
Metatranscriptomic Analysis Reveals Disordered Alterations in Oropharyngeal Microbiome during the Infection and Clearance Processes of SARS-CoV-2: A Warning for Secondary Infections
 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Design
2.2. Patient and Public Involvement
2.3. Sample Collection
2.4. RNA Extraction
2.5. Metatranscriptomic Sequencing
2.6. RT-qPCR Assay
2.7. Bioinformatics Analysis
2.8. Statistical Analyses
3. Results
3.1. Study Cohort
