
Developmental Inhibitory Changes in the Primary Somatosensory Cortex of the Stargazer Mouse Model of Absence Epilepsy


Department of Anatomy, School of Biomedical Sciences, Brain Health Research Centre, University of Otago, P.O. Box 913, Dunedin 9054, New Zealand
*
Author to whom correspondence should be addressed.
Biomolecules 2023, 13(1), 186; https://doi.org/10.3390/biom13010186
Submission received: 18 December 2022
/
Revised: 8 January 2023
/
Accepted: 11 January 2023
/
Published: 16 January 2023
(This article belongs to the Special Issue GABA Receptors in Pharmacology and Neurobiology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Childhood absence epilepsy seizures arise in the cortico-thalamocortical network due to multiple cellular and molecular mechanisms, which are still under investigation. Understanding the precise mechanisms is imperative given that treatment fails in ~30% of patients while adverse neurological sequelae remain common. Impaired GABAergic neurotransmission is commonly reported in research models investigating these mechanisms. Recently, we reported a region-specific reduction in the whole-tissue and synaptic GABAA receptor (GABAAR) α1 subunit and an increase in whole-tissue GAD65 in the primary somatosensory cortex (SoCx) of the adult epileptic stargazer mouse compared with its non-epileptic (NE) littermate. The current study investigated whether these changes occurred prior to the onset of seizures on postnatal days (PN) 17–18, suggesting a causative role. Synaptic and cytosolic fractions were biochemically isolated from primary SoCx lysates followed by semiquantitative Western blot analyses for GABAAR α1 and GAD65. We found no significant changes in synaptic GABAAR α1 and cytosolic GAD65 in the primary SoCx of the stargazer mice at the critical developmental stages of PN 7–9, 13–15, and 17–18. This indicates that altered levels of GABAAR α1 and GAD65 in adult mice do not directly contribute to the initial onset of absence seizures but are a later consequence of seizure activity.
1. Introduction
A developing perinatal brain is highly vulnerable to genetic, neurological, mental, and behavioral disorders due to the immense rapid structural and functional changes at critical stages [1,2,3,4,5,6,7]. Evidence indicates that this developing brain is also more susceptible to seizures than that of an adult [8,9,10]. Indeed, childhood absence epilepsy (CAE) develops early on during childhood, with 3–8 years as the peak age of onset. CAE is a genetic generalized epileptic syndrome, characterized by absence seizures clinically detected as 2.5–4 Hz spike–wave discharges (SWDs). These seizures are believed to arise in the cortico-thalamocortical (CTC) network due to complex underlying mechanisms. The network comprises the cortex and ventroposterior (VP) relay thalamus reciprocally connected via excitatory projections and modulated via inhibitory interneurons in the cortex and reticular thalamic nucleus (RTN) through feedforward and feedback inhibition. Normal development ensures the important excitatory/inhibitory (E/I) balance within the network, disruption of which predisposes individuals towards the development of seizures.
The rapid development of the human brain continues during the first few postnatal years [11,12,13,14], during which thalamocortical relay activity is crucial for the development of cortical networks [15]. The association cortical areas are the fastest growing regions during this period, making them the most vulnerable as well [16,17]. The complexity of cortical neurons rapidly increases, reaching a peak at 2–4 years [18]. Given the time period of absence seizures onset in CAE, an inevitable relationship may exist between an abnormal perinatal development of the CTC network and the pathogenesis of absence seizures [19,20].
Interestingly, the common rodent models for absence seizures used to study human absence epilepsy also suggest a causative correlation with cellular and molecular changes in postnatal brain development, given the onset of seizures in later postnatal stages [21]. In the stargazer mouse model for absence epilepsy used in the current study, seizures were detected at PN 17–18 [22], which, in terms of the brain development time course in mice, is roughly equivalent to the time of onset in humans [8,22,23]. The primary genetic defect of the stargazin gene in the mouse leads to the reduced expression and function of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in various brain regions with evidence of downstream changes in GABAergic neurotransmission [24,25,26,27,28,29,30,31,32]. We have previously reported that such loss of AMPA receptors occurs both in post-seizure adults and young postnatal stargazers prior to the age of seizure onset [33,34,35]. Recently, we reported a reduced expression of the synaptic GABAA receptor (GABAAR) α1 subunit [36], an increase in global whole-tissue GABA neurotransmitter content but reduced GABA levels in individual inhibitory synaptic terminals [37], and an increase in global whole-tissue GAD65 but not GAD67 in adult stargazer primary SoCx, which was suggestive of altered inhibition [38].
Given that we have found no change in the number of GABA-positive neurons and immunoreactive profiles [39], one implication of the higher tissue levels of GABA despite lower levels within terminals is a persistent increase in GABA in the extracellular space. Such an increase has previously been linked to absence seizure generation [40,41]. The inference of increased extracellular GABA is also substantiated by the increased GAD65, but not GAD67, in adult stargazer primary SoCx [38]. GAD65 is an enzyme with a preferential localization in the axon terminals for on-demand GABA synthesis and release [42]. Its activity is crucial during intense synaptic activity [43]. Since GADs are the rate-limiting enzyme in the synthesis of GABA [44], there appears to be a good correlation between the demand for GABA and GAD65 levels in the neuronal tissue. Hence, GAD65 is an acceptable representative enzyme for GABA levels in stargazer primary SoCx. Given that it is unknown whether the altered expressions of synaptic GABAAR α1, the GABA neurotransmitter, and GAD65 in adult stargazer primary SoCx are a potential cause or a downstream consequence of absence seizures, the current study investigated changes in synaptic GABAAR α1 and GAD65 at critical postnatal stages prior to the onset of seizures.
Most studies describing developmental changes in rodent GABAergic neurotransmission have been conducted in rats. GABA and GABAAR’s developmental patterns are crucial for the physiological regulation of the rapidly proliferating and migrating neural and glial progenitors [45,46]. GABAergic neurotransmission during early rodent perinatal development is excitatory and crucial for normal neural development [47], with the switch to inhibitory neurotransmission in the CTC network occurring after the first postnatal week [47]. The GABA neurotransmitter is present in the neural system perinatally [45], with its synthesis being dependent on the GAD65 and GAD67 enzymes. GAD67, which is present during birth, achieves adult expression by PN 13, while GAD65 shows a prolonged increase from PN 6 into adulthood [48,49]. Any disruptions in GABAergic neurotransmission during these stages adversely affect normal development. For instance, the GAD65 knock-out mouse model, which displays a high susceptibility to seizures, shows normal cerebral GABA content at birth attributed to normal GAD67, but the GABA levels do not increase significantly after birth, increasing the brain’s susceptibility to seizures. This is unlike normal wild-type mice in which GABA content increases significantly and rapidly as the brain develops [50,51].
Similarly, GABAARs, which are expressed during embryonic stages [52,53,54], show prolonged postnatal changes in subunit expression, which continues well into adulthood. For instance, GABAAR α1 shows a steady increase in expression into the adult stages, while α2 and α3, which are abundant in the rodent neonatal brain, show a steady parallel decrease in the rat brain [49,55,56,57,58] due to a developmental switch. A similar developmental switch from α2 and α3 to α1 has also been reported in the mouse brain [59,60]. The type of α subunit expressed in a GABAAR affects its physiological properties. Naturally, any disruption in the normal composition of GABAARs can disrupt the physiological E/I balance. Interestingly, a recent publication reported impairment of the brain’s inhibitory networks and development of seizures due to GABAAR α1 loss of function in a juvenile zebrafish model [61].
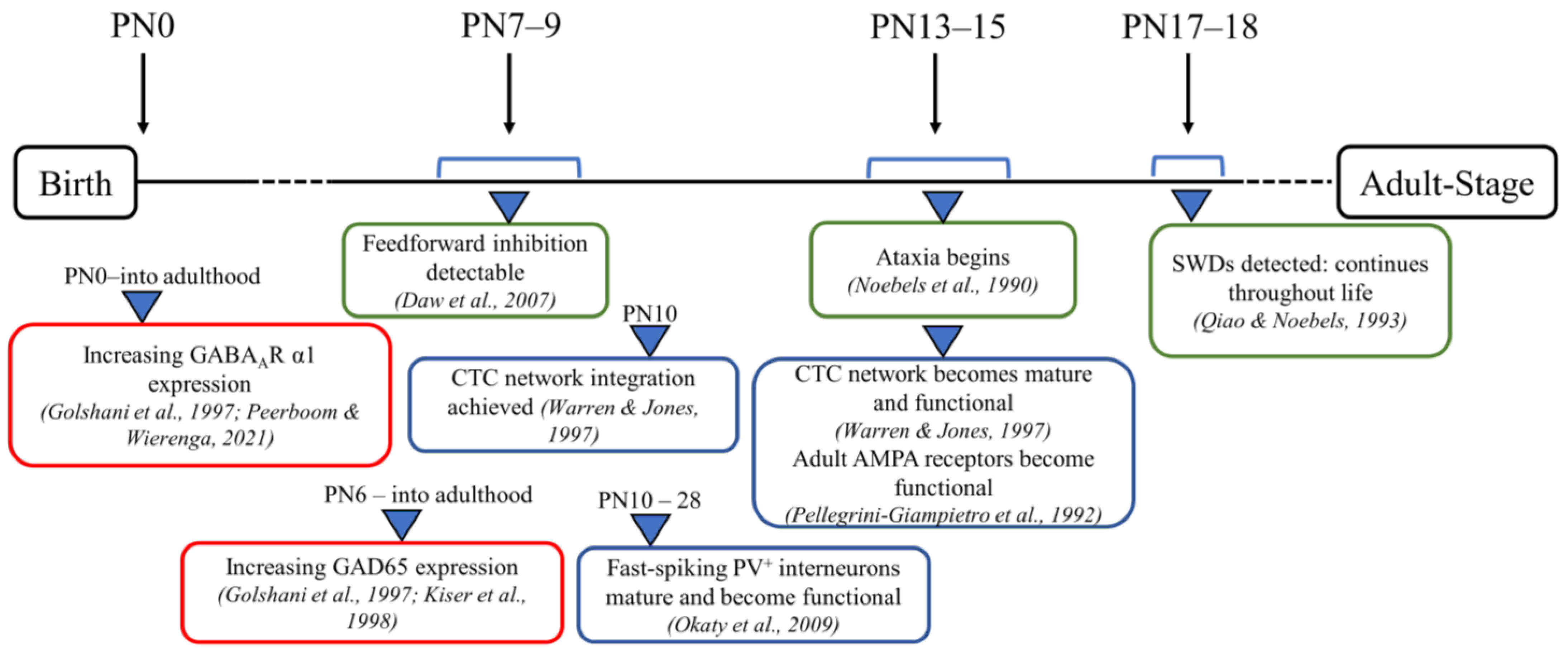
It is conceivable that the GABAergic changes observed in adult stargazer primary SoCx could have an onset at critical time points during the development of the CTC network prior to the onset of seizures that could contribute to seizure generation. Hence, the current study was conducted to test our hypothesis that modified expression of the stargazin gene leads to downstream changes in synaptic GABAAR subunit α1 and GAD65 that take effect during the developmental stages of a stargazer’s neuronal development. To assess this, we selected three developmental stages in juveniles (Figure 1). At PN 7–9, feed-forward inhibition is detectable, while GABAergic transmission switches from excitatory to inhibitory, and the GABAAR subunit switch to α1 has occurred [47,58,62]. At PN 13–15, adult AMPA receptors become functionally active, and the CTC network is operational [63,64]. At PN 17–18, seizures are first detected in stargazers [65]. Biochemical fractionation was first utilized to isolate the synaptic and cytosolic subcellular components of the primary SoCx from NE control littermates and stargazers at the three juvenile stages followed by probing for synaptic GABAAR subunit α1 and cytosolic GAD65.
