
Bioinformatic Analyses of Peripheral Blood Transcriptome Identify Altered Neutrophil-Related Pathway and Different Transcriptomic Profiles for Acute Pancreatitis in Patients with and without Chylomicronemia Syndrome

Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Differential Expression Analysis
2.3. Gene Set Enrichment Analysis (GSEA)
2.4. Over-Representation Analysis (ORA)
2.5. Construction of Regulatory Gene Network and Identification of Hub Genes
2.6. Identification of Target miRNAs
2.7. Single Sample Gene Set Enrichment Analysis (ssGSEA)
2.8. Immune Cell Infiltration Analysis
2.9. Prediction of Associated Genes and Biological Pathways
2.10. Construction of Diagnostic Models for AP
2.11. Statistical Analysis
3. Results
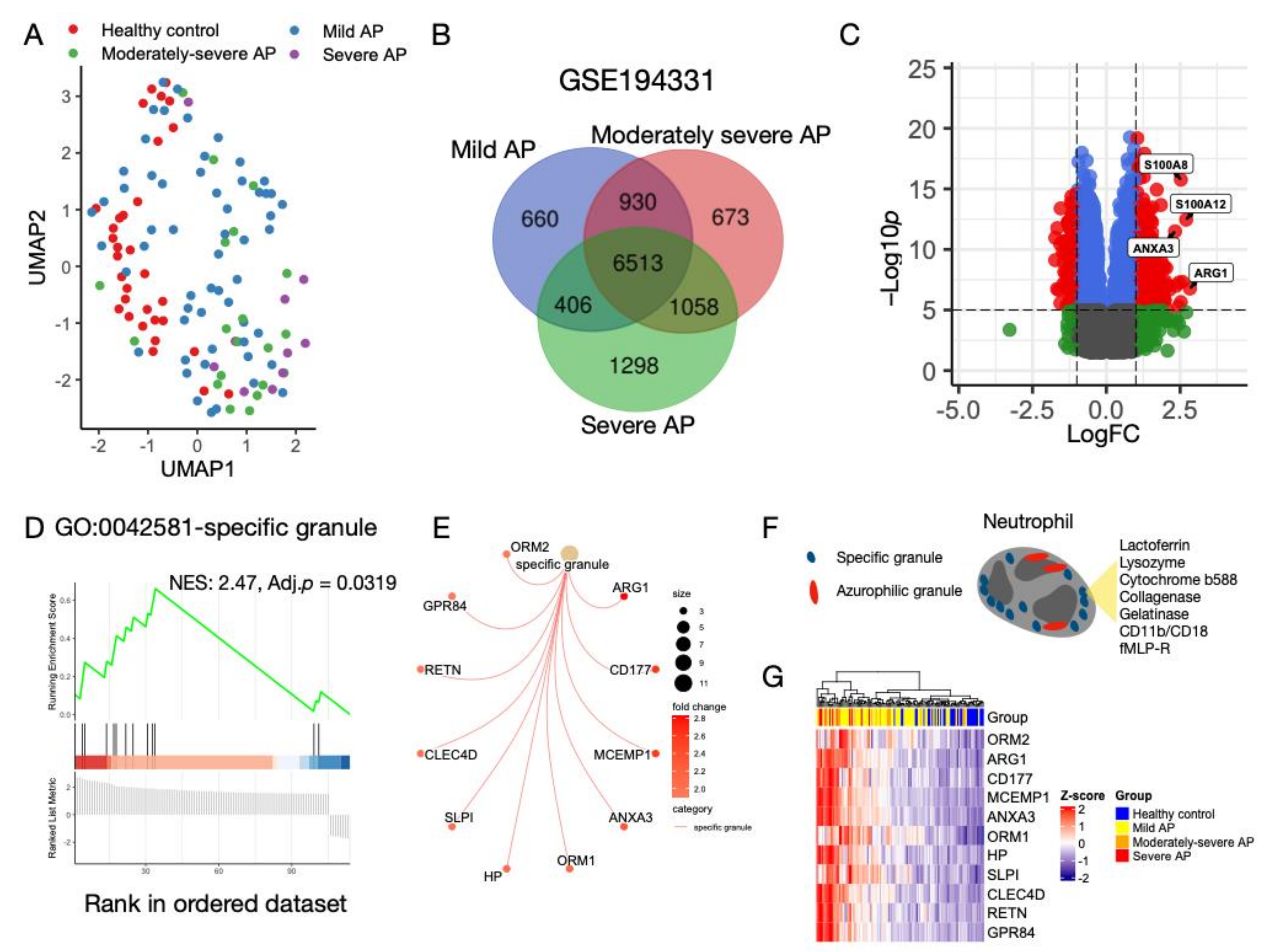
3.1. Characterization of Transcriptomic Profiles in AP without CS
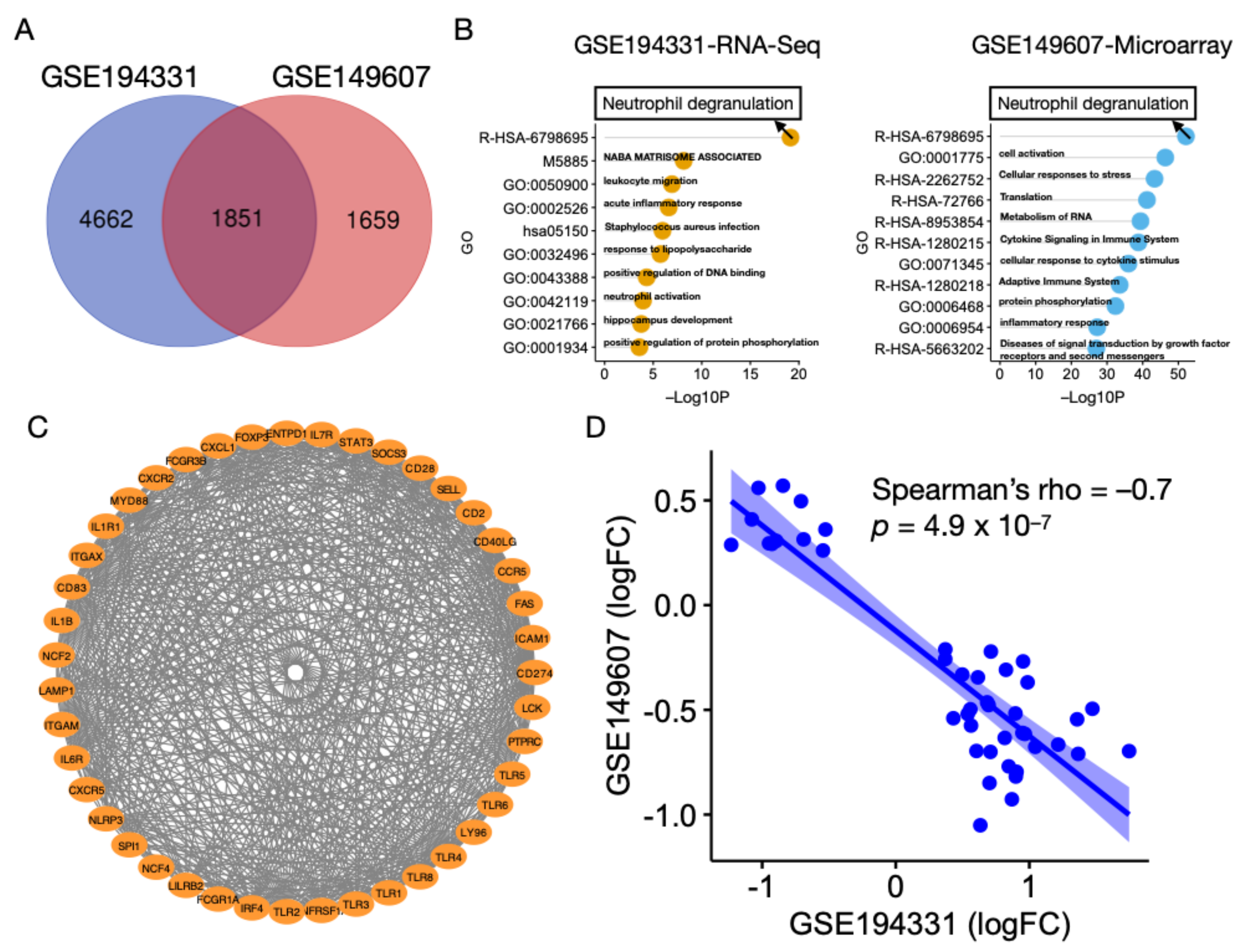
3.2. Neutrophil Degranulation Pathway Is Altered in AP with and without CS
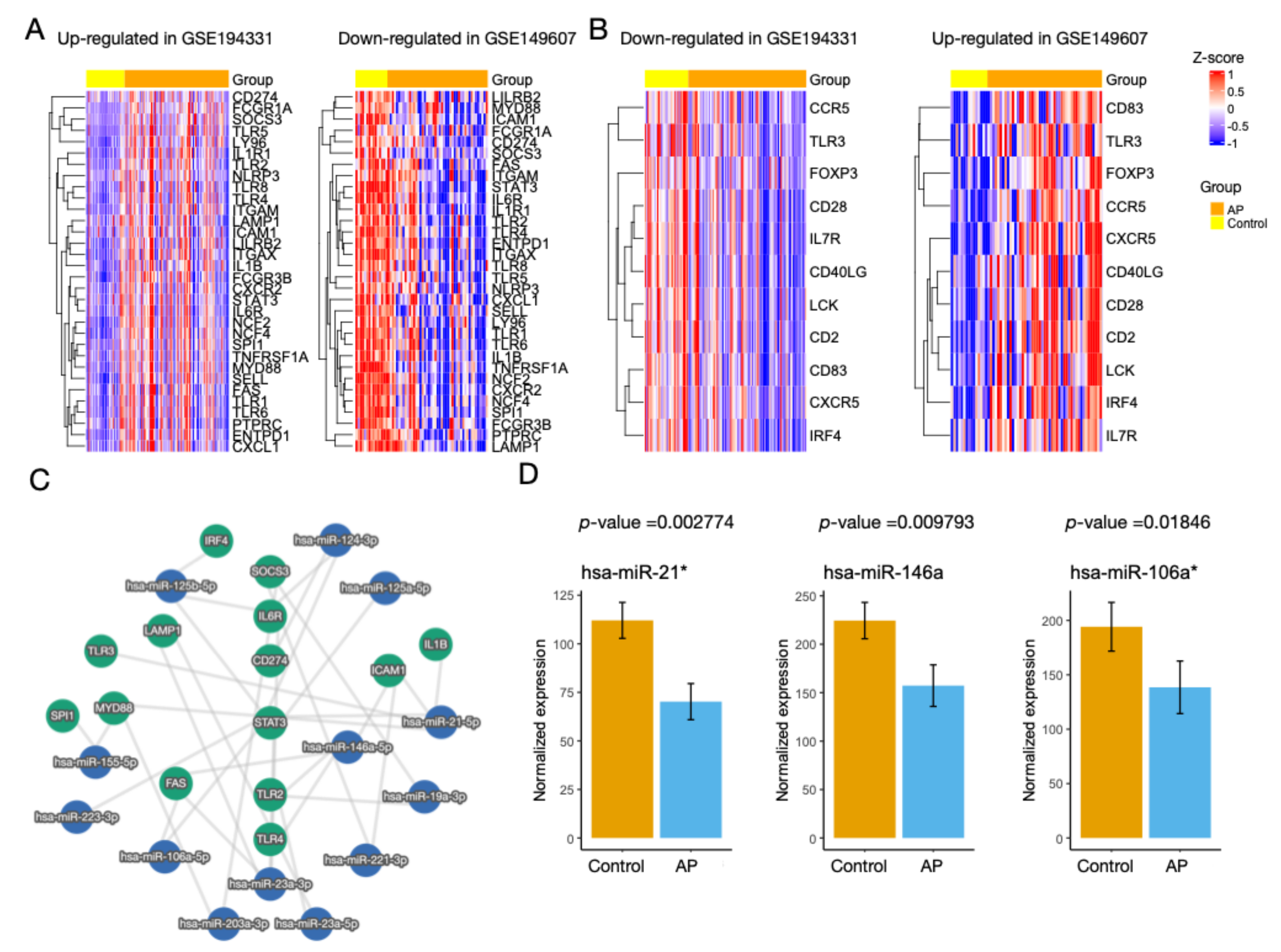
3.3. Identification of miRNAs
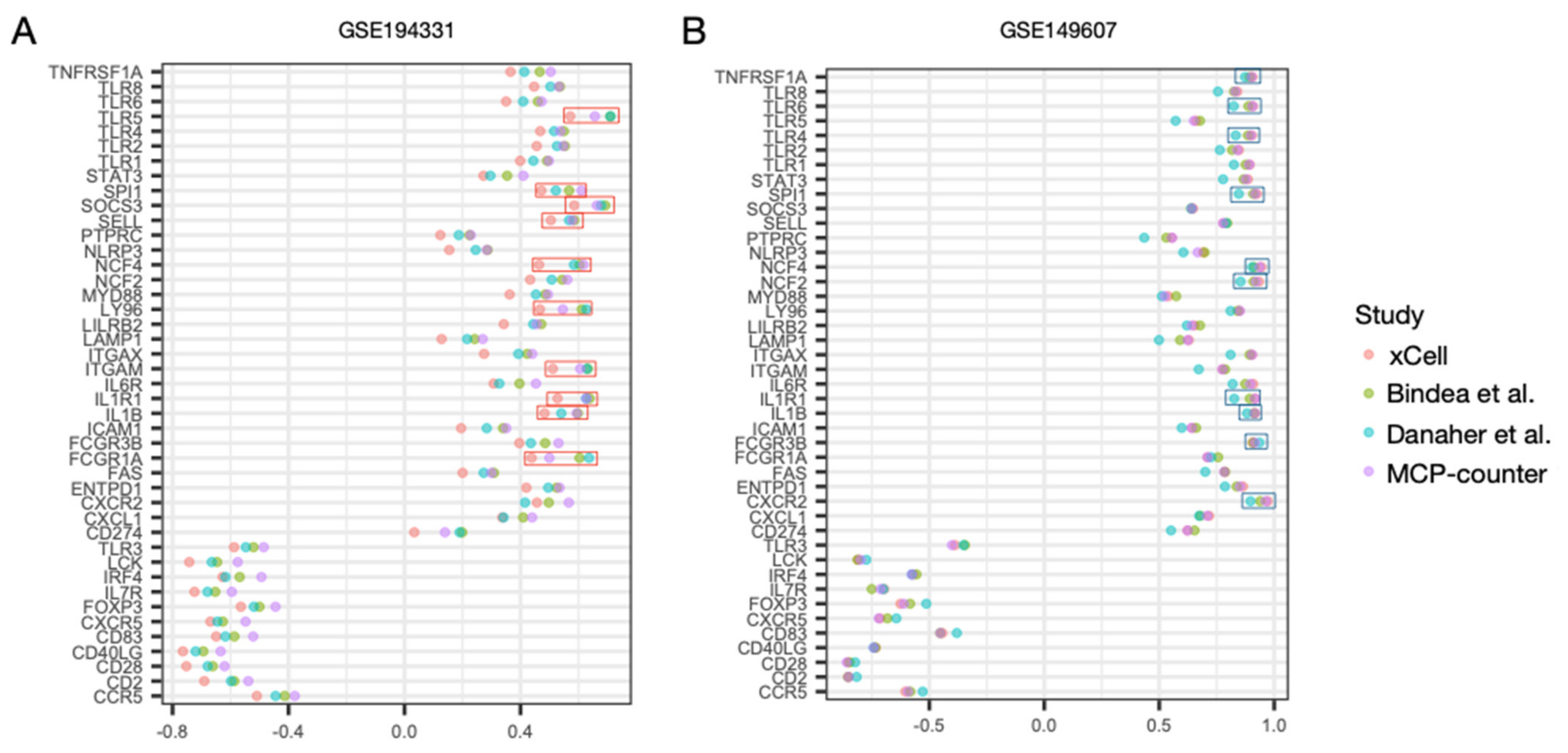
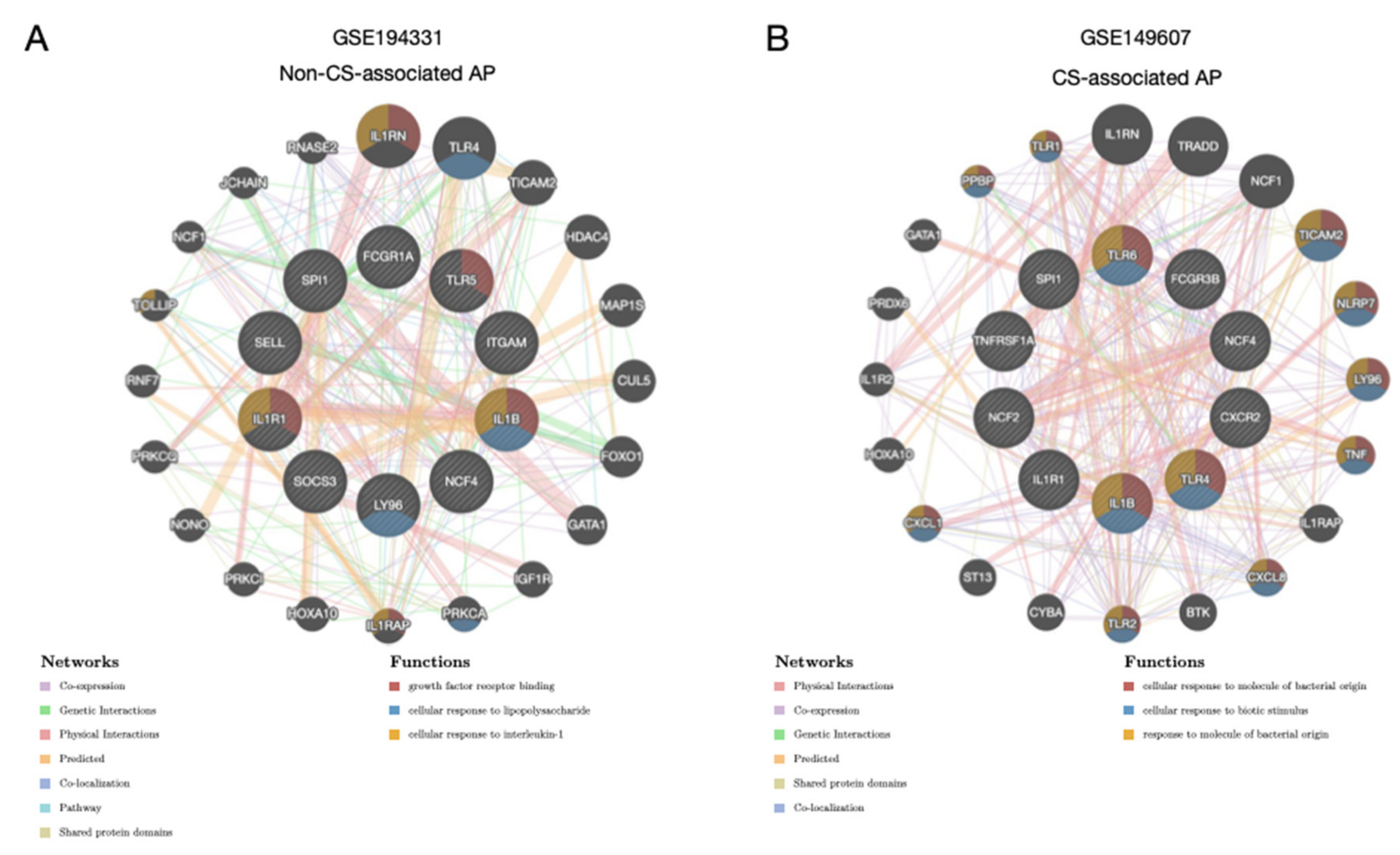
3.4. Regulatory Activity of the Hub Genes Was Different in Patients with CS
3.5. Correlation Profiles of Hub Genes in Patients with CS- and Non-CS-Associated AP
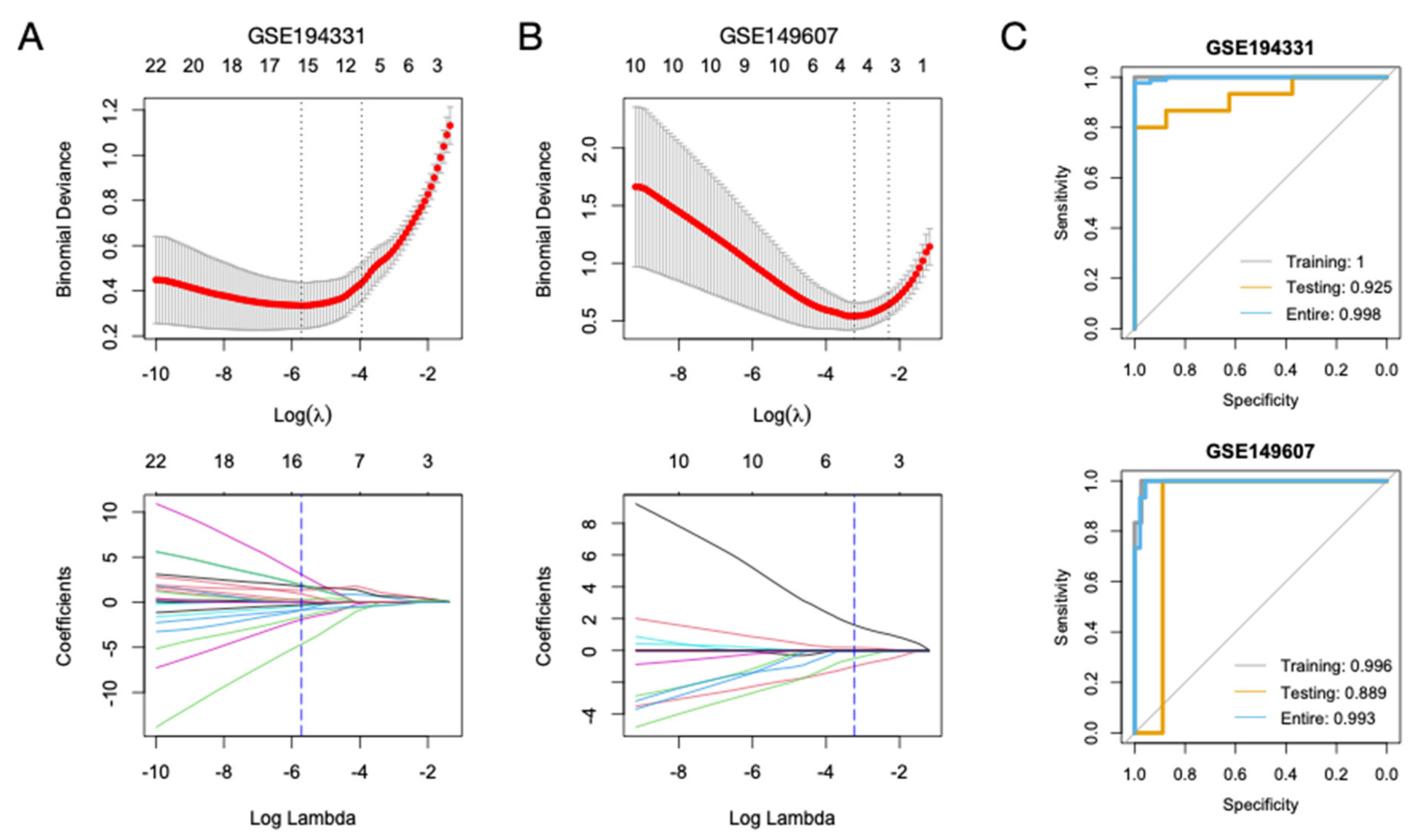
3.6. Diagnostic Models for CS- and Non-CS-Associated AP Based on Hub Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peery, A.F.; Crockett, S.D.; Murphy, C.C.; Lund, J.L.; Dellon, E.S.; Williams, J.L.; Jensen, E.T.; Shaheen, N.J.; Barritt, A.S.; Lieber, S.R.; et al. Burden and Cost of Gastrointestinal, Liver, and Pancreatic Diseases in the United States: Update 2018. Gastroenterology 2019, 156, 254–272.e11. [Google Scholar] [CrossRef] [PubMed]
- Forsmark, C.E.; Baillie, J. AGA Institute Technical Review on Acute Pancreatitis. Gastroenterology 2007, 132, 2022–2044. [Google Scholar] [CrossRef]
- Yang, A.L.; Vadhavkar, S.; Singh, G.; Omary, M.B. Epidemiology of Alcohol-Related Liver and Pancreatic Disease in the United States. Arch. Intern. Med. 2008, 168, 649–656. [Google Scholar] [CrossRef]
- Rosendahl, J.; Witt, H.; Szmola, R.; Bhatia, E.; Ózsvári, B.; Landt, O.; Schulz, H.-U.; Gress, T.M.; Pfützer, R.; Löhr, M.; et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 2007, 40, 78–82. [Google Scholar] [CrossRef]
- Kahaleh, M.; Freeman, M. Prevention and Management of Post-Endoscopic Retrograde Cholangiopancreatography Complications. Clin. Endosc. 2012, 45, 305–312. [Google Scholar] [CrossRef]
