1. Introduction
RNA and its functions play a significant role in a variety of biological operations. DNA molecules, where the genetic information is stored, are transcribed into mRNA, which carries the information into the cytoplasm, where translation takes place and leads to the production of a protein. Due to its utmost importance, this procedure is also called the “central dogma” of molecular biology [
1]. Apart from that major functionality, RNA has been proven to be involved in a wide range of central biological phenomena, such as gene expression regulation, site recognition, and catalysis [
2,
3]. All these RNAs, except the mRNA, are called noncoding because they fulfill functions other than encoding proteins, also elaborating the necessity of the detailed analysis of these molecules. In this context, it is vital to predict the structure of RNA, specifically its 3D structure, to understand its functions. This tertiary structure can be determined using techniques such as X-ray crystallography [
4] and nuclear magnetic resonance [
5]. However, researchers have focused on the development of a methodology toward the prediction of a simpler representation of an RNA structure in a two-dimensional space, named a secondary structure, which is a collection of A (Adenine), U (Uracil), G (Guanine), and C (Cytosine) bases that form duplex regions and unpaired ones that form important motifs around them, such as loops, bulges, and hairpins. Therefore, the secondary structure is this collection of pairs (A–U, C–G, and G–U pairs) that form different motifs. The accurate location of the base pairs and motifs is a useful milestone and starting point for the enlightenment of the 3D structure and, consequently, the understanding of RNA operations.
Recent RNA secondary structure prediction methods have been based mainly on a scoring function that may rely on a thermodynamic, probabilistic, or Artificial Intelligence (AI)-based algorithm. The majority of the methods have utilized or adopted a partially minimum free energy algorithm introduced by Zuker, which facilitates dynamic programming enhanced with parameters from experiments [
6]. The Nussinov algorithm is also a widely used method that has succeeded in predicting the largest number of base pairings using dynamic programming [
7], which performed even better when it was combined or incorporated as an internal component in other more sophisticated algorithms, as in [
8]. Other current approaches have leveraged stochastic methods, syntactic pattern recognition, machine learning, statistical techniques, integer programming, or other heuristic algorithms to tackle the prediction task.