2. Materials and Methods
2.1. Animals
Male epileptic (E) stargazers (stg/stg) and their male non-epileptic (NE: heterozygous [Het, +/stg] and wild-type [WT, +/+]) control littermates were used in all experiments. These offspring were obtained from stargazer (B6C3Fe a/a-Cacng2stg/J) breeding stocks obtained from the Jackson Laboratory (Bar Harbor, ME, USA). All animals used in the study were raised at the University of Otago’s Animal Resource Unit. The mice had ad libitum access to food and were housed in well-ventilated cages with optimum environmental conditions (12 h light/dark cycle, ~21 °C temperature, and ~50% humidity). All animal procedures were carried out according to the University of Otago Animal Ethics Committee approved protocols (32/17). Animals selected were juvenile pups from 3 age groups, PN 7–9, 13–15, and 17–18, marking 3 stages of neuronal development in mice. After obtaining ear-notches post-sacrifice, genotypes were confirmed with PCR-amplified DNA using the primer sequences oIMR9601 (TAC TTC ATC CGC CAT CCT TC), oIMR9602 (TGG CTT TCA CTG TCT GTT GC), and oIMR8983 (GAG CAA GCA GGT TTC AGG C). A total of 82 animals (41 NE control littermates and 41 epileptic stargazers) were used for the current study with n numbers representing the sample numbers used. Subcellular fractions from PN 7–9 brains were pooled based on the same age and genotype to increase the protein content. The GAD65 group n numbers are one less for PN 13–15 and 17–18 because of a lack of enough protein in the excluded samples.
2.2. Biochemical Fractionation
Juvenile mice were weaned before being brought in for decapitation, followed immediately by brain extraction and snap-freezing on dry ice. The brains were coronally sectioned in a rostral-to-caudal direction at 200 μm thickness using a vibratome (Leica VT1200, Nussloch, Germany). Sections obtained were thaw-mounted on glass slides before micro-dissecting the full depth of the primary SoCx region. Tissue samples were homogenized in a fractionation buffer (320 mM sucrose, 10 mM tris-base, and 1 mM EDTA; pH 7.37) and supplemented with 1% PMSF (phenylmethyl sulfonyl fluoride) and a 1% protease inhibitor (P8340, Sigma-Aldrich, St. Louis, MO, USA), using clean sterilized plastic pestles coupled with ultrasonication. The subcellular components (total lysate, cytosol, and extra-synaptic and synaptic fractions) were obtained by taking each of the homogenized tissue samples through a previously established multi-step centrifugation fractionation technique [34,67,68]. Centrifugation at 1000× g for 10 min pelleted unhomogenized debris and nuclei. The supernatant (total lysate) was centrifuged for 15 min at 10,000× g, pelleting the cell membrane and yielding the cytosol fraction as a supernatant. The cell membrane pellet was resuspended in an ice-cold homogenization buffer (50 mM Tris, 2 nM EDTA, and 3% SDS; pH 6.8), supplemented with 0.5% Triton X-100, incubated for 40 min on ice, and centrifuged at 16,000× g for 30 min at 4 °C. This yielded a pelleted synaptic fraction due to its insolubility in Triton X-100 with a supernatant containing the extra-synaptic fraction. The synaptic fraction was resuspended in the homogenization buffer, while acetone was added to the extra-synaptic fraction and incubated overnight at −20 °C followed by centrifugation at 3000× g for 15 min. The resulting extra-synaptic pellet was resuspended in the homogenization buffer.
2.3. Western Blotting of Subcellular Fractions
The protein content of each sample was assessed using a detergent-compatible protein assay (DC Protein Assay, 500, 0116, Bio-Rad, Hercules, CA, USA). Western blot analyses were carried out as per previously established protocols [33,34]. In brief, the proteins were separated in 8.5% resolving gels using SDS-PAGE. After transferring them onto nitrocellulose membranes, the protein expression was probed using relevant antibodies. The synaptic component was probed for the principal protein of interest GABAAR α1 (1:500; AGA-001, Alomone, Jerusalem, Israel) and marker proteins with PanC (1:1000; 4068P, Cell Signaling Technology, Danver, MA, USA) for normalization and analyses, and PSD-95 (1:1000; 124011, Synaptic Systems, Goettingen, Germany) and β-actin (1:1000; ab8226, Abcam, Cambridge, UK) were used as additional controls. The cytosol component was probed for the principal protein of interest GAD65 (1:1000, ab26113, Abcam, Cambridge, UK) and the marker proteins. Although this study was focused on the synaptic and cytosol components, all subcellular components were taken through Western blot runs to establish the specificities of the fractions. Protein standard (Novex Sharp Pre-stained Protein Standard, Life Technologies, LC5800) was added to each Western blot run for molecular weight reference. Protein expression was detected using secondary antibodies IRDye 800CW (1:10,000; 926-3221, LI-COR Biosciences, Lincoln, NE, USA) and IRDye 680 (1:10,000; 926-32210, LI-COR Biosciences, Lincoln, USA). An Odyssey infrared imager (Li-COR Biosciences, Lincoln, NE, USA) was used to detect the immunofluorescence, while Odyssey v3.1 (Li-COR Biosciences, Lincoln, NE, USA) software was used to estimate the intensities of each band. The intensities of the protein of interest were normalized against PanC followed by a statistical analysis comparing stargazers with their NE control littermates.
2.4. Statistical Tests
A non-parametric Kruskal–Wallis one-way ANOVA test followed by a post hoc Dunn’s multiple comparison test for pairwise comparison between the groups were used to probe the data for developmental changes across the three age groups in NE control littermates and stargazers. An unpaired non-parametric Mann–Whitney U test was used for the data analysis and comparison between the two independent groups of NE control littermates and stargazers from the same population. The independent variable between the two groups was the existence of a genetic mutation-based phenotype in the stargazers and the lack thereof in the control group. The data obtained from each experiment conducted represent dependent variables. The statistical comparison between the NE control littermates and stargazers was conducted in GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA). Significance was defined as p < 0.05 as *; p < 0.01 as **; and p < 0.001 as ***. The data are presented as means ± standard error of the mean (SEM).
3. Results
3.1. Biochemical Fractionation Technique Verification
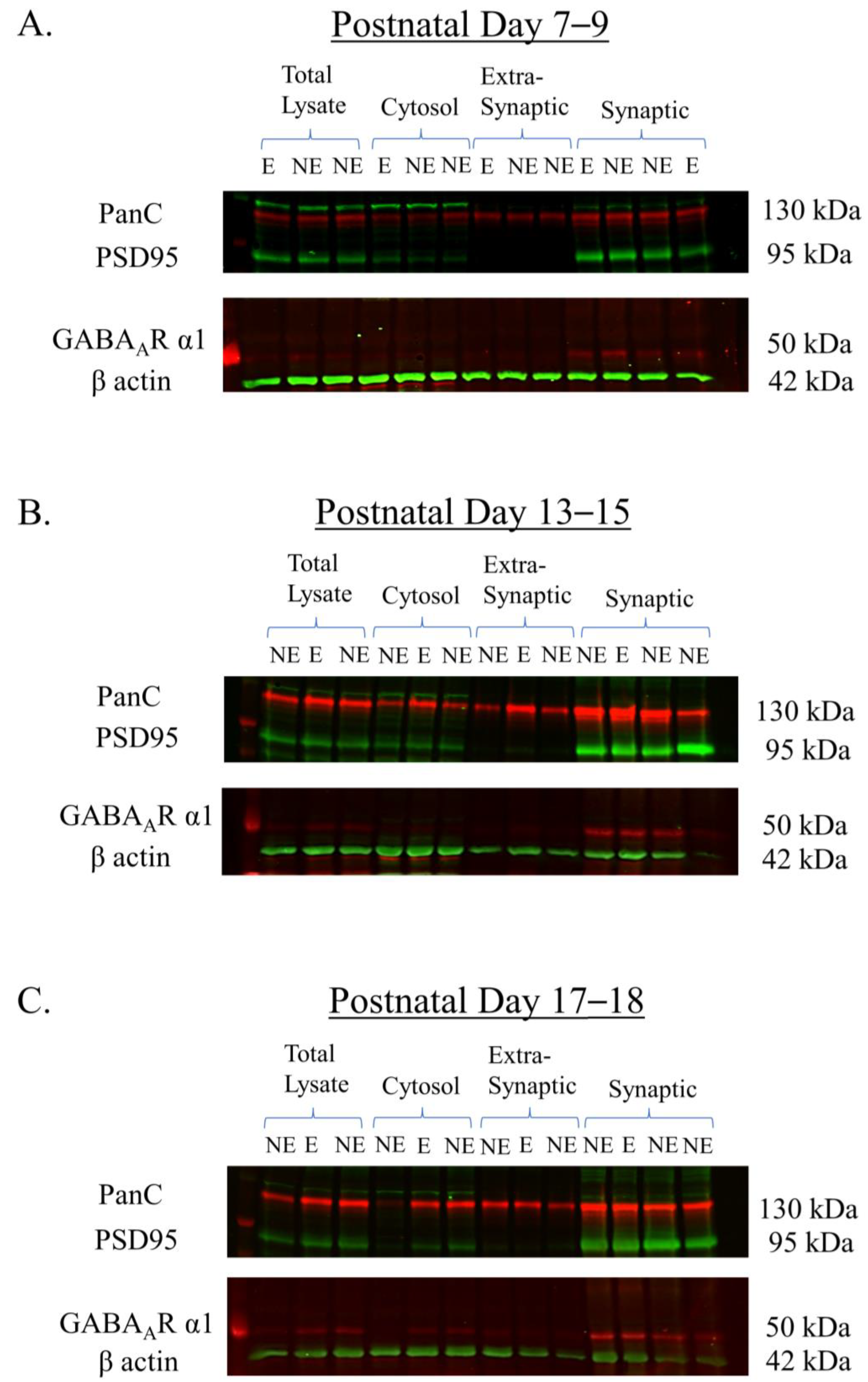
A pilot Western blotting study was first conducted to validate the purity of the subcellular fractions derived from the primary SoCx using biochemical fractionation in the 3 juvenile age groups PN 7–9, 13–15, and 17–18. The proteins on the Western blots were probed using antibodies for the PanC, PSD95, and β-actin marker proteins, and GABAAR α1. Figure 2 shows the representative Western blots with immuno-positive fluorescence bands detected at the manufacturers’ specified molecular weights as follows: PanC at ~130 kDa, PSD95 at 95 kDa, GABAAR α1 at ~50 kDa, and β-actin at 42 kDa. PanC, a transmembrane glycoprotein, was detected in all fractions [69] and was used as the normalization protein because its expression pattern is not dependent on the stargazin gene and its expression remains unaltered between NE control littermates and stargazers, as previously reported in [68,70]. PSD95 is a component of the post-synaptic density [71,72], and, as expected, showed intense enrichment in the synaptic fraction with no bands seen in the extra-synaptic fraction, demonstrating the successful isolation of the synaptic subcellular component due to its insolubility in the non-ionic detergent Triton X-100. β-actin was seen in all fractions but was not used for the normalization analysis because of its inconsistent expression in membranous components as well as its association with other membranous proteins [34,70,73]. GABAAR α1 displayed enrichment in synaptic fractions, which was expected given its predominant expression at GABAergic synapses. Conversely, extra-synaptic fractions displayed no visible bands. GABAAR α1 also showed faint bands in the total lysate and cytosolic fractions, given that this protein is synthesized in the cytoplasm.