- Nawaz, H.; Koutroumpakis, E.; Easler, J.; Slivka, A.; Whitcomb, D.C.; Singh, V.P.; Yadav, D.; I Papachristou, G. Elevated Serum Triglycerides are Independently Associated With Persistent Organ Failure in Acute Pancreatitis. Am. J. Gastroenterol. 2015, 110, 1497–1503. [Google Scholar] [CrossRef]
- Simons-Linares, C.R.; Elkhouly, M.A.; Salazar, M.J. Drug-Induced Acute Pancreatitis in Adults. Pancreas 2019, 48, 1263–1273. [Google Scholar] [CrossRef]
- Olesen, S.S.; Harakow, A.; Krogh, K.; Drewes, A.M.; Handberg, A.; Christensen, P.A. Hypertriglyceridemia is often under recognized as an aetiologic risk factor for acute pancreatitis: A population-based cohort study. Pancreatology 2021, 21, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Yadav, D.; Pitchumoni, C.S. Association of Diabetic Ketoacidosis and Acute Pancreatitis: Observations in 100 Consecutive Episodes of Dka. Am. J. Gastroenterol. 2000, 95, 2795–2800. [Google Scholar] [CrossRef] [PubMed]
- Rivellese, A.A.; De Natale, C.; Di Marino, L.; Patti, L.; Iovine, C.; Coppola, S.; Del Prato, S.; Riccardi, G.; Annuzzi, G. Exogenous and Endogenous Postprandial Lipid Abnormalities in Type 2 Diabetic Patients with Optimal Blood Glucose Control and Optimal Fasting Triglyceride Levels. J. Clin. Endocrinol. Metab. 2004, 89, 2153–2159. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, R.B.; Chait, A. A Comprehensive Update on the Chylomicronemia Syndrome. Front. Endocrinol. 2020, 11, 593931. [Google Scholar] [CrossRef]
- Vipperla, K.; Somerville, C.; Furlan, A.; Koutroumpakis, E.; Saul, M.; Chennat, J.; Rabinovitz, M.; Whitcomb, D.C.; Slivka, A.; Papachristou, G.I.; et al. Clinical Profile and Natural Course in a Large Cohort of Patients With Hypertriglyceridemia and Pancreatitis. J. Clin. Gastroenterol. 2017, 51, 77–85. [Google Scholar] [CrossRef]
- Brahm, A.J.; Hegele, R.A. Chylomicronaemia—current diagnosis and future therapies. Nat. Rev. Endocrinol. 2015, 11, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.L.; McNabb-Baltar, J. Hypertriglyceridemia and acute pancreatitis. Pancreatology 2020, 20, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Phillip, V.J.V.; Steiner, J.M.H.A.R.M.; Algül, H. Early phase of acute pancreatitis: Assessment and management. World J. Gastrointest. Pathophysiol. 2014, 5, 158–168. [Google Scholar] [CrossRef]