Section 2 contains a detailed analysis of the related literature.
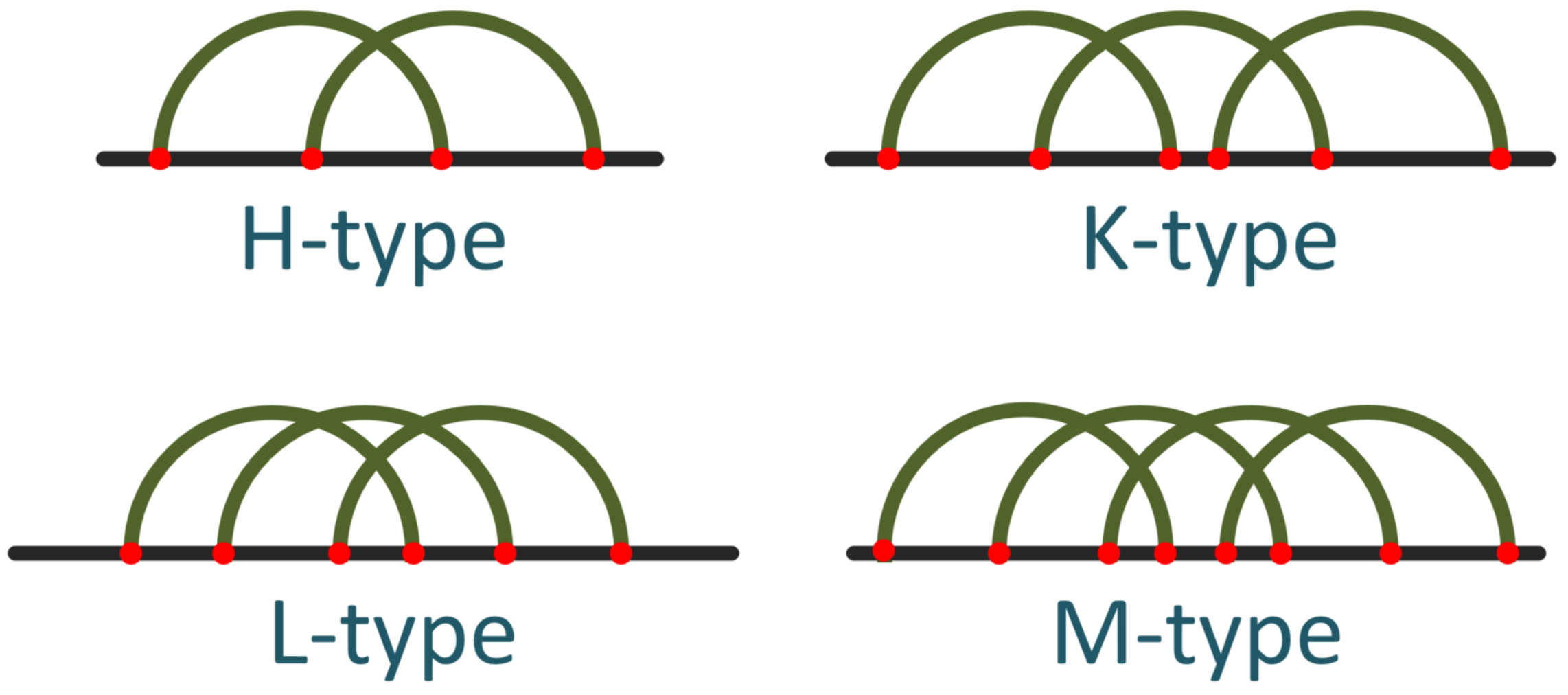
In an RNA secondary structure, the pseudoknot’s prediction is the most demanding task in terms of prediction. Other common motifs are stems, hairpins, bulges, internal loops, and multibranch loops, which a variety of algorithms are able to predict with high accuracy. On the other hand, the prediction of a pseudoknot is quite complicated because dynamic programming and minimum free energy algorithms are not constructed in such a way as to facilitate the interconnection of a pseudoknot. Another important reason is that with the increase in the length of the RNA, these algorithms need an exponential execution time. Thus, the need to achieve an accurate prediction for pseudoknots led our research towards constructing a platform that predicts H-type pseudoknots combined with bulges and internal loops with an accuracy similar to well-known methods and, at the same time, an efficiency in terms of execution time, called Knotify+. The H-type pseudoknot [
9] consists of two stems and two loops of arbitrary lengths.
Bulge loops or bulges form when a helix is interrupted by unpaired nucleotides on one strand, and they are frequently observed in the secondary structures of RNA [
10,
11], as they appear in a universal distribution in all types of structured functional RNAs [
12]. Base–base mismatches, shaping internal loops, also appear often in RNA, affecting the stability of the molecule [
10]. Specifically, researchers have focused on the study of bulges, due to the frequency with which bulged adenosine residues occur at protein binding sites in RNA [
10,
13], while they also operate as contact points in the tertiary folding of RNA [
11,
14]. Bulges construct unique recognition sites in RNA tertiary structures in two ways, first by acting as molecular handles within the helical regions and second, in an indirect way, by distorting the RNA backbone and allowing access to base pairs in a widened deep groove [
12]. Additionally, helical elements separated by bulges frequently undergo transitions between unstacked and coaxially stacked conformations during the folding and function of noncoding RNAs [
12]. All the above references show the importance of the identification of bulges and internal loops as key structural elements in a wide range of RNAs and emphasize their significance and pluralism in RNA architecture and molecular recognition.