3.2. Synaptic GABAAR α1 Is Unaltered in the Stargazer Primary SoCx
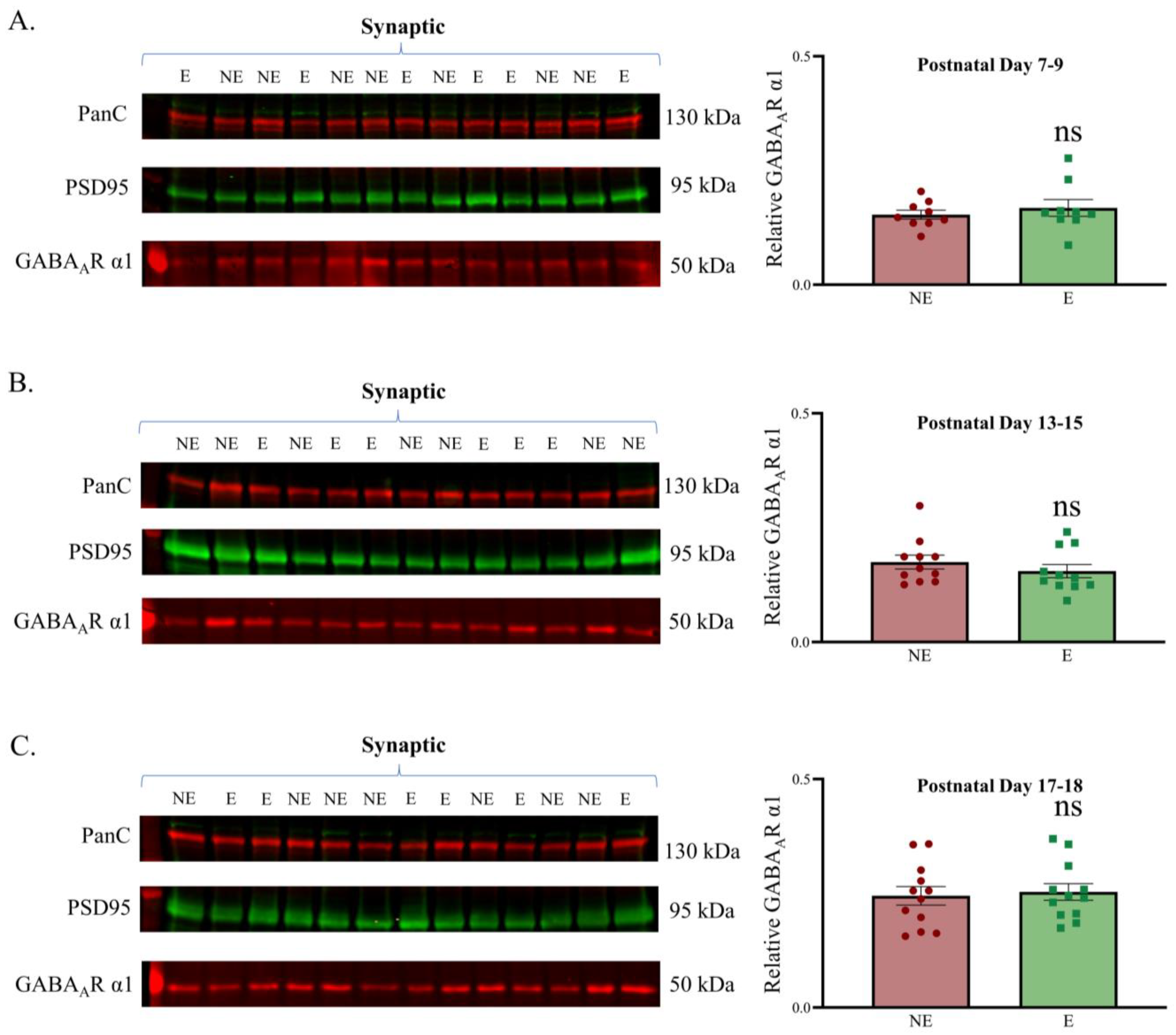
Having established the purity of subcellular fractions, we next assessed the levels of synaptic GABAAR α1 in the juvenile stargazer primary SoCx compared with their NE counterparts before the onset of seizures by probing Western blots containing synaptic fractions with GABAAR α1 antibodies. GABAAR α1 showed good enrichment in the synaptic fractions. The analyses demonstrated that synaptic GABAAR α1 expression in the stargazer primary SoCx was not significantly different from that in the NE control littermates at each development time point (Figure 3). Representative original Western blots for all subcellular fractions are provided in Supplementary Figure S1 (PN 7–9), Supplementary Figure S2 (PN 13–15), and Supplementary Figure S3 (PN 17–18).
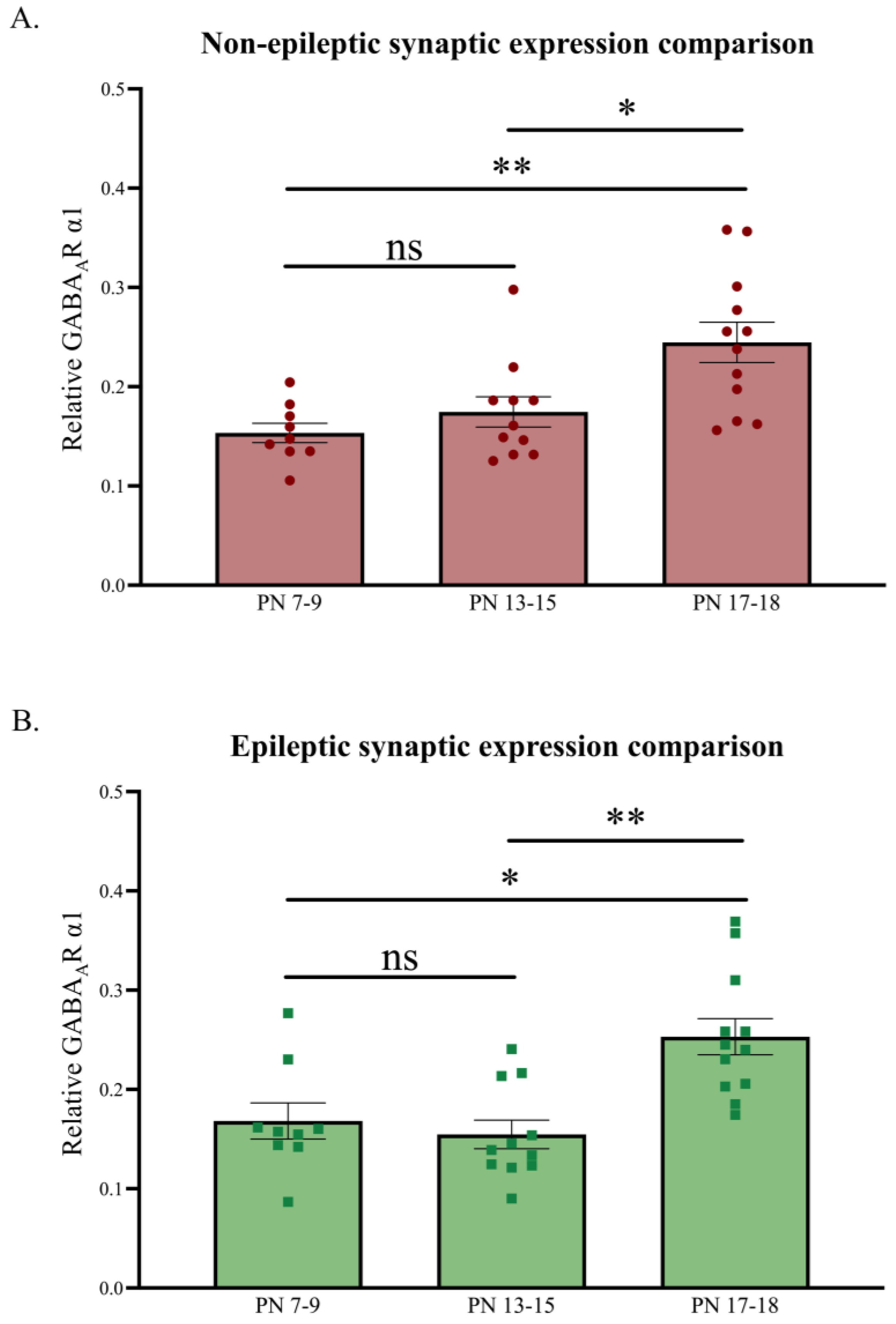
The relative synaptic GABAAR α1 levels, normalized against PanC, were also used to analyze developmental changes across the three juvenile age groups for both the NE control littermates and stargazers (Figure 4). As expected, synaptic GABAAR α1 revealed a significant increase across the three developmental stages [49,74]. Synaptic GABAAR α1 expression showed a ~123% increase in NE control littermates (n = 32; p = 0.0032) and a ~77% increase in stargazers (n = 32; p = 0.0032) from PN 7–9 to 17–18, with the increase being notably significant between PN 13–15 and 17–18.
3.3. GAD65 Is Unaltered in the Stargazer Primary SoCx
Previously, we reported that the GABA neurotransmitter was significantly increased globally throughout the primary SoCx (as revealed with HPLC whole-tissue analyses) but reduced within each inhibitory terminal (as revealed with immunogold ICC-EM analyses) of adult epileptic stargazers [37]. We further found that GABAergic neuron population density and immunoreactive profile density within the neuropil were not significantly different in the adult stargazer primary SoCx compared with their NE control littermates, which suggested that the likely increase in tissue GABA levels was extracellular rather than a consequence of the increased branching of GABAergic axons within the neuropil during development [39]. The adult stargazer primary SoCx also showed a significant increase in GAD65 expression [38]. Given the correlation between GAD65 levels and changes in the synthesis/release of GABA at presynaptic terminals, GAD65 was assessed during the stargazers’ development using a Western blot analysis of the cytosol fractions at PN 7–9, 13–15, and 17–18. Bands were detected at the expected molecular weight of 65 kDa. Analyses after normalization against PanC showed no difference between GAD65 expression levels in the NE control littermate and stargazer primary SoCx (Figure 5). Representative original Western blots for all subcellular fractions are provided in Supplementary Figure S1 (PN 7–9), Supplementary Figure S2 (PN 13–15), and Supplementary Figure S3 (PN 17–18).