- Chaussabel, D.; Pascual, V.; Banchereau, J. Assessing the human immune system through blood transcriptomics. BMC Biol. 2010, 8, 84. [Google Scholar] [CrossRef]
- Meher, S.; Mishra, T.S.; Sasmal, P.K.; Rath, S.; Sharma, R.; Rout, B.; Sahu, M.K. Role of Biomarkers in Diagnosis and Prognostic Evaluation of Acute Pancreatitis. J. Biomarkers 2015, 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-W.; Dai, S.-J.; Kong, H.-R.; Fan, J.-X.; Yang, F.-Y.; Dai, J.-Q.; Jin, Y.-P.; Yu, G.-Z.; Chen, B.-C.; Shi, K.-Q. Accurate prediction of acute pancreatitis severity based on genome-wide cell free DNA methylation profiles. Clin. Epigenetics 2021, 13, 1–12. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, M.; Zhang, Y.; Xia, C.; Peng, L.; Li, K.; Yin, H.; Li, S.; Yang, X.; Su, X.; et al. Novel insight on marker genes and pathogenic peripheral neutrophil subtypes in acute pancreatitis. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Keller, A.; Leidinger, P.; Bauer, A.; ElSharawy, A.; Haas, J.; Backes, C.; Wendschlag, A.; Giese, N.; Tjaden, C.; Ott, K.; et al. Toward the blood-borne miRNome of human diseases. Nat. Methods 2011, 8, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, K.; Gaudet, D.; Khoury, E.; Brisson, D. Dissection of Clinical and Gene Expression Signatures of Familial versus Multifactorial Chylomicronemia. J. Endocr. Soc. 2020, 4, bvaa056. [Google Scholar] [CrossRef] [PubMed]
- Nesvaderani, M.M.; Dhillon, B.K.; Chew, T.; Tang, B.F.; Baghela, A.; Hancock, R.E.O.; Eslick, G.D.F.; Cox, M.F. Gene Expression Profiling: Identification of Novel Pathways and Potential Biomarkers in Severe Acute Pancreatitis. J. Am. Coll. Surg. 2022, 234, 803–815. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef]
- Xiang, H.; Tao, X.; Xia, S.; Qu, J.; Song, H.; Liu, J.; Shang, D. Targeting MicroRNA Function in Acute Pancreatitis. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Sánchez, A.; Cast, O.; Miller, M.L. Comprehensive Benchmarking and Integration of Tumor Microenvironment Cell Estimation Methods. Cancer Res 2019, 79, 6238–6246. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Danaher, P.; Warren, S.; Dennis, L.; D’Amico, L.; White, A.; Disis, M.L.; Geller, M.A.; Odunsi, K.; Beechem, J.; Fling, S.P. Gene expression markers of Tumor Infiltrating Leukocytes. J. Immunother. Cancer 2017, 5, 18. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Takenaka, T.; Kawabori, S.; Ogami, Y. Ultrastructural study of the specific granule of the human eosinophil. . J. Submicrosc. Cytol. 1981, 13. [Google Scholar]