In this work, we suggest a new version of Knotify [
15], which is capable of predicting bulges and internal loops [
12] in an H-type pseudoknot. The sequence of the RNA is imported to a parser which produces the entire set of the possible core stems of a pseudoknot. Next, all these trees are decorated with possible base pairs close to the two stems (core stems) that form the pseudoknot, with the difference that the algorithm is searching for possible bulges and internal loops around it. Towards the prediction of the optimal tree, a set of candidates is created according to the greatest number of base pairs, and finally, the structure with the minimum free energy is chosen. The current update enhances Knotify+’s ability to recognize and predict even more complex motifs, while it maintains the same level of complexity. In practice, the additional computations increase the execution time of the algorithm but are slight enough to be considered acceptable.
2. Related Work
Most of the algorithms have encapsulated dynamic programming in their pipeline process in order to determine the most likely secondary structure of an RNA, trying to minimize the free energy [
16,
17]. Other approaches that have focused on pseudoknot prediction, e.g., [
18], have enforced entropy, stability, and minimum free energy. The proof that this problem is NP (nondeterministic polynomial time)-complete [
19] has encouraged the development of stochastic and heuristic methods [
20,
21,
22]. A typical example is Knotty [
23], which predicts pseudoknots, with a CCJ (Chen–Condon–Jabbari) algorithm [
24] with sparsification. Additionally, ProbKnot [
25] computes base pair probabilities of non-pseudoknotted substructures, building the secondary structure based on the maximum expected accuracy. IPknot [
26] also leverages the advantages of integer programming and base pair probabilities, performing better than the previous methods. Its extension [
27] calculates secondary structures with pseudoknots in linear time using the LinearPartition model and pseudo-expected accuracy. This improved version can handle long sequences in a reasonable execution time, but there is still room for improvement in terms of accuracy.
Other approaches such as Pfold [
28,
29], PPfold [
30], and RNA-Decoder [
31] predict the secondary structure by applying Stochastic Context-Free Grammar (SCFG). All these approaches are specialized in pattern recognition, so they reveal similarities in structures, and in turn, they can be fine-tuned by assigning appropriate weights to the rules. Other typical SCFG-based frameworks are Contrafold [
32], Evfold [
33], Infernal [
34], and Oxfold [
35]. The extensive research on SCFG-based methods reveals the need for the efficient collaboration of grammar and computation methods, heuristic and probabilistic algorithms, minimum free energy computation, maximum base pairing, base pairing probabilities, and other algorithmic and biological concepts. Therefore, it is crucial to find the optimal match between these concepts, to succeed in predicting the RNA secondary structure. In this direction, we propose a grammar-based framework, which leverages maximum base pairing and minimum free energy, creating an efficient prediction pipeline. However, the underlying model of the proposed methodology is that of Context-Free Grammar (CFG).
Machine learning algorithms have also been proposed in the literature. They endeavor to unveil hidden patterns by applying supervised and unsupervised methods in training datasets. The majority of these need large datasets because they use deep learning techniques which require a significant amount of data for the training process to avoid overfitting. In [
36], for example, the authors used deep learning and tertiary constraints to tackle this task, while others, e.g., [
8], have constructed bidirectional-LSTM (long short-term memory) networks and the IBPMP (improved base-pair maximization principle) to select the correct base pairs to then predict the optimal structure. Similarly, 2dRNA [
37] applies a coupled two-staged deep neural network that provides data to a U-net architecture. A bidirectional LSTM encodes the data in a higher dimension, and at the final stage, a fully connected network decodes them, producing the dot-bracket structure. To predict the secondary structure, including pseudoknots, ATTfold [
38] also adopts deep learning models by incorporating an attention mechanism as an encoder. It encodes a base pairing score matrix; then, a CNN (Convolutional Neural Network) decodes the data in an appropriate format. The training process takes place according to hard biological concepts, aiming to reduce structures that do not exist in nature in agreement with the folding rules.