Changes in the relative cytosolic GAD65 levels, normalized against PanC, across the three developmental age groups were also analyzed for both NE control littermates and epileptic stargazers (Figure 6). As expected, GAD65 revealed a significant increase across the three developmental stages [49,75]. Expression patterns showed a ~220% increase in NE control littermates (n = 30; p = 0.0008) and a ~175% increase in stargazers (n = 30; p = 0.001) from PN 7–9 to 17–18, with the relatively significant increase occurring between PN 7–9 and 13–15.
4. Discussion
The current study investigated whether the altered expressions of the GABAAR α1 subunit and GAD65, seen in adult stargazer primary SoCx, were established prior to the onset of seizures (at PN18) by analyzing their expressions at 3 development stages between PN 7–18. Both the NE control littermates and stargazers showed significant increases in the expressions of synaptic GABAAR α1 and cytosolic GAD65 in the primary SoCx during development from PN7–9 to PN17–18. However, no statistically significant difference was found in synaptic GABAAR α1 and cytosolic GAD65 levels in epileptic stargazers compared with their age-matched NE littermate controls, at any of the three developmental stages. These data suggest that aberrant GABAergic expression found in adult stargazer primary SoCx does not occur prior to the onset of seizures, and hence may not contribute directly to the pathogenesis of seizures but perhaps is a secondary compensatory change that could either play a role in limiting seizure generation or alternatively potentially contribute to the maintenance of seizures.
The first three postnatal weeks (in rodent models) are the most important in terms of the morphological and functional maturation of neurons and synaptogenesis. Hence, we first determined the developmental patterns for synaptic GABAAR α1 and cytosolic GAD65 during PN 7–9, 13–15, and 17–18. Our results showed a steady overall increase in both GABAAR α1 and GAD65, which is consistent with previous reports [76,77]. A similar expression pattern for GABAAR α1 has been reported in rats where α1 is low at birth but shows a gradual increase thereafter up until about three weeks of age when the expression is widespread in most brain areas, including the neocortex [55,58]. Fritschy et al. (1994) also reported the region-specific maturation of α1 with, for instance, the primary SoCx preceding association areas by several days, which is perhaps one explanation for why the increase in expression levels was slower between PN 7–15 (Figure 4). A similar pattern of change in the expression of postnatal GABAAR α1 has also been reported in another mouse model, wherein the gene expressions of GABAAR subunits and GADs in the primary SoCx were studied using in situ hybridization histochemistry [49]. In the current study, GAD65 also showed a marked developmental increase from PN 7–9, but the increase became comparatively slower from PN 13–18, which was likely due to reaching adult levels at around the 3 weeks mark, as previously reported in [48]. Kiser et al. (1998) reported that in rats, there was a minimal presence of GAD65 prior to PN 6 but a rapid increase thereafter. When comparing the developmental expression pattern of GAD65 between stargazers and NE control littermates, a relative decrease in GAD65 at PN 17–18 in the stargazer primary SoCx was seen (Figure 6). This coincides with the stage at which fast-spiking properties of parvalbumin-positive (PV+) interneurons become active and absence seizures SWDs are detected [78,79,80]. It is possible that the dysfunctional engagement of fast-spiking PV+ feed-forward inhibitory interneurons in stargazer primary SoCx results in a transient reduction in GAD65.
Multiple studies implicate impaired GABAergic neurotransmission in the CTC network pathognomonic of absence seizures [81,82,83,84,85,86]. Similarly, we reported altered GABAergic neurotransmission in the adult stargazer primary SoCx in the form of reduced synaptic GABAAR α1 expression and a potential increase in ambient GABA levels supplemented by the findings of increased GAD65 [36,38]. Such disruptions could contribute to absence seizure generation in the CTC network [82]. Thus far, many studies have reported impaired GABAergic neurotransmission in the adult CTC network, but very few have investigated the developing CTC network in epilepsy models prior to the onset of seizures. One such study conducted at PN 25 in the Genetic Absence Epilepsy Rats from Strasbourg (GAERS), prior to the age when absence seizures are fully established in this rodent model, reported an increase in the density of GABAergic interneurons in the primary SoCx and RTN and an increased GABAergic innervation of relay neurons in the ventroposterior (VP) thalamus [41]. A two-fold increase in tonic currents in the VP relay thalamus at PN 17 has also been reported in these GAERS [40]. Conversely, in the stargazer mouse, direct impairment of GABA-mediated neurotransmission, in the form of altered GABAAR subunit expression in the primary SoCx (the current study) and VP relay thalamus [30,31,32], and altered tonic inhibition in the VP relay thalamus [40,87], occurs after seizure onset and is secondary to dysfunctional feedforward inhibition [33,34,82,88,89].
In stargazers, the onset of seizures is related to dysfunctional feedforward inhibitory microcircuits within the CTC network [90,91] as a result of AMPA receptor deficits in the GABAergic fast-spiking PV+ interneurons [35]. Chemogenetic inactivation of these feedforward inhibitory interneurons using Designer Receptor Exclusively Activated by Designer Drug (DREADD) technology induced absence seizures in normal mice [89], whereas activation of the same neurons during absence seizures reduced them [90]. The genetic defect in the stargazers results in the loss of stargazin, a transmembrane AMPA receptor regulatory protein (TARP) that regulates synaptic AMPA receptor expression. Stargazin is the only TARP expressed in PV+ interneurons in the CTC network, and it is expressed in the perinatal stages [92]. While evidence shows AMPA receptor subunit reduction in stargazers as far back as PN 13–15, a stage at which mature calcium-permeable AMPA receptors are expressed [63], the onset of seizures themselves occurs at PN 17–18. Given that PV+ interneurons begin engaging their fast-spiking properties during this stage [78,79,80], a correlation between dysfunctional fast-spiking PV+ interneuron-mediated phasic inhibition and the initiation of seizures is indicated. Our findings indicate that it is past this point that altered GABAergic neurotransmission (i.e., altered GABA receptor and neurotransmitter expression) occurs, as the neural system continues to functionally and structurally mature while affected by SWDs. This could be a compensatory response to overcome overexcitations in the cortical circuitry or downstream effects from overexcitations that may further potentiate or maintain SWDs.
Structural and functional plasticity in neural circuits regulates and maintains the homeostatic E/I balance [93], failure of which results in neurological disorders including epilepsy [94,95,96]. In stargazers, chronic loss of key proteins (the stargazin and AMPA receptor subunits) and the consequential dysfunctional inhibitory CTC microcircuits and resultant downstream changes permit the generation and maintenance of absence seizures. Given that pyramidal neurons comprise around 80% of the cortical neurons, it is possible that hyperexcitations in the pyramidal neurons lead to intense firing in other GABAergic interneurons causing a release of GABA from multiple sites in adult stargazer primary SoCx [97], which may explain the increased levels of GAD65 and extracellular GABA. Increased extracellular GABA-mediated enhanced tonic inhibition in thalamocortical relay neurons due to compromised GABA re-uptake by GABA transporter-1 (GAT-1) has been reported by Cope et al. (2009) in GAERS, stargazers, and lethargic genetic models for absence epilepsy [40]. Cope et al. (2009) postulated that this could be the mechanism driving absence seizures; however, these changes were reported in adult animals with fully established seizures and not in juveniles. Whether a similar mechanism operates in the primary SoCx of stargazer mice remains to be investigated. Future studies should be conducted to confirm if this is indeed the case. The microdialysis method, for instance, can be used to measure the levels of GABA before and after the onset of seizures in stargazers, while intracellular electrophysiological recordings can give insights into the activity of cortical neurons. Although an increase in extracellular GABA in adult stargazer primary SoCx might be expected to suppress seizures, it could in fact lead to their enhancement via disinhibitory circuits or enhanced tonic-inhibition-mediated T-type calcium channel burst firing [40,87,98,99]. The fact that the use of GABAergic inhibition-enhancing drugs aggravates absence seizures supports the concept that secondary GABAergic changes past the age of seizure onset may have pro-seizure effects rather than the opposite [100,101,102,103,104].
5. Conclusions
The current study has clearly shown that changes in synaptic GABAAR α1 and cytosolic GAD65 expression do not occur prior to absence seizure onset in the stargazer primary SoCx. This would indicate that changes in synaptic GABAAR α1 and GAD65 are not responsible for initiating seizures but are secondary compensatory changes. However, developmental changes in neuronal networks followed by secondary compensatory changes and the pathogenesis of absence seizures are clearly inter-dependent [105]. Therefore, these post-seizure compensatory changes could clearly be involved in the ongoing maintenance of seizures, even if they are not causative of their initiation. The timeline for such changes should be taken into account when designing targeted therapeutics, as the outcome of treatment could be affected. It is important to note that our current study assessed changes in all layers of the primary SoCx collectively, while pre-seizure layer-specific changes may be present that could be important in causing absence seizures. Recent evidence indicates the presence of cortical layer cell-specific changes in stargazer primary SoCx [106]. Future investigations could use techniques, such as laser microdissection, to procure layer-specific tissue, coupled with quantitative protein expression analyses (for instance, Western blotting or liquid chromatography/mass spectrometry) to identify any layer-specific changes that may have been masked when the whole-tissue primary SoCx were assessed in our study.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/biom13010186/s1. Figure S1: representative Western blots for subcellular fractions at PN 7–9, Figure S2: representative Western blots for subcellular fractions at PN 13–15, and Figure S3: representative Western blots for subcellular fractions at PN 17–18.
Author Contributions
B.L. conceptualized and designed the research, supervised the project, and secured the funding. D.R.G. provided advice and reviewed the results and discussion. M.H. conducted the experiments and wrote the first draft of the manuscript. Analyses and interpretation of data were conducted jointly. All authors contributed to the manuscript writing and approved the final version. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by funding from the University of Otago to B.L.
Institutional Review Board Statement
This study was conducted according to the guidelines approved on 20 November 2017 by the University of Otago Animal Ethics Committee under the AEC no. DET 32/17.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available upon reasonable request.