- Almkvist, J.; Fäldt, J.; Dahlgren, C.; Leffler, H.; Karlsson, A. Lipopolysaccharide-Induced Gelatinase Granule Mobilization Primes Neutrophils for Activation by Galectin-3 and Formylmethionyl-Leu-Phe. Infect. Immun. 2001, 69, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.-C.; Girard, D.; Tessier, P.A. Induction of neutrophil degranulation by S100A9 via a MAPK-dependent mechanism. J. Leukoc. Biol. 2010, 87, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Radom-Aizik, S.; Zaldivar, F.; Oliver, S.; Galassetti, P.; Cooper, D.M. Evidence for microRNA involvement in exercise-associated neutrophil gene expression changes. J. Appl. Physiol. 2010, 109, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liang, Y.; Han, H.; Qin, H. Identification of a miRNA signature in neutrophils after traumatic injury. Acta Biochim. et Biophys. Sin. 2013, 45, 938–945. [Google Scholar] [CrossRef]
- Meisgen, F.; Landén, N.X.; Wang, A.; Réthi, B.; Bouez, C.; Zuccolo, M.; Gueniche, A.; Ståhle, M.; Sonkoly, E.; Breton, L.; et al. MiR-146a Negatively Regulates TLR2-Induced Inflammatory Responses in Keratinocytes. J. Investig. Dermatol. 2014, 134, 1931–1940. [Google Scholar] [CrossRef]
- Hutcheson, R.; Terry, R.; Hutcheson, B.; Jadhav, R.; Chaplin, J.; Smith, E.; Barrington, R.; Proctor, S.D.; Rocic, P. miR-21-mediated decreased neutrophil apoptosis is a determinant of impaired coronary collateral growth in metabolic syndrome. . Am. J. Physiol. Circ. Physiol. 2015, 308, H1323–H1335. [Google Scholar] [CrossRef]
- Arroyo, A.B.; Fernández-Pérez, M.P.; del Monte, A.; Águila, S.; Méndez, R.; Hernández-Antolín, R.; García-Barber, N.; Reyes-García, A.M.D.L.; González-Jiménez, P.; Arcas, M.I.; et al. miR-146a is a pivotal regulator of neutrophil extracellular trap formation promoting thrombosis. Haematologica 2020, 106, 1636–1646. [Google Scholar] [CrossRef]
- Hsieh, Y.-T.; Chou, Y.-C.; Kuo, P.-Y.; Tsai, H.-W.; Yen, Y.-T.; Shiau, A.-L.; Wang, C.-R. Down-regulated miR-146a expression with increased neutrophil extracellular traps and apoptosis formation in autoimmune-mediated diffuse alveolar hemorrhage. J. Biomed. Sci. 2022, 29, 1–20. [Google Scholar] [CrossRef]