4. Proposed Methodology
In this section, the methodology proposed by the Knotify+ platform is presented. Knotify+ is an extension of the Knotify platform presented in [
15], including the pruning technique presented in [
42], capable of predicting bulges and internal loops around the core stems of the pseudoknot. Knotify manages to predict a pseudoknot in an RNA sequence making use of three main tasks: (a) a CFG parser analyzes the RNA sequence so that all trees in which a pseudoknot pattern is detected are generated; (b) the produced trees are parsed to detect the core stems that form the pseudoknot and the possible base pairs around the core stems of the pseudoknot, (c) the optimal tree is selected using two well-known criteria, that of the maximum number of base pairs and the minimum free energy of the sequence. A thorough analysis of the abovementioned tasks (see
Figure 3) is provided in the next subsections. Knotify+ adds a new task (see the blue box in
Figure 3) before the selection of the pseudoknot, which is responsible for the identification of bulges or internal loops around the core stems.
Consequently, the proposed implementation receives as input a string representing an RNA sequence of nitrogenous bases and produces the base pairing of the given RNA sequence in extended dot-bracket notation. The Knotify+ source code and implementation details are available as a public repository on GitHub [
55].
4.1. CFG to Identify Pseudoknots
Knotify+’s methodology is based on the platform proposed in [
15]. Hence, Knotify+ makes use of an efficient CFG parser. Therefore, initially, the appropriate primitive patterns should be selected. With regard to the RNA sequence representation, the obvious choice was to assign the nitrogenous bases A, C, G, and U to the characters “A”, “C”, “G”, and “U”, respectively, which also formed the set T of the terminal symbols of the grammar. The sequences of those four characters, such as AAUCCGG or CCGAAAUACG, formed a string that represents an RNA. After the primitive patterns were selected, a convenient grammar was defined, so as to syntactically analyze the linguistic representation of the original patterns.
The proposed platform makes use of the CFG
that was initially presented and extensively described in [
15]. Knotify+ initially executed the space elimination proposed in [
42] aiming to dramatically decrease the substrings to be parsed by our sliding-windows technique. Then, the CGF parser analyzed the RNA sequence so that all trees in which a pseudoknot pattern was detected were generated. The main contribution of this paper is the creation of a new module that predicts bulges and internal loops around the core stems when the pseudoknot is decorated. This process is presented in
Section 4.2. Finally, the last task of pseudoknot selection is executed as described in [
42] and presented in
Section 4.3.
4.2. Decorate Core Stems
During the first task, all parse trees were constructed by the parser. By the use of these trees, all possible pseudoknots and their core stems were allocated. The second task dealt with the traversing of all these trees to locate further stems. The CFG proposed in [
15] was dedicated to detecting the initially crossing stems of the pseudoknot, in our notation the core stems, trying to amend the CFG parser’s efficiency. Consequently, all parse trees were evaluated for the possible detection of base pairs surrounding the pseudoknot’s core stems. All bases located in each of the two loops were consecutively checked for their ability to create a pair with a base in an appropriate position.