Acknowledgments
The authors acknowledge the University of Otago’s research funding and publishing bursary awarded to B.L. and M.H., respectively.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vasung, L.; Abaci Turk, E.; Ferradal, S.L.; Sutin, J.; Stout, J.N.; Ahtam, B.; Lin, P.Y.; Grant, P.E. Exploring early human brain development with structural and physiological neuroimaging. Neuroimage 2019, 187, 226–254. [Google Scholar] [CrossRef] [PubMed]
- Jamuar, S.S.; Lam, A.T.; Kircher, M.; D’Gama, A.M.; Wang, J.; Barry, B.J.; Zhang, X.; Hill, R.S.; Partlow, J.N.; Rozzo, A.; et al. Somatic mutations in cerebral cortical malformations. N. Engl. J. Med. 2014, 371, 733–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlotz, W.; Phillips, D.I. Fetal origins of mental health: Evidence and mechanisms. Brain Behav. Immun. 2009, 23, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Raznahan, A.; Greenstein, D.; Lee, N.R.; Clasen, L.S.; Giedd, J.N. Prenatal growth in humans and postnatal brain maturation into late adolescence. Proc. Natl. Acad. Sci. USA 2012, 109, 11366–11371. [Google Scholar] [CrossRef] [Green Version]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, J.H.; Knickmeyer, R.C.; Gao, W. Imaging structural and functional brain development in early childhood. Nat. Rev. Neurosci. 2018, 19, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Haartsen, R.; Jones, E.J.H.; Johnson, M.H. Human brain development over the early years. Curr. Opin. Behav. Sci. 2016, 10, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ben-Ari, Y. Basic developmental rules and their implications for epilepsy in the immature brain. Epileptic Disord. 2006, 8, 91–102. [Google Scholar]
- Velísková, J.; Claudio, O.I.; Galanopoulou, A.S.; Lado, F.A.; Ravizza, T.; Velísek, L.; Moshé, S.L. Seizures in the developing brain. Epilepsia 2004, 45, 6–12. [Google Scholar] [CrossRef]
- Krsnik, Ž.; Majić, V.; Vasung, L.; Huang, H.; Kostović, I. Growth of Thalamocortical Fibers to the Somatosensory Cortex in the Human Fetal Brain. Front. Neurosci. 2017, 11, 233. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, G.M.; Price, D.J. Exuberance in the development of cortical networks. Nat. Rev. Neurosci. 2005, 6, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Kostović, I.; Judas, M. Correlation between the sequential ingrowth of afferents and transient patterns of cortical lamination in preterm infants. Anat. Rec. 2002, 267, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, J.; Wei, H.; Han, V.; Zhu, W.Z.; Liu, C. Neonate and infant brain development from birth to 2 years assessed using MRI-based quantitative susceptibility mapping. Neuroimage 2019, 185, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.G. The Thalamus; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Yu, Q.; Ouyang, A.; Chalak, L.; Jeon, T.; Chia, J.; Mishra, V.; Sivarajan, M.; Jackson, G.; Rollins, N.; Liu, S.; et al. Structural Development of Human Fetal and Preterm Brain Cortical Plate Based on Population-Averaged Templates. Cereb. Cortex 2016, 26, 4381–4391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Wang, L.; Shi, F.; Lyall, A.E.; Lin, W.; Gilmore, J.H.; Shen, D. Mapping longitudinal development of local cortical gyrification in infants from birth to 2 years of age. J. Neurosci. 2014, 34, 4228–4238. [Google Scholar] [CrossRef] [Green Version]
- Conel, J.L. The Postnatal Development of the Human Cerebral Cortex, V. 7: The Cortex of the Four-year Child; Harvard University Press: Cambridge, MA, USA, 1963. [Google Scholar]
- Drenthen, G.S.; Fonseca Wald, E.L.A.; Backes, W.H.; Aldenkamp, A.P.; Vermeulen, R.J.; Debeij-van Hall, M.; Klinkenberg, S.; Jansen, J.F.A. Constructing an Axonal-Specific Myelin Developmental Graph and its Application to Childhood Absence Epilepsy. J. Neuroimaging 2020, 30, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Crunelli, V.; Lorincz, M.L.; McCafferty, C.; Lambert, R.C.; Leresche, N.; Di Giovanni, G.; David, F. Clinical and experimental insight into pathophysiology, comorbidity and therapy of absence seizures. Brain 2020, 143, 2341–2368. [Google Scholar] [CrossRef]
- Jarre, G.; Altwegg-Boussac, T.; Williams, M.S.; Studer, F.; Chipaux, M.; David, O.; Charpier, S.; Depaulis, A.; Mahon, S.; Guillemain, I. Building Up Absence Seizures in the Somatosensory Cortex: From Network to Cellular Epileptogenic Processes. Cereb. Cortex 2017, 27, 4607–4623. [Google Scholar] [CrossRef] [Green Version]
- Qiao, X.; Noebels, J.L. Developmental analysis of hippocampal mossy fiber outgrowth in a mutant mouse with inherited spike-wave seizures. J. Neurosci. 1993, 13, 4622. [Google Scholar] [CrossRef] [Green Version]
- Letts, V.A. Stargazer—A mouse to seize! Epilepsy Curr. 2005, 5, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chetkovich, D.M.; Petralia, R.S.; Sweeney, N.T.; Kawasaki, Y.; Wenthold, R.J.; Bredt, D.S.; Nicoll, R.A. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 2000, 408, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Fukaya, M.; Qiao, X.; Sakimura, K.; Watanabe, M.; Kano, M. Impairment of AMPA receptor function in cerebellar granule cells of ataxic mutant mouse stargazer. J. Neurosci. 1999, 19, 6027–6036. [Google Scholar] [CrossRef] [Green Version]
- Shevtsova, O.; Leitch, B. Selective loss of AMPA receptor subunits at inhibitory neuron synapses in the cerebellum of the ataxic stargazer mouse. Brain Res. 2012, 1427, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.A.; Leitch, B. Cerebellar golgi, purkinje, and basket cells have reduced gamma-aminobutyric acid immunoreactivity in stargazer mutant mice. J. Comp. Neurol. 2002, 453, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.L.; Tehrani, M.H.J.; Barnes, E.M.; Stephenson, F.A. Decreased expression of GABA(A) receptor alpha 6 and beta 3 subunits in stargazer mutant mice: A possible role for brain-derived neurotrophic factor in the regulation of cerebellar GABA(A) receptor expression? Mol. Brain. Res. 1998, 60, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Payne, H.L. Aberrant GABA(A) receptor expression in the dentate gyrus of the epileptic mutant mouse stargazer. J. Neurosci. 2006, 26, 8600–8608. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.; Leitch, B. Altered thalamic GABAA-receptor subunit expression in the stargazer mouse model of absence epilepsy. Epilepsia 2014, 55, 224–232. [Google Scholar] [CrossRef]
- Seo, S.; Leitch, B. Synaptic changes in GABAA receptor expression in the thalamus of the stargazer mouse model of absence epilepsy. Neuroscience 2015, 306, 28–38. [Google Scholar] [CrossRef]
- Seo, S.; Leitch, B. Postnatal expression of thalamic GABAA receptor subunits in the stargazer mouse model of absence epilepsy. Neuroreport 2017, 28, 1255–1260. [Google Scholar] [CrossRef]
- Adotevi, N.K.; Leitch, B. Alterations in AMPA receptor subunit expression in cortical inhibitory interneurons in the epileptic stargazer mutant mouse. Neuroscience 2016, 339, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Adotevi, N.K.; Leitch, B. Synaptic Changes in AMPA Receptor Subunit Expression in Cortical Parvalbumin Interneurons in the Stargazer Model of Absence Epilepsy. Front. Mol. Neurosci. 2017, 10, 434. [Google Scholar] [CrossRef] [Green Version]
- Adotevi, N.K.; Leitch, B. Cortical expression of AMPA receptors during postnatal development in a genetic model of absence epilepsy. Int. J. Dev. Neurosci. 2018, 73, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Adotevi, N.K.; Leitch, B. Altered GABAA Receptor Expression in the Primary Somatosensory Cortex of a Mouse Model of Genetic Absence Epilepsy. Int. J. Mol. Sci. 2022, 23, 15685. [Google Scholar] [CrossRef]
- Adotevi, N.; Su, A.; Peiris, D.; Hassan, M.; Leitch, B. Altered Neurotransmitter Expression in the Corticothalamocortical Network of an Absence Epilepsy Model with impaired Feedforward Inhibition. Neuroscience 2021, 467, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Panthi, S.; Lyons, N.M.A.; Leitch, B. Impact of Dysfunctional Feed-Forward Inhibition on Glutamate Decarboxylase Isoforms and γ-Aminobutyric Acid Transporters. Int. J. Mol. Sci. 2021, 22, 7740. [Google Scholar] [CrossRef]
- Hassan, M. Changes in GABAergic Neurotransmission in the Primary Somatosensory Cortex of the Stargazer Mouse Model of Absence Epilepsy; University of Otago: Dunedin, New Zealand, 2022. [Google Scholar]
- Cope, D.W.; Di Giovanni, G.; Fyson, S.J.; Orban, G.; Errington, A.C.; Lorincz, M.L.; Gould, T.M.; Carter, D.A.; Crunelli, V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med. 2009, 15, 1392–1398. [Google Scholar] [CrossRef] [Green Version]
- Bombardi, C.; Venzi, M.; Crunelli, V.; Di Giovanni, G. Developmental changes of GABA immunoreactivity in cortico-thalamic networks of an absence seizure model. Neuropharmacology 2018, 136, 56–67. [Google Scholar] [CrossRef]
- Tian, N.; Petersen, C.; Kash, S.; Baekkeskov, S.; Copenhagen, D.; Nicoll, R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc. Natl. Acad. Sci. USA 1999, 96, 12911–12916. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.B.; de Graaf, R.A.; Martin, D.L.; Battaglioli, G.; Behar, K.L. Evidence that GAD(65) mediates increased GABA synthesis during intense neuronal activity in vivo. J. Neurochem. 2006, 97, 385–396. [Google Scholar] [CrossRef]