- Weitz, G.; Woitalla, J.; Wellhöner, P.; Schmidt, K.; Büning, J.; Fellermann, K. Does Etiology of Acute Pancreatitis Matter? A Review of 391 Consecutive Episodes. JOP. J. Pancreas 2015, 16, 171–175. [Google Scholar] [CrossRef]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S.; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Al-Bahrani, A.Z.; Ammori, B.J. Clinical laboratory assessment of acute pancreatitis. Clin. Chim. Acta 2005, 362, 26–48. [Google Scholar] [CrossRef]
- Sigounas, D.E.; Tatsioni, A.; Christodoulou, D.K.; Tsianos, E.V.; Ioannidis, J.P.A. New Prognostic Markers for Outcome of Acute Pancreatitis. Pancreas 2011, 40, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Rau, B.M.; Kruger, C.M.; Schilling, M.K. Anti-cytokine strategies in acute pancreatitis: Pathophysiological in-sights and clinical implications. Rocz. Akad. Med. Bialymst. 2005, 50, 106–115. [Google Scholar]
- Bhatia, M.; Brady, M.; Shokuhi, S.; Christmas, S.; Neoptolemos, J.P.; Slavin, J. Inflammatory mediators in acute pancreatitis. J. Pathol. 2000, 190, 117–125. [Google Scholar] [CrossRef]
- Bhatia, M.; Neoptolemos, J.; Slavin, J. Inflammatory mediators as therapeutic targets in acute pancreatitis. Curr. Opin. Investig. Drugs 2001, 2. [Google Scholar]
- Bhatia, M. Novel Therapeutic Targets for Acute Pancreatitis and Associated Multiple Organ Dysfunction Syndrome. Curr. Drug Targets Inflamm. Allergy 2002, 1, 343–351. [Google Scholar] [CrossRef]
- Bhatia, M.; Wong, F.L.; Cao, Y.; Lau, H.Y.; Huang, J.; Puneet, P.; Chevali, L. Pathophysiology of acute pancreatitis. Pancreatology 2005, 5, 132–144. [Google Scholar] [CrossRef]
- Brady, M.; Bhatia, M.; Christmas, S.; Boyd, M.T.; Neoptolemos, J.; Slavin, J. Expression of the Chemokines MCP-1/JE and Cytokine-Induced Neutrophil Chemoattractant in Early Acute Pancreatitis. Pancreas 2002, 25, 260–269. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, P.; Röckel, M.; Nees, M.; Röder, C.; Kienle, P.; Doeberitz, M.V.K.; Kalthoff, H.; Neumaier, M. Systematic identification and validation of candidate genes for detection of circulating tumor cells in peripheral blood specimens of colorectal cancer patients. Int. J. Oncol. 1992, 33, 1001–1010. [Google Scholar] [CrossRef]
- Pankla, R.; Buddhisa, S.; Berry, M.; Blankenship, D.M.; Bancroft, G.J.; Banchereau, J.; Lertmemongkolchai, G.; Chaussabel, D. Genomic transcriptional profiling identifies a candidate blood biomarker signature for the diagnosis of septicemic melioidosis. Genome Biol. 2009, 10, R127. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, M.; Repsilber, D.; Gutschmidt, A.; Neher, A.; Feldmann, K.; Mollenkopf, H.J.; Ziegler, A.; Kaufmann, S.H.E. Candidate biomarkers for discrimination between infection and disease caused by Mycobacterium tuberculosis. J. Mol. Med. 2007, 85, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.P.; McLean, A.S.; Dawes, I.W.; Huang, S.J.; Lin, R.C.Y. Gene-expression profiling of peripheral blood mononuclear cells in sepsis*. Crit. Care Med. 2009, 37, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Gukovskaya, A.S.; Vaquero, E.; Zaninovic, V.; Gorelick, F.; Lusis, A.J.; Brennan, M.; Holland, S.; Pandol, S.J. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 2002, 122, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, A.; Awla, D.; Thorlacius, H.; Regnér, S. Role of neutrophils in the activation of trypsinogen in severe acute pancreatitis. J. Leukoc. Biol. 2011, 90, 975–982. [Google Scholar] [CrossRef]
- Oiva, J.; Mustonen, H.; Kylänpää, M.-L.; Kuuliala, K.; Siitonen, S.; Kemppainen, E.; Puolakkainen, P.; Repo, H. Patients with acute pancreatitis complicated by organ dysfunction show abnormal peripheral blood polymorphonuclear leukocyte signaling. Pancreatology 2013, 13, 118–124. [Google Scholar] [CrossRef]
- Paulino, E.C.; de Souza, L.J.; Molan, N.A.; Machado, M.C.; Jancar, S. Neutrophils From Acute Pancreatitis Patients Cause More Severe In Vitro Endothelial Damage Compared With Neutrophils From Healthy Donors and Are Differently Regulated by Endothelins. Pancreas 2007, 35, 37–41. [Google Scholar] [CrossRef]
- Panopoulos, A.; Zhang, L.; Snow, J.W.; Jones, D.M.; Smith, A.M.; El Kasmi, K.C.; Liu, F.; Goldsmith, M.A.; Link, D.C.; Murray, P.J.; et al. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood 2006, 108, 3682–3690. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Jackson, H.; Panopoulos, A.D.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 controls the neutrophil migratory response to CXCR2 ligands by direct activation of G-CSF–induced CXCR2 expression and via modulation of CXCR2 signal transduction. Blood 2010, 115, 3354–3363. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-W.; Zeng, H.-S. Regulation of JAK/STAT signal pathway by miR-21 in the pathogenesis of juvenile idiopathic arthritis. World J. Pediatr. 2019, 16, 502–513. [Google Scholar] [CrossRef]
- Tang, Y.; Luo, X.; Cui, H.; Ni, X.; Yuan, M.; Guo, Y.; Huang, X.; Zhou, H.; de Vries, N.; Tak, P.P.; et al. MicroRNA-146a contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009, 60, 1065–1075. [Google Scholar] [CrossRef]