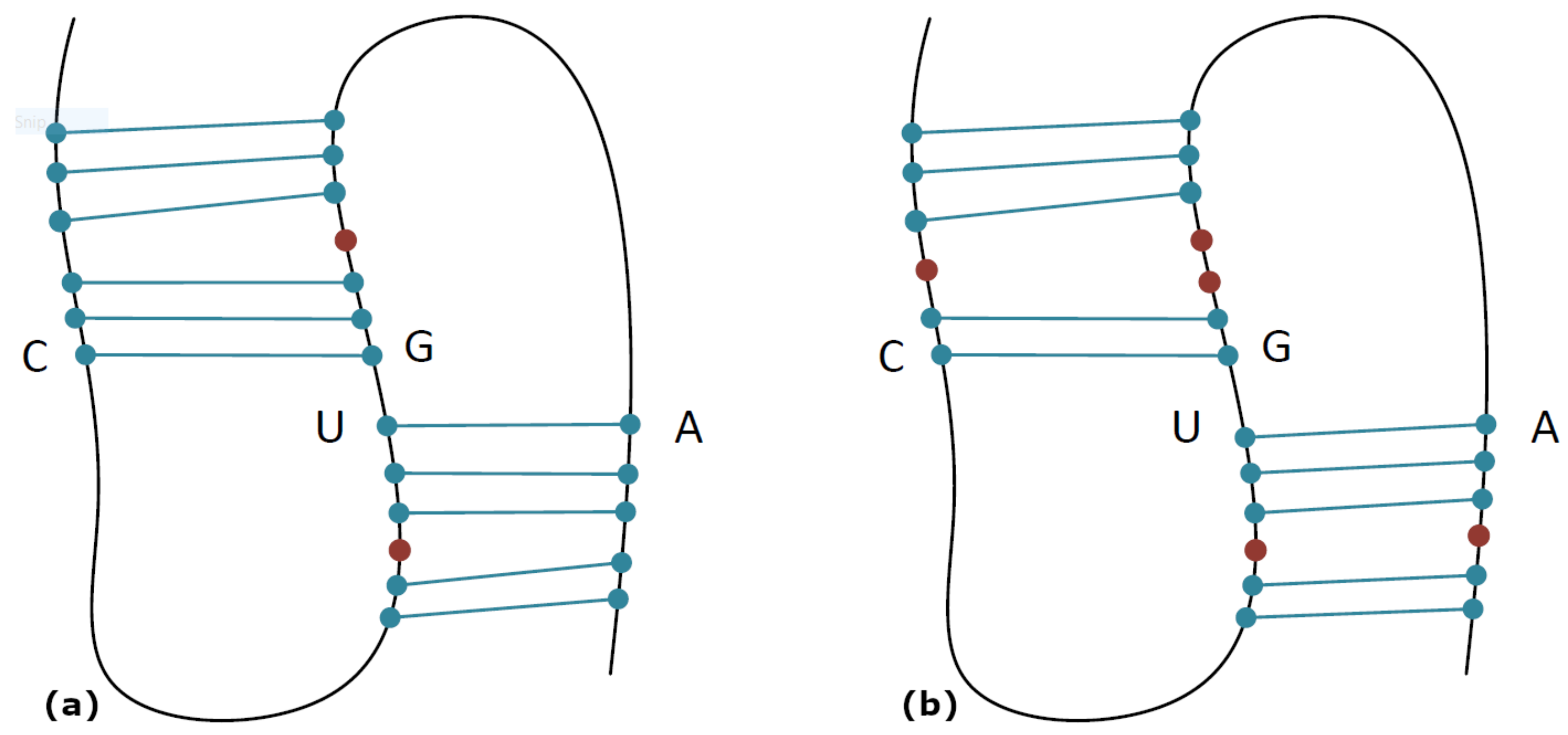
In
Table 1, the process of the core stems decoration is presented. After the parser detected the core stems U–A and C–G at positions 10–17 and 5–11, the two pseudoknot loops were specified. The left loop was at positions 6 to 9, and the right loop was at positions 12 to 16. The bases in these loops were initially examined for whether they might pair with bases outside the pseudoknot’s loops. The base pairs in the left loop were tested for a match with bases at positions 18 to 22, while bases in the right were tested for a match with bases at positions 18 to 22.
In both loops of the pseudoknot, the base pairs at positions 9–18, 8–19, 4–12, and 3–14 were sequentially detected during stages 1 to 4, respectively.