- Wei, J.; Wu, J.Y. Post-translational regulation of L-glutamic acid decarboxylase in the brain. Neurochem. Res. 2008, 33, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Haydar, T.F.; Wang, F.; Schwartz, M.L.; Rakic, P. Differential modulation of proliferation in the neocortical ventricular and subventricular zones. J. Neurosci. 2000, 20, 5764–5774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanopoulou, A.S. GABA(A) receptors in normal development and seizures: Friends or foes? Curr. Neuropharmacol. 2008, 6, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peerboom, C.; Wierenga, C.J. The postnatal GABA shift: A developmental perspective. Neurosci. Biobehav. Rev. 2021, 124, 179–192. [Google Scholar] [CrossRef]
- Kiser, P.J.; Cooper, N.G.; Mower, G.D. Expression of two forms of glutamic acid decarboxylase (GAD67 and GAD65) during postnatal development of rat somatosensory barrel cortex. J. Comp. Neurol. 1998, 402, 62–74. [Google Scholar] [CrossRef]
- Golshani, P.; Truong, H.; Jones, E.G. Developmental expression of GABA(A) receptor subunit and GAD genes in mouse somatosensory barrel cortex. J. Comp. Neurol. 1997, 383, 199–219. [Google Scholar] [CrossRef]
- Stork, O.; Ji, F.Y.; Kaneko, K.; Stork, S.; Yoshinobu, Y.; Moriya, T.; Shibata, S.; Obata, K. Postnatal development of a GABA deficit and disturbance of neural functions in mice lacking GAD65. Brain Res. 2000, 865, 45–58. [Google Scholar] [CrossRef]
- Asada, H.; Kawamura, Y.; Maruyama, K.; Kume, H.; Ding, R.; Ji, F.Y.; Kanbara, N.; Kuzume, H.; Sanbo, M.; Yagi, T.; et al. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem. Biophys. Res. Commun. 1996, 229, 891–895. [Google Scholar] [CrossRef]
- Laurie, D.J.; Wisden, W.; Seeburg, P.H. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 1992, 12, 4151–4172. [Google Scholar] [CrossRef]
- Ma, W.; Barker, J.L. Complementary expressions of transcripts encoding GAD67 and GABAA receptor alpha 4, beta 1, and gamma 1 subunits in the proliferative zone of the embryonic rat central nervous system. J. Neurosci. 1995, 15, 2547–2560. [Google Scholar] [CrossRef] [Green Version]
- Serafini, R.; Ma, W.; Maric, D.; Maric, I.; Lahjouji, F.; Sieghart, W.; Barker, J.L. Initially expressed early rat embryonic GABA(A) receptor Cl- ion channels exhibit heterogeneous channel properties. Eur. J. Neurosci. 1998, 10, 1771–1783. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.Y.; Wang, W.; Fritschy, J.M.; Witte, O.W.; Redecker, C. Changes in neocortical and hippocampal GABAA receptor subunit distribution during brain maturation and aging. Brain Res. 2006, 1099, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Heinen, K.; Bosman, L.W.; Spijker, S.; van Pelt, J.; Smit, A.B.; Voorn, P.; Baker, R.E.; Brussaard, A.B. GABAA receptor maturation in relation to eye opening in the rat visual cortex. Neuroscience 2004, 124, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.M.; Mohler, H. GABAA-receptor heterogeneity in the adult rat brain: Differential regional and cellular distribution of seven major subunits. J. Comp. Neurol. 1995, 359, 154–194. [Google Scholar] [CrossRef]
- Fritschy, J.; Paysan, J.; Enna, A.; Mohler, H. Switch in the expression of rat GABAA-receptor subtypes during postnatal development: An immunohistochemical study. J. Neurosci. 1994, 14, 5302–5324. [Google Scholar] [CrossRef]
- Henneberger, C.; Jüttner, R.; Schmidt, S.A.; Walter, J.; Meier, J.C.; Rothe, T.; Grantyn, R. GluR- and TrkB-mediated maturation of GABA receptor function during the period of eye opening. Eur. J. Neurosci. 2005, 21, 431–440. [Google Scholar] [CrossRef]
- Chen, L.; Yang, C.; Mower, G.D. Developmental changes in the expression of GABA(A) receptor subunits (alpha(1), alpha(2), alpha(3)) in the cat visual cortex and the effects of dark rearing. Brain Res. Mol. Brain Res. 2001, 88, 135–143. [Google Scholar] [CrossRef]
- Samarut, É.; Swaminathan, A.; Riché, R.; Liao, M.; Hassan-Abdi, R.; Renault, S.; Allard, M.; Dufour, L.; Cossette, P.; Soussi-Yanicostas, N.; et al. γ-Aminobutyric acid receptor alpha 1 subunit loss of function causes genetic generalized epilepsy by impairing inhibitory network neurodevelopment. Epilepsia 2018, 59, 2061–2074. [Google Scholar] [CrossRef] [Green Version]
- Daw, M.I.; Ashby, M.C.; Isaac, J.T. Coordinated developmental recruitment of latent fast spiking interneurons in layer IV barrel cortex. Nat. Neurosci. 2007, 10, 453–461. [Google Scholar] [CrossRef]
- Pellegrini-Giampietro, D.E.; Bennett, M.V.; Zukin, R.S. Are Ca2+-permeable kainate/AMPA receptors more abundant in immature brain? Neurosci. Lett. 1992, 144, 65–69. [Google Scholar] [CrossRef]
- Warren, R.A.; Jones, E.G. Maturation of Neuronal Form and Function in a Mouse Thalamo-Cortical Circuit. J. Neurosci. 1997, 17, 277. [Google Scholar] [CrossRef] [PubMed]
- Noebels, J.L.; Qiao, X.; Bronson, R.T.; Spencer, C.; Davisson, M.T. Stargazer: A new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res. 1990, 7, 129–135. [Google Scholar] [CrossRef]
- Okaty, B.W.; Miller, M.N.; Sugino, K.; Hempel, C.M.; Nelson, S.B. Transcriptional and Electrophysiological Maturation of Neocortical Fast-Spiking GABAergic Interneurons. J. Neurosci. 2009, 29, 7040–7052. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.D.; Alvestad, R.M.; Coultrap, S.J.; Browning, M.D. alphaCaMKII autophosphorylation levels differ depending on subcellular localization. Brain Res. 2007, 1158, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barad, Z.; Grattan, D.R.; Leitch, B. NMDA Receptor Expression in the Thalamus of the Stargazer Model of Absence Epilepsy. Sci. Rep. 2017, 7, 42926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beesley, P.W.; Mummery, R.; Tibaldi, J. N-cadherin is a major glycoprotein component of isolated rat forebrain postsynaptic densities. J. Neurochem. 1995, 64, 2288–2294. [Google Scholar] [CrossRef]
- Tomita, S.; Chen, L.; Kawasaki, Y.; Petralia, R.S.; Wenthold, R.J.; Nicoll, R.A.; Bredt, D.S. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J. Cell. Biol. 2003, 161, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Petersen, J.D.; Chen, X.B.; Vinade, L.; Dosemeci, A.; Lisman, J.E.; Reese, T.S. Distribution of postsynaptic density (PSD)-95 and Ca2+/calmodulin-dependent protein kinase II at the PSD. J. Neurosci. 2003, 23, 11270–11278. [Google Scholar] [CrossRef] [Green Version]
- Heller, E.A.; Zhang, W.; Selimi, F.; Earnheart, J.C.; Ślimak, M.A.; Santos-Torres, J.; Ibañez-Tallon, I.; Aoki, C.; Chait, B.T.; Heintz, N. The biochemical anatomy of cortical inhibitory synapses. PLoS ONE 2012, 7, e39572. [Google Scholar] [CrossRef] [Green Version]
- Schubert, V.; Dotti, C.G. Transmitting on actin: Synaptic control of dendritic architecture. J. Cell Sci. 2007, 120, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Gambarana, C.; Pittman, R.; Siegel, R.E. Developmental Expression of the Gaba-a Receptor Alpha-1 Subunit Messenger-Rna in the Rat-Brain. J. Neurobiol. 1990, 21, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Popp, A.; Urbach, A.; Witte, O.W.; Frahm, C. Adult and embryonic GAD transcripts are spatiotemporally regulated during postnatal development in the rat brain. PLoS ONE 2009, 4, e4371. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kaplan, I.V.; Cooper, N.G.F.; Mower, G.D. Expression of two forms of glutamic acid decarboxylase (GAD67 and GAD65) during postnatal development of cat visual cortex. Dev. Brain Res. 1997, 103, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wong-Riley, M.T. Developmental changes in the expression of GABAA receptor subunits alpha1, alpha2, and alpha3 in the rat pre-Botzinger complex. J. Appl. Physiol. 2004, 96, 1825–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardin, J.A. Inhibitory Interneurons Regulate Temporal Precision and Correlations in Cortical Circuits. Trends Neurosci. 2018, 41, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Pangratz-Fuehrer, S.; Hestrin, S. Synaptogenesis of electrical and GABAergic synapses of fast-spiking inhibitory neurons in the neocortex. J. Neurosci. 2011, 31, 10767–10775. [Google Scholar] [CrossRef]
- Goldberg, E.M.; Jeong, H.-Y.; Kruglikov, I.; Tremblay, R.; Lazarenko, R.M.; Rudy, B. Rapid Developmental Maturation of Neocortical FS Cell Intrinsic Excitability. Cerebral Cortex 2011, 21, 666–682. [Google Scholar] [CrossRef] [Green Version]
- Crunelli, V.; Leresche, N.; Cope, D.W. GABA-A Receptor Function in Typical Absence Seizures. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Library of Medicine: Bethesda, MD, USA, 2012. [Google Scholar]
- Maheshwari, A.; Noebels, J.L. Monogenic models of absence epilepsy: Windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. Prog. Brain Res. 2014, 213, 223–252. [Google Scholar] [CrossRef]
- Bessaih, T.; Bourgeais, L.; Badiu, C.I.; Carter, D.A.; Toth, T.I.; Ruano, D.; Lambolez, B.; Crunelli, V.; Leresche, N. Nucleus-specific abnormalities of GABAergic synaptic transmission in a genetic model of absence seizures. J. Neurophysiol. 2006, 96, 3074–3081. [Google Scholar] [CrossRef]
- Maljevic, S.; Krampfl, K.; Rebstock, J.; Tilgen, N.; Weber, Y.G.; Cossette, P.; Rouleau, G.; Bufler, J.; Lerche, H.; Heils, A. A de novo mutation of the GABA(A) receptor alpha1-subunit associated with childhood absence epilepsy in a single patient. Epilepsia 2004, 45, 119. [Google Scholar]
- Wallace, R.H.; Marini, C.; Petrou, S.; Harkin, L.A.; Bowser, D.N.; Panchal, R.G.; Williams, D.A.; Sutherland, G.R.; Mulley, J.C.; Scheffer, I.E.; et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 2001, 28, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, M. Genetics of idiopathic generalized epilepsies. Epilepsia 2005, 46, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Cope, D.W.; Hughes, S.W.; Crunelli, V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J. Neurosci. 2005, 25, 11553–11563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barad, Z.; Shevtsova, O.; Arbuthnott, G.W.; Leitch, B. Selective loss of AMPA receptors at corticothalamic synapses in the epileptic stargazer mouse. Neuroscience 2012, 217, 19–31. [Google Scholar] [CrossRef]
