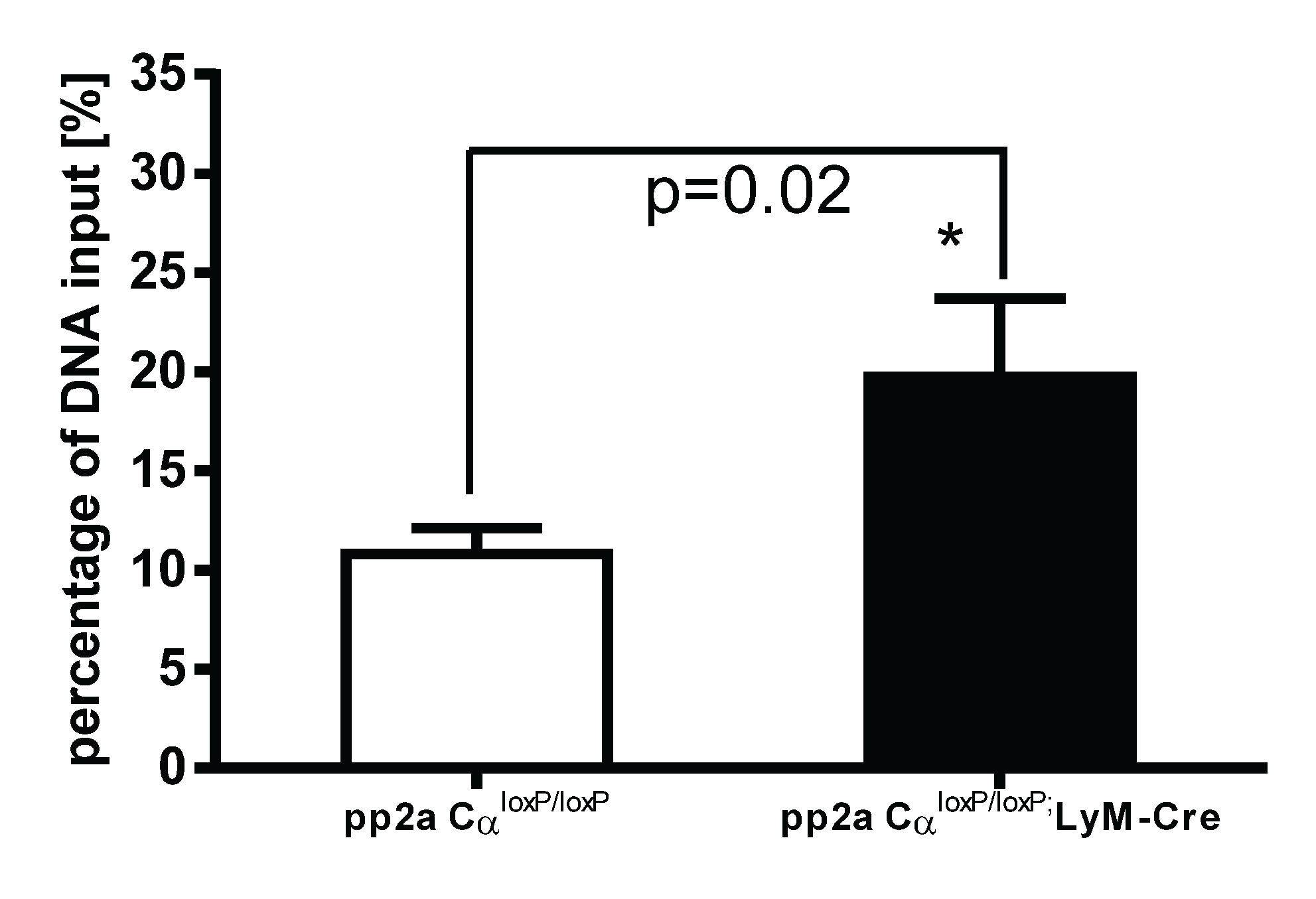
3.7. PP2ACα Knockout Induced Increased Binding of H3K4me3 to the Promoter Region of TNF-α after LPS Tolerance


4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interests
References
- Mumby, M.C.; Walter, G. Protein phosphatases and DNA tumor viruses: Transformation through the back door? Cell. Regul. 1991, 2, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Shanley, T.P.; Vasi, N.; Denenberg, A.; Wong, H.R. The serine/threonine phosphatase, PP2A: Endogenous regulator of inflammatory cell signaling. J. Immunol. 2001, 166, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Wang, J. Role of serine/threonine protein phosphatase in Alzheimer’s disease. Neurosignals 2002, 11, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Deng, X. Protein phosphatase 2A enhances the proapoptotic function of Bax through dephosphorylation. J. Biol. Chem. 2006, 281, 18859–18867. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, B.; Grundke-Iqbal, I.; Iqbal, K. I1PP2A affects tau phosphorylation via association with the catalytic subunit of protein phosphatase 2A. J. Biol. Chem. 2008, 283, 10513–10521. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin tolerance: New mechanisms, molecules and clinical significance. Trends Immunol. 2009, 30, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.C.; Chen, G.H.; Newstead, M.W.; Moore, T.; Zeng, X.; Tateda, K.; Standiford, T.J. Alveolar macrophage deactivation in murine septic peritonitis: Role of interleukin 10. Infect. Immun. 2001, 69, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Sly, L.M.; Rauh, M.J.; Kalesnikoff, J.; Song, C.H.; Krystal, G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity 2004, 21, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Classification of protein-serine/threonine phosphatases: Identification and quantitation in cell extracts. Methods Enzymol. 1991, 201, 389–398. [Google Scholar] [PubMed]
- Klumpp, S.; Krieglstein, J. Serine/threonine protein phosphatases in apoptosis. Curr. Opin. Pharmacol. 2002, 2, 458–462. [Google Scholar] [CrossRef]
- Garriga, J.; Jayaraman, A.L.; Limon, A.; Jayadeva, G.; Sotillo, E.; Truongcao, M.; Patsialou, A.; Wadzinski, B.E.; Grana, X. A dynamic equilibrium between CDKs and PP2A modulates phosphorylation of pRB, p107 and p130. Cell cycle 2004, 3, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Forster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Arino, J.; Woon, C.W.; Brautigan, D.L.; Miller, T.B., Jr.; Johnson, G.L. Human liver phosphatase 2A: cDNA and amino acid sequence of two catalytic subunit isotypes. Proc. Natl. Acad. Sci. USA 1988, 85, 4252–4256. [Google Scholar] [CrossRef] [PubMed]
- Shakhov, A.N.; Collart, M.A.; Vassalli, P.; Nedospasov, S.A.; Jongeneel, C.V. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J. Exp. Med. 1990, 171, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Trede, N.S.; Tsytsykova, A.V.; Chatila, T.; Goldfeld, A.E.; Geha, R.S. Transcriptional activation of the human TNF-α promoter by superantigen in human monocytic cells: Role of NF-κB. J. Immunol. 1995, 155, 902–908. [Google Scholar] [PubMed]
- Krappmann, D.; Scheidereit, C. A pervasive role of ubiquitin conjugation in activation and termination of IκB kinase pathways. EMBO Rep. 2005, 6, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Baltimore, D. Activation in vitro of NF-κB by phosphorylation of its inhibitor IκB. Nature 1990, 344, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Huxford, T.; Huang, D.B.; Malek, S.; Ghosh, G. The crystal structure of the IκBα/NF-κB complex reveals mechanisms of NF-κB inactivation. Cell 1998, 95, 759–770. [Google Scholar] [CrossRef]