- Wang, J.; Yan, Y.; Song, D.; Liu, B. Reduced Plasma miR-146a Is a Predictor of Poor Coronary Collateral Circulation in Patients with Coronary Artery Disease. BioMed Res. Int. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.-B.; Li, Z.-H.; Gao, S. Circulating miR-146a/b correlates with inflammatory cytokines in COPD and could predict the risk of acute exacerbation COPD. Medicine 2018, 97, e9820. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Hong, H.; Liu, J.; Zheng, X.; Huang, M.; Li, C.; Xia, J. miR-106a Is Downregulated in Peripheral Blood Mononuclear Cells of Chronic Hepatitis B and Associated with Enhanced Levels of Interleukin-8. Mediat. Inflamm. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- De Vries, M.A.; Klop, B.; Alipour, A.; van de Geijn, G.-J.M.; Prinzen, L.; Liem, A.H.; Valdivielso, P.; Villodres, J.R.; Ramírez-Bollero, J.; Cabezas, M.C. In vivo evidence for chylomicrons as mediators of postprandial inflammation. Atherosclerosis 2015, 243, 540–545. [Google Scholar] [CrossRef]
- Genkel, V.; Dolgushin, I.; Baturina, I.; Savochkina, A.; Kuznetsova, A.; Pykhova, L.; Shaposhnik, I. Associations between Hypertriglyceridemia and Circulating Neutrophil Subpopulation in Patients with Dyslipidemia. Int. J. Inflamm. 2021, 2021, 1–8. [Google Scholar] [CrossRef]
- Zhang, Y.; He, W.; He, C.; Wan, J.; Lin, X.; Zheng, X.; Li, L.; Li, X.; Yang, X.; Yu, B.; et al. Large triglyceride-rich lipoproteins in hypertriglyceridemia are associated with the severity of acute pancreatitis in experimental mice. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.-H.; Xue, P.; Xia, Q.; Yang, X.-N.; Wan, M.-H. Effect of admission hypertriglyceridemia on the episodes of severe acute pancreatitis. World J. Gastroenterol. 2008, 14, 4558–4561. [Google Scholar] [CrossRef]
- Arroyo, J.C.C.; Zavaleta, M.J.C.; Villasante, E.J.G.; Lam, M.K.; Urteaga, L.A.C.; Gutiérrez, F.E.Z. Familial Chylomicronemia Syndrome-Induced Acute Necrotizing Pancreatitis during Pregnancy. Rev. Bras. Ginecol. Obs. 2021, 43, 220–224. [Google Scholar] [CrossRef]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell. Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Westra, J.; Rutgers, A.; Der Meer, B.D.-V.; Huitema, M.G.; A Stegeman, C.; Abdulahad, W.H.; Satchell, S.C.; Mathieson, P.W.; Heeringa, P.; et al. Decreased CXCR1 and CXCR2 expression on neutrophils in anti-neutrophil cytoplasmic autoantibody-associated vasculitides potentially increases neutrophil adhesion and impairs migration. Thromb. Haemost. 2011, 13, R201. [Google Scholar] [CrossRef] [PubMed]