- Menuz, K.; Nicoll, R.A. Loss of inhibitory neuron AMPA receptors contributes to ataxia and epilepsy in stargazer mice. J. Neurosci. 2008, 28, 10599–10603. [Google Scholar] [CrossRef] [Green Version]
- Panthi, S.; Leitch, B. The impact of silencing feed-forward parvalbumin-expressing inhibitory interneurons in the cortico-thalamocortical network on seizure generation and behaviour. Neurobiol. Dis. 2019, 132, 1. [Google Scholar] [CrossRef]
- Panthi, S.; Leitch, B. Chemogenetic Activation of Feed-Forward Inhibitory Parvalbumin-Expressing Interneurons in the Cortico-Thalamocortical Network During Absence Seizures. Front. Cell. Neurosci. 2021, 15, 688905. [Google Scholar] [CrossRef]
- Fukaya, M.; Yamazaki, M.; Sakimura, K.; Watanabe, M. Spatial diversity in gene expression for VDCCγ subunit family in developing and adult mouse brains. Neurosci. Res. 2005, 53, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Tien, N.-W.; Kerschensteiner, D. Homeostatic plasticity in neural development. Neural. Dev. 2018, 13, 9. [Google Scholar] [CrossRef]
- Ramocki, M.B.; Zoghbi, H.Y. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 2008, 455, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Vislay, R.L.; Martin, B.S.; Olmos-Serrano, J.L.; Kratovac, S.; Nelson, D.L.; Corbin, J.G.; Huntsman, M.M. Homeostatic responses fail to correct defective amygdala inhibitory circuit maturation in fragile X syndrome. J. Neurosci. 2013, 33, 7548–7558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lignani, G.; Baldelli, P.; Marra, V. Homeostatic Plasticity in Epilepsy. Front. Cell. Neurosci. 2020, 14, 197. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.G.; Masuoka, T.; Gong, X.D.; Chen, K.S.; Yanagawa, Y.; Law, S.K.; Konishi, S. NMDA receptor activation enhances inhibitory GABAergic transmission onto hippocampal pyramidal neurons via presynaptic and postsynaptic mechanisms. J. Neurophysiol. 2011, 105, 2897–2906. [Google Scholar] [CrossRef] [PubMed]
- Errington, A.C.; Cope, D.W.; Crunelli, V. Augmentation of Tonic GABA(A) Inhibition in Absence Epilepsy: Therapeutic Value of Inverse Agonists at Extrasynaptic GABA(A) Receptors. Adv. Pharmacol. Sci. 2011, 2011, 790590. [Google Scholar] [CrossRef] [Green Version]
- McCafferty, C.; David, F.; Venzi, M.; Lorincz, M.L.; Delicata, F.; Atherton, Z.; Recchia, G.; Orban, G.; Lambert, R.C.; Di Giovanni, G.; et al. Cortical drive and thalamic feed-forward inhibition control thalamic output synchrony during absence seizures. Nat. Neurosci. 2018, 21, 744–756. [Google Scholar] [CrossRef]
- Pires, N.M.; Bonifácio, M.J.; Soares-da-Silva, P. Carbamazepine aggravates absence seizures in two dedicated mouse models. Pharmacol. Rep. 2015, 67, 986–995. [Google Scholar] [CrossRef]
- Liu, L.; Zheng, T.; Morris, M.J.; Wallengren, C.; Clarke, A.L.; Reid, C.A.; Petrou, S.; O’Brien, T.J. The mechanism of carbamazepine aggravation of absence seizures. J. Pharmacol. Exp. Ther. 2006, 319, 790–798. [Google Scholar] [CrossRef] [Green Version]
- Wallengren, C.; Li, S.; Morris, M.J.; Jupp, B.; O’Brien, T.J. Aggravation of absence seizures by carbamazepine in a genetic rat model does not induce neuronal c-Fos activation. Clin. Neuropharmacol. 2005, 28, 60–65. [Google Scholar] [CrossRef]
- Coenen, A.M.; Blezer, E.H.; van Luijtelaar, E.L. Effects of the GABA-uptake inhibitor tiagabine on electroencephalogram, spike-wave discharges and behaviour of rats. Epilepsy Res. 1995, 21, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Knake, S.; Hamer, H.M.; Schomburg, U.; Oertel, W.H.; Rosenow, F. Tiagabine-induced absence status in idiopathic generalized epilepsy. Seizure 1999, 8, 314–317. [Google Scholar] [CrossRef] [Green Version]
- Kozák, G.; Földi, T.; Berényi, A. Spike-and-Wave Discharges Are Not Pathological Sleep Spindles, Network-Level Aspects of Age-Dependent Absence Seizure Development in Rats. Eneuro 2020, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-Y.; Maheshwari, A.; Noebels, J.L.; Jiang, X. (Abstract) Layer- and pathway-specific disruption of perisomatic inhibition in the somatosensory cortex of the stargazer model of absence epilepsy. In Proceedings of the Neuroscience 2021 50th Annual Meeting, Chicago, IL, USA, 8–11 November 2008; p. 5. [Google Scholar]
Figure 1.
Timeline of critical postnatal development time points in stargazer mice. Schematic representation of developmental timepoints related to the CTC network with green-bordered boxes showing phenotypic changes specifically in stargazer mice, blue-bordered boxes showing CTC network maturation, and red-bordered boxes briefly pointing out developmental patterns of GABAAR α1 subunit and GAD65 [22,47,48,49,62,63,64,65,66].
Figure 1.
Timeline of critical postnatal development time points in stargazer mice. Schematic representation of developmental timepoints related to the CTC network with green-bordered boxes showing phenotypic changes specifically in stargazer mice, blue-bordered boxes showing CTC network maturation, and red-bordered boxes briefly pointing out developmental patterns of GABAAR α1 subunit and GAD65 [22,47,48,49,62,63,64,65,66].

Figure 2.
Representative Western blots showing the expression of GABAAR α1 in subcellular components (total lysate, cytosol, and extra-synaptic and synaptic). Subcellular fractions were derived from the primary SoCx of NE control littermates and epileptic (E) stargazers at PN 7–9 (A), 13–15 (B), and 17–18 (C). Note that the synaptic fraction shows intense signals for PSD95 while the extra-synaptic shows the complete absence of PSD95. PanC shows expression in all subcellular fractions. GABAAR α1 shows intense enrichment in synaptic fractions.
Figure 2.
Representative Western blots showing the expression of GABAAR α1 in subcellular components (total lysate, cytosol, and extra-synaptic and synaptic). Subcellular fractions were derived from the primary SoCx of NE control littermates and epileptic (E) stargazers at PN 7–9 (A), 13–15 (B), and 17–18 (C). Note that the synaptic fraction shows intense signals for PSD95 while the extra-synaptic shows the complete absence of PSD95. PanC shows expression in all subcellular fractions. GABAAR α1 shows intense enrichment in synaptic fractions.

Figure 3.
Relative synaptic expression of GABAAR α1 in the developing primary SoCx. GABAAR α1 remains unchanged in the epileptic (E) stargazer primary SoCx compared with their NE control littermates in all 3 age groups: (A) PN 7–9 (NE 0.153 ± 0.009, n = 9; E 0.168 ± 0.018, n = 9; p = 0.546), (B) PN 13–15 (NE 0.174 ± 0.015, n = 11; E 0.155 ± 0.014, n = 11; p = 0.235), and (C) PN 17–18 (NE 0.245 ± 0.020, n = 12; E 0.253 ± 0.018, n = 12; p = 0.671). The significance threshold was set at 0.05. ‘ns’ is indicative of no significant change found.
Figure 3.
Relative synaptic expression of GABAAR α1 in the developing primary SoCx. GABAAR α1 remains unchanged in the epileptic (E) stargazer primary SoCx compared with their NE control littermates in all 3 age groups: (A) PN 7–9 (NE 0.153 ± 0.009, n = 9; E 0.168 ± 0.018, n = 9; p = 0.546), (B) PN 13–15 (NE 0.174 ± 0.015, n = 11; E 0.155 ± 0.014, n = 11; p = 0.235), and (C) PN 17–18 (NE 0.245 ± 0.020, n = 12; E 0.253 ± 0.018, n = 12; p = 0.671). The significance threshold was set at 0.05. ‘ns’ is indicative of no significant change found.

Figure 4.
Relative developmental changes in synaptic GABAAR α1 expression levels in the primary SoCx. Synaptic GABAAR α1 expression levels increase with increasing postnatal age in both NE littermate controls and epileptic stargazers. (A) Kruskal–Wallis ANOVA results show a significant increase from PN 7–9 to 17–18 (H(2) = 11.49; p = 0.0032; **); post hoc Dunn’s multiple comparison test, shown on the graph, reveals a significant increase in synaptic GABAAR α1 from PN 7–9 to 17–18 (p = 0.031), PN 13–15 to 17–18 (p = 0.001), and PN 7–9 to 13–15 (p > 0.999). (B) Kruskal–Wallis ANOVA results show a significant increase from PN 7–9 to 17–18 (H(2) = 13.72; p = 0.0032; **); post hoc Dunn’s multiple comparison test, shown on the graph, reveals a significant increase in synaptic GABAAR α1 from PN 7–9 to 17–18 (p = 0.031), PN 13–15 to 17–18 (p = 0.001), and PN 7–9 to 13–15 (p > 0.999). The significance threshold was set at 0.05 with * and ** indicating p < 0.05 and p < 0.01, respectively. ‘ns’ is indicative of no significant change found.
Figure 4.
Relative developmental changes in synaptic GABAAR α1 expression levels in the primary SoCx. Synaptic GABAAR α1 expression levels increase with increasing postnatal age in both NE littermate controls and epileptic stargazers. (A) Kruskal–Wallis ANOVA results show a significant increase from PN 7–9 to 17–18 (H(2) = 11.49; p = 0.0032; **); post hoc Dunn’s multiple comparison test, shown on the graph, reveals a significant increase in synaptic GABAAR α1 from PN 7–9 to 17–18 (p = 0.031), PN 13–15 to 17–18 (p = 0.001), and PN 7–9 to 13–15 (p > 0.999). (B) Kruskal–Wallis ANOVA results show a significant increase from PN 7–9 to 17–18 (H(2) = 13.72; p = 0.0032; **); post hoc Dunn’s multiple comparison test, shown on the graph, reveals a significant increase in synaptic GABAAR α1 from PN 7–9 to 17–18 (p = 0.031), PN 13–15 to 17–18 (p = 0.001), and PN 7–9 to 13–15 (p > 0.999). The significance threshold was set at 0.05 with * and ** indicating p < 0.05 and p < 0.01, respectively. ‘ns’ is indicative of no significant change found.

Figure 5.