- Jin Jung, K.; Hyun Kim, D.; Kyeong Lee, E.; Woo Song, C.; Yu, B.P.; Chung, H.Y. Oxidative stress induces inactivation of protein phosphatase 2A, promoting proinflammatory NF-κB in aged rat kidney. Free Radic. Biol. Med. 2013, 61C, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, L.; Berman, M.A.; Zhang, Y.; Dorf, M.E. RNAi screen in mouse astrocytes identifies phosphatases that regulate NF-κB signaling. Mol. Cell 2006, 24, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.G. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J. Periodontol. 2008, 79, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Bannister, A.J.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004, 6, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Stoecklin, G.; van Way, S.; Hinkovska-Galcheva, V.; Guo, R.F.; Anderson, P.; Shanley, T.P. Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2A and stabilizes tumor necrosis factor-α mRNA. J. Biol. Chem. 2007, 282, 3766–3777. [Google Scholar] [CrossRef] [PubMed]
- Wegner, A.M.; McConnell, J.L.; Blakely, R.D.; Wadzinski, B.E. An automated fluorescence-based method for continuous assay of PP2A activity. Methods Mol. Biol. 2007, 365, 61–69. [Google Scholar] [PubMed]
- Wera, S.; Hemmings, B.A. Serine/threonine protein phosphatases. Biochem. J. 1995, 311, 17–29. [Google Scholar] [PubMed]
- Goldberg, J.; Huang, H.B.; Kwon, Y.G.; Greengard, P.; Nairn, A.C.; Kuriyan, J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 1995, 376, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Favre, B.; Turowski, P.; Hemmings, B.A. Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J. Biol. Chem. 1997, 272, 13856–13863. [Google Scholar] [CrossRef] [PubMed]
- Katayose, Y.; Li, M.; Al-Murrani, S.W.; Shenolikar, S.; Damuni, Z. Protein phosphatase 2A inhibitors, I1PP2A and I2PP2A, associate with and modify the substrate specificity of protein phosphatase 1. J. Biol. Chem. 2000, 275, 9209–9214. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.J.; Yoon, J.G.; Yi, A.K. Myeloid differentiation factor 88-dependent post-transcriptional regulation of cyclooxygenase-2 expression by CpG DNA: Tumor necrosis factor-α receptor-associated factor 6, a diverging point in the Toll-like receptor 9-signaling. J. Biol. Chem. 2003, 278, 40590–40600. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Davis, R.J.; Flavell, R.A. MAP kinases in the immune response. Ann. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E.; Lentschat, A.; Wahl, L.M.; Golenbock, D.T.; Vogel, S.N. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J. Immunol. 2002, 169, 5209–5216. [Google Scholar] [CrossRef] [PubMed]
- Heagy, W.; Hansen, C.; Nieman, K.; Rodriguez, J.L.; West, M.A. Impaired mitogen-activated protein kinase activation and altered cytokine secretion in endotoxin-tolerant human monocytes. J. Trauma 2000, 49, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Wahlstrom, K.; Bellingham, J.; Rodriguez, J.L.; West, M.A. Inhibitory kappaBalpha control of nuclear factor-kappaB is dysregulated in endotoxin tolerant macrophages. Shock 1999, 11, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Calvano, J.E.; Agnese, D.M.; Um, J.Y.; Goshima, M.; Singhal, R.; Coyle, S.M.; Reddell, M.T.; Kumar, A.; Calvano, S.E.; Lowry, S.F. Modulation of the lipopolysaccharide receptor complex (CD14, TLR4, MD-2) and toll-like receptor 2 in systemic inflammatory response syndrome-positive patients with and without infection: Relationship to tolerance. Shock 2003, 20, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Medvedev, A.E.; Thomas, K.E.; Cuesta, N.; Toshchakov, V.; Ren, T.; Cody, M.J.; Michalek, S.M.; Rice, N.R.; Vogel, S.N. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: Effects of TLR “homotolerance” versus “heterotolerance” on NF-κB signaling pathway components. J. Immunol. 2003, 170, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Sarkisian, M.R.; Rakic, P.; Flavell, R.A. Loss of mitogen-activated protein kinase kinase kinase 4 (MEKK4) results in enhanced apoptosis and defective neural tube development. Proc. Natl. Acad. Sci. USA 2005, 102, 3846–3851. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Fan, G.H.; Wadzinski, B.E.; Sakurai, H.; Richmond, A. Protein phosphatase 2A interacts with and directly dephosphorylates RelA. J. Biol. Chem. 2001, 276, 47828–47833. [Google Scholar] [PubMed]
- Kanarek, N.; London, N.; Schueler-Furman, O.; Ben-Neriah, Y. Ubiquitination and degradation of the inhibitors of NF-κB. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef]
- Crispin, J.C.; Apostolidis, S.A.; Rosetti, F.; Keszei, M.; Wang, N.; Terhorst, C.; Mayadas, T.N.; Tsokos, G.C. Cutting edge: Protein phosphatase 2A confers susceptibility to autoimmune disease through an IL-17-dependent mechanism. J. Immunol. 2012, 188, 3567–3571. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, S.A.; Rauen, T.; Hedrich, C.M.; Tsokos, G.C.; Crispin, J.C. Protein phosphatase 2A enables expression of interleukin 17 (IL-17) through chromatin remodeling. J. Biol. Chem. 2013, 288, 26775–26784. [Google Scholar] [CrossRef] [PubMed]
- Escobar, J.; Pereda, J.; Arduini, A.; Sandoval, J.; Sabater, L.; Aparisi, L.; Vento, M.; Lopez-Rodas, G.; Sastre, J. Role of redox signaling, protein phosphatases and histone acetylation in the inflammatory cascade in acute pancreatitis. Therapeutic implications. Inflamm. Allergy Drug Targets 2010, 9, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.A. New approaches to the study of sepsis. EMBO Mol. Med. 2012, 4, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Lyn-Kew, K.; Rich, E.; Zeng, X.; Wen, H.; Kunkel, S.L.; Newstead, M.W.; Bhan, U.; Standiford, T.J. IRAK-M regulates chromatin remodeling in lung macrophages during experimental sepsis. PLoS ONE 2010, 5, e11145. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Pappy, A.L., II; Pham, T.T.; Shanley, T.P. Study of Protein Phosphatase 2A (PP2A) Activity in LPS-Induced Tolerance Using Fluorescence-Based and Immunoprecipitation-Aided Methodology. Biomolecules 2015, 5, 1284-1301. https://doi.org/10.3390/biom5031284
Sun L, Pappy AL II, Pham TT, Shanley TP. Study of Protein Phosphatase 2A (PP2A) Activity in LPS-Induced Tolerance Using Fluorescence-Based and Immunoprecipitation-Aided Methodology. Biomolecules. 2015; 5(3):1284-1301. https://doi.org/10.3390/biom5031284
Chicago/Turabian StyleSun, Lei, Adlai L. Pappy, II, Tiffany T. Pham, and Thomas P. Shanley. 2015. "Study of Protein Phosphatase 2A (PP2A) Activity in LPS-Induced Tolerance Using Fluorescence-Based and Immunoprecipitation-Aided Methodology" Biomolecules 5, no. 3: 1284-1301. https://doi.org/10.3390/biom5031284