- Narrandes, S.; Xu, W. Gene Expression Detection Assay for Cancer Clinical Use. J. Cancer 2018, 9, 2249–2265. [Google Scholar] [CrossRef] [PubMed]
- Orgogozo, V.; Morizot, B.; Martin, A. The differential view of genotype–phenotype relationships. Front. Genet. 2015, 6, 179. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Nielsen, J.B.; Fritsche, L.G.; Dey, R.; Gabrielsen, M.E.; Wolford, B.N.; LeFaive, J.; VandeHaar, P.; Gagliano, S.A.; Gifford, A.; et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018, 50, 1335–1341. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNAs | Condition | Neutrophil Source | Literature | PMID |
|---|---|---|---|---|
| hsa-miR-106a | Down-regulated after acute exercise | Peripheral blood | Radom-Aizik et al. | [44] |
| hsa-miR-21 | Down-regulated after traumatic injury | Peripheral blood | Yang et al. | [45] |
| Down-regulation induces apoptosis in neutrophil | Peripheral blood | Hutcheson et al. | [46] | |
| hsa-miR-146a | Down-regulated in diffuse alveolar hemorrhage | Peripheral blood | Hsieh et al. | [47] |
| Down-regulation induces neutrophil extracellular traps | Peripheral blood | Arroyo et al. | [48] | |
| Up-regulation represses neutrophil attraction | Peripheral blood | Meisgen et al. | [49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.-L.; Dai, Y.-H. Bioinformatic Analyses of Peripheral Blood Transcriptome Identify Altered Neutrophil-Related Pathway and Different Transcriptomic Profiles for Acute Pancreatitis in Patients with and without Chylomicronemia Syndrome. Biomolecules 2023, 13, 284. https://doi.org/10.3390/biom13020284
Liu C-L, Dai Y-H. Bioinformatic Analyses of Peripheral Blood Transcriptome Identify Altered Neutrophil-Related Pathway and Different Transcriptomic Profiles for Acute Pancreatitis in Patients with and without Chylomicronemia Syndrome. Biomolecules. 2023; 13(2):284. https://doi.org/10.3390/biom13020284
Chicago/Turabian StyleLiu, Chia-Lun, and Yang-Hong Dai. 2023. "Bioinformatic Analyses of Peripheral Blood Transcriptome Identify Altered Neutrophil-Related Pathway and Different Transcriptomic Profiles for Acute Pancreatitis in Patients with and without Chylomicronemia Syndrome" Biomolecules 13, no. 2: 284. https://doi.org/10.3390/biom13020284