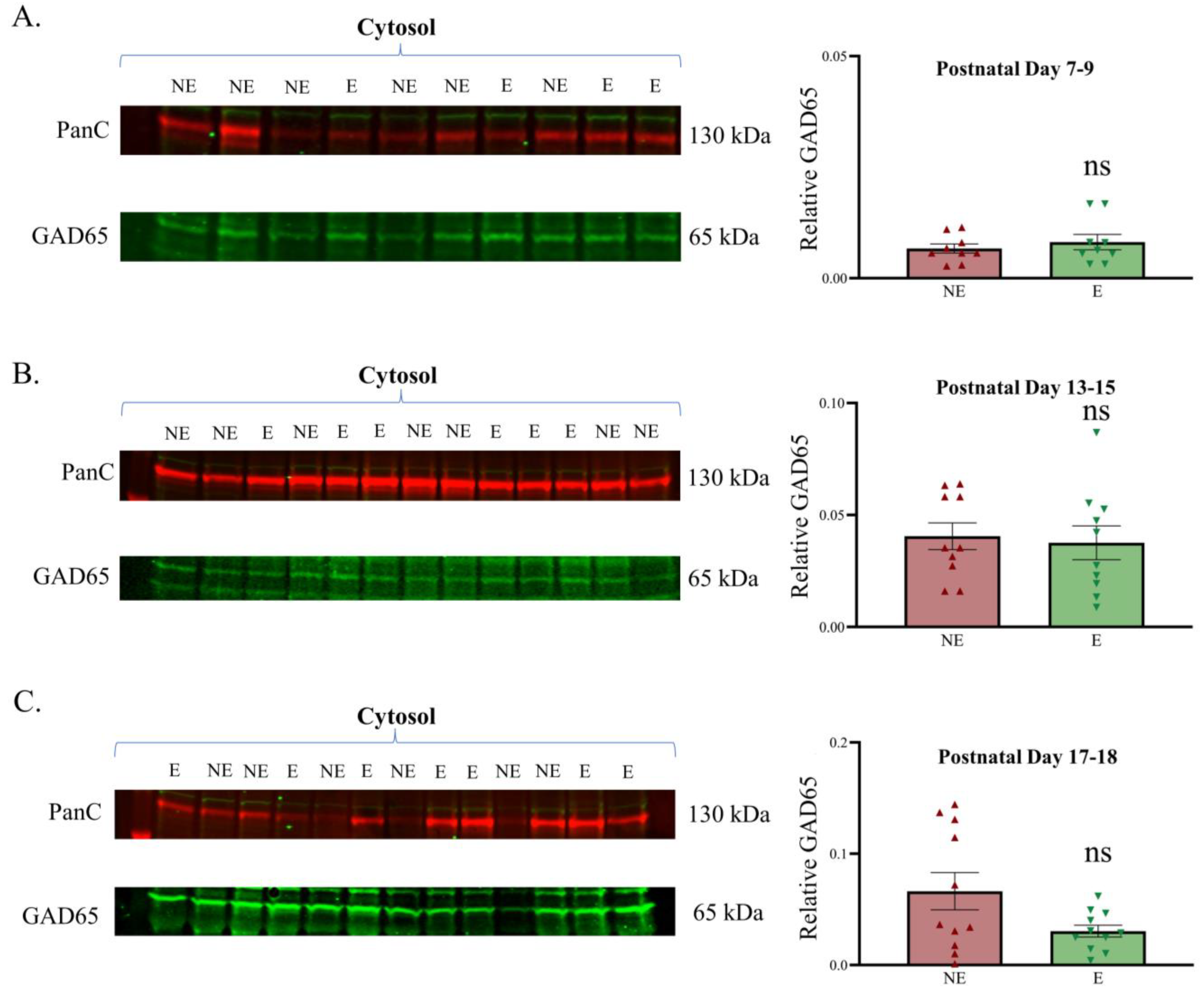
Relative expression of GAD65 in the developing primary SoCx. Western blot analyses of GAD65 reveal a lack of significant change in the epileptic (E) stargazer primary SoCx prior to the onset of seizures compared with NE control littermates. Assessments were conducted in cytosol fraction at PN 7–9, 13–15, and 17–18. (A) PN 7–9 (NE 0.007 ± 0.001, n = 9; E 0.008 ± 0.002, n = 9; p = 0.779), (B) PN 13–15 (NE 0.040 ± 0.006, n = 10; E 0.037 ± 0.007, n = 10; p = 0.517), and (C) PN 17–18 (NE 0.066 ± 0.017, n = 11; E 0.030 ± 0.005, n = 11; p = 0.217). The significance threshold was set at 0.05. ‘ns’ is indicative of no significant change found.
Figure 5.
Relative expression of GAD65 in the developing primary SoCx. Western blot analyses of GAD65 reveal a lack of significant change in the epileptic (E) stargazer primary SoCx prior to the onset of seizures compared with NE control littermates. Assessments were conducted in cytosol fraction at PN 7–9, 13–15, and 17–18. (A) PN 7–9 (NE 0.007 ± 0.001, n = 9; E 0.008 ± 0.002, n = 9; p = 0.779), (B) PN 13–15 (NE 0.040 ± 0.006, n = 10; E 0.037 ± 0.007, n = 10; p = 0.517), and (C) PN 17–18 (NE 0.066 ± 0.017, n = 11; E 0.030 ± 0.005, n = 11; p = 0.217). The significance threshold was set at 0.05. ‘ns’ is indicative of no significant change found.

Figure 6.
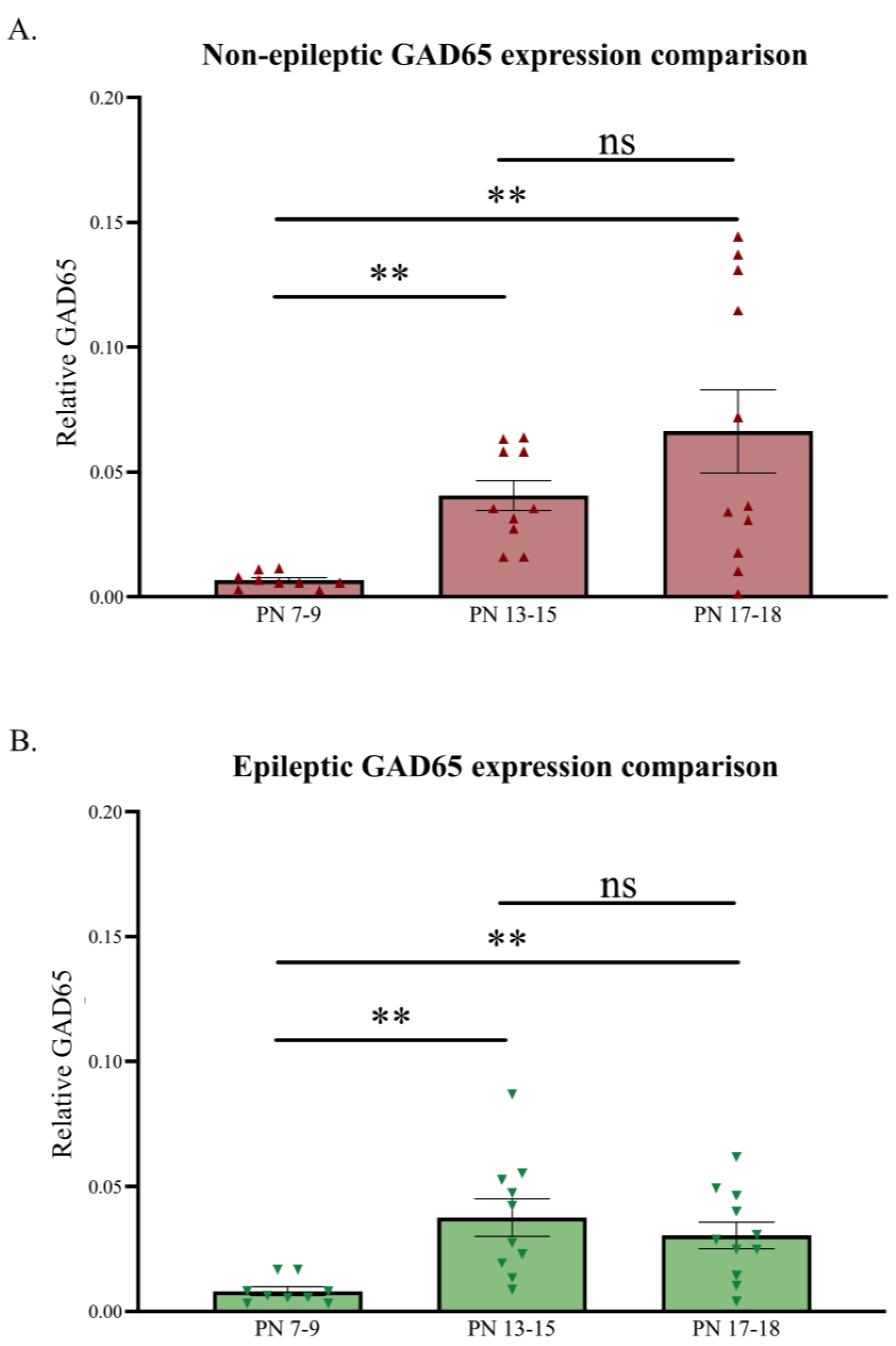
Relative developmental changes in GAD65 levels in the primary SoCx. GAD65 expression levels increase with increasing postnatal age in the NE control littermates and epileptic (E) stargazers, although the stargazers show a sudden dip at PN 17–18. (A) Kruskal–Wallis ANOVA results show a significant increase from PN 7–9 to 17–18 (H(2) = 14.36; p = 0.0008; **); post hoc Dunn’s multiple comparison test results, shown on the graph, reveal a significant increase in GAD65 from PN 7–9 to 17–18 (p = 0.002), PN 13–15 to 17–18 (p > 0.999), and PN 7–9 to 13–15 (p = 0.005). (B) Kruskal–Wallis ANOVA results show a significant increase from PN 7–9 to 17–18 (H(2) = 13.20; p = 0.001; **); post hoc Dunn’s multiple comparison test reveals a significant increase in GAD65 from PN 7–9 to 17–18 (p = 0.009), PN 13–15 to 17–18 (p > 0.999), and PN 7–9 to 13–15 (p = 0.002). The significance threshold was set at 0.05, with ** indicating p < 0.01. ‘ns’ is indicative of no significant change found.
Figure 6.
Relative developmental changes in GAD65 levels in the primary SoCx. GAD65 expression levels increase with increasing postnatal age in the NE control littermates and epileptic (E) stargazers, although the stargazers show a sudden dip at PN 17–18. (A) Kruskal–Wallis ANOVA results show a significant increase from PN 7–9 to 17–18 (H(2) = 14.36; p = 0.0008; **); post hoc Dunn’s multiple comparison test results, shown on the graph, reveal a significant increase in GAD65 from PN 7–9 to 17–18 (p = 0.002), PN 13–15 to 17–18 (p > 0.999), and PN 7–9 to 13–15 (p = 0.005). (B) Kruskal–Wallis ANOVA results show a significant increase from PN 7–9 to 17–18 (H(2) = 13.20; p = 0.001; **); post hoc Dunn’s multiple comparison test reveals a significant increase in GAD65 from PN 7–9 to 17–18 (p = 0.009), PN 13–15 to 17–18 (p > 0.999), and PN 7–9 to 13–15 (p = 0.002). The significance threshold was set at 0.05, with ** indicating p < 0.01. ‘ns’ is indicative of no significant change found.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hassan, M.; Grattan, D.R.; Leitch, B. Developmental Inhibitory Changes in the Primary Somatosensory Cortex of the Stargazer Mouse Model of Absence Epilepsy. Biomolecules 2023, 13, 186. https://doi.org/10.3390/biom13010186
AMA Style
Hassan M, Grattan DR, Leitch B. Developmental Inhibitory Changes in the Primary Somatosensory Cortex of the Stargazer Mouse Model of Absence Epilepsy. Biomolecules. 2023; 13(1):186. https://doi.org/10.3390/biom13010186
Chicago/Turabian StyleHassan, Muhammad, David R. Grattan, and Beulah Leitch. 2023. "Developmental Inhibitory Changes in the Primary Somatosensory Cortex of the Stargazer Mouse Model of Absence Epilepsy" Biomolecules 13, no. 1: 186. https://doi.org/10.3390/biom13010186
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.