Table 1 presents this procedure in detail. Once no more sequential base pairs could be formed, the existence of bulges and internal loops was checked (stage 5). For each side, left or right, the unpaired bases were examined for whether they could form a base pair after creating a bulge or an internal loop. In our example, on the left side, the set at positions 6–7 may form base pairs with a set at positions 20–22 after creating bulges; those two sets were called the left pair of sets. On the right side, the set at positions 1–2 may form base pairs with a set at positions 14–16 after creating bulges; those two sets were called the right pair of sets. Users may define the maximum bulge size, which is given as an argument when the program is executed. This parameter is called the
maximum_bulge_size. For each pair of sets, there may be a bulge of length 0 to
maximum_bulge_size at one set and 0 to
maximum_bulge_size at the other set. In the case where the bulge’s length is zero on one side and greater than zero on the other side, then a bulge is located. Otherwise, if the bulge’s length is greater than zero on both sides, then an internal loop is located. The Cartesian product of those cases was executed, and multiple dot-brackets strings were produced. By applying the criteria of the minimum free energy and the greatest number of base pairs of the pseudoknot, the optimal case was selected. The result of this procedure is shown in stage 5 of
Table 1. Regarding the left pair of sets, there may be a base pair at positions 7–21 after creating a bulge at position 20. Regarding the right pair of sets, there may be a base pair at positions 2–16 after creating a bulge at positions 14–15, there may be a base pair at positions 1–14 after creating a bulge at position 2, or there may be a base pair at positions 1–15 after creating a bulge at position 2 and another one at position 14, creating in this way an internal loop. The last case was the one that was finally selected, as shown in stage 5, where the internal loop is highlighted in red.
Knotify+ allows the user to choose the option of the base pairs, U–G, as an argument from the command line, as well as the value of the maximum_bulge_size.
4.3. Optimal Tree Selection
Knotify+ incorporated a hybrid model to choose the optimal tree among the trees that were produced from the CFG. This task facilitated the maximum base pairing and the MFE (Minimum Free Energy) principles. In the first stage, it ranked all the produced trees according to the count of base pairs around the pseudoknot, excluding the stems formed after the bulges or internal loops. The next stage consisted of the application of the MFE in the trees that were ranked at the top in the first stage, i.e., the trees with the most base pairs around the pseudoknot. After extensive experiments, we observed that including all the possible detected stems after the bulges or internal loops may lead to excluding the correct RNA sequence from the top-ranking sequences (regarding base pairs count) that were promoted to the second stage of selection, that of the MFE calculation. Consequently, the first task of the proposed tree selection, that of maximum pairing, was applying it to the RNA sequences without taking into consideration the stems detected after the bulges or internal loops.
Finally, the secondary structure with the minimum free energy was selected. A module derived from HotKnots [
56] calculated each candidate’s energy and, in turn, provided the energy scores to Knotify+ to make the final selection. The energy calculation algorithm was introduced by Mathews [
57], but we used a variation based on [
58] presented in the following relation:
where
is the weight or cost of the existence of a pseudoknot;
is the total number of core stems;
is the total number of unpaired bases inside the pseudoknot. Following the experimental evaluation in [
56], we set the parameters
(cost for the core stems) and
(cost for the unpaired bases) equal to 0.1 and
equal to 9.6 (see
Figure 4).
6. Conclusions
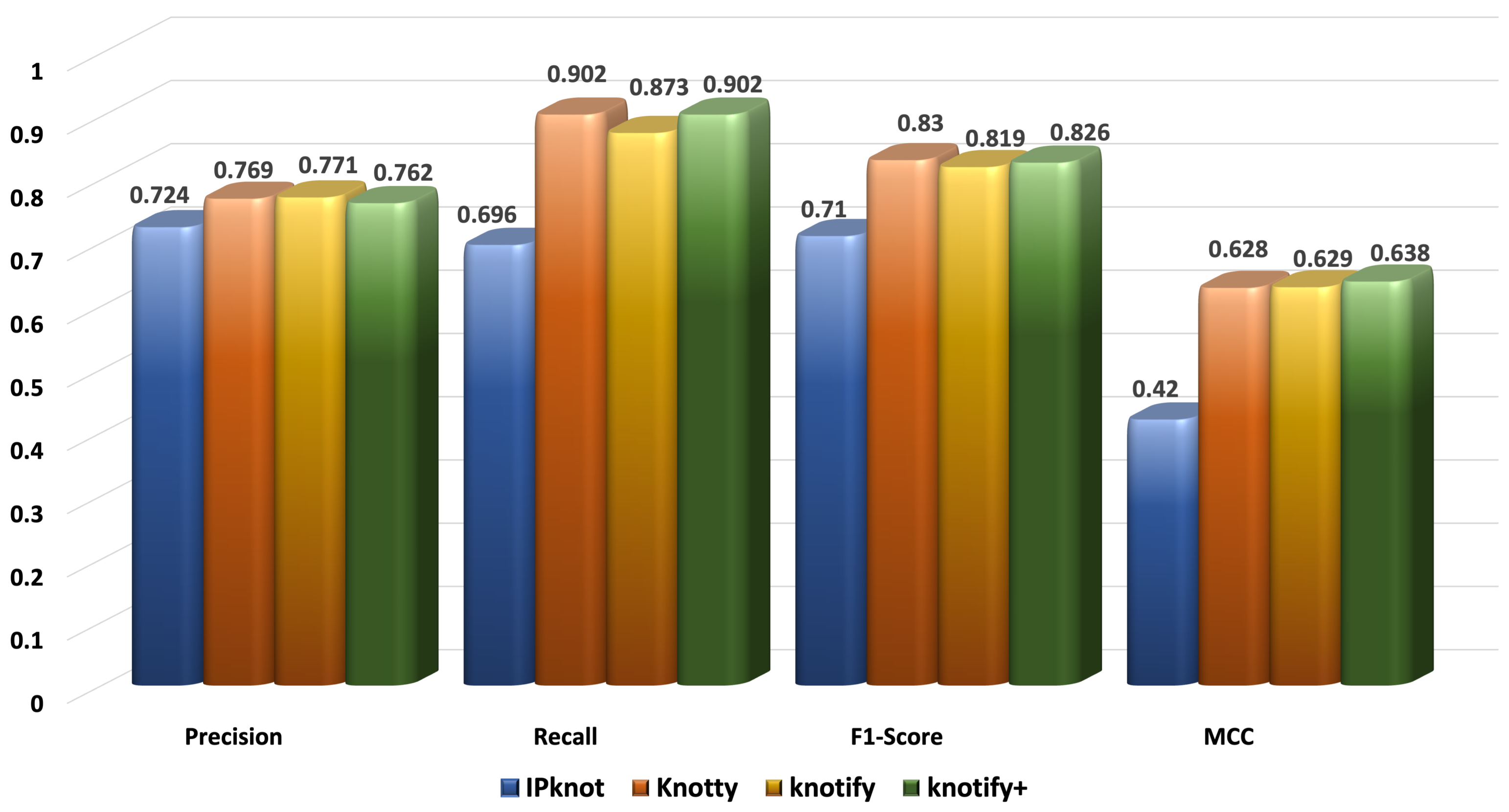
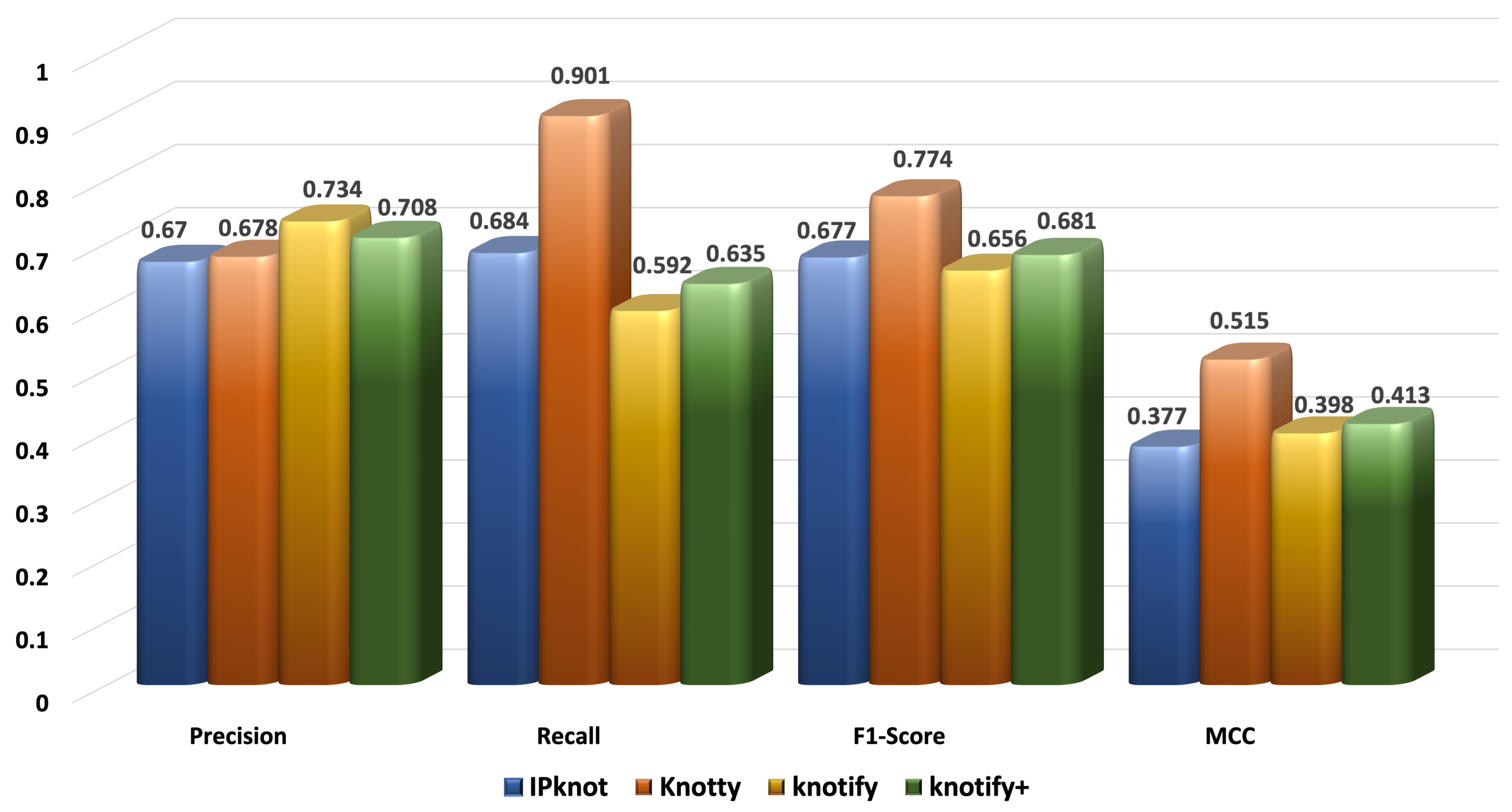
The prediction of the RNA secondary structure is quite a challenging task, especially for pseudoknotted structures. In this context, we proposed an intelligent grammar-based algorithm that predicted H-type pseudoknots with bulges and internal loops. It detected the secondary structure performant, and its accuracy was comparable to well-known platforms. Especially for sequences smaller than 30 bases, it outperformed all the examined methods, showing that the enhancement of its expressiveness led to an important advancement of our previous version. The most notable finding was that the proposed methodology outperformed our previous version Knotify regarding the recall, F1-score, and MCC in all sets, showing a significant improvement for sequences larger than 40. In addition, Knotify+ continued to outperform Knotty for small sequences, while it was comparable for sequences between 30 and 50, and significantly decreased the gap with Knotty for sequences larger than 50 bases. Meanwhile, Knotify+ maintained the highest percentage of core stems prediction compared to all the examined methods and was approximately eight times faster than Knotty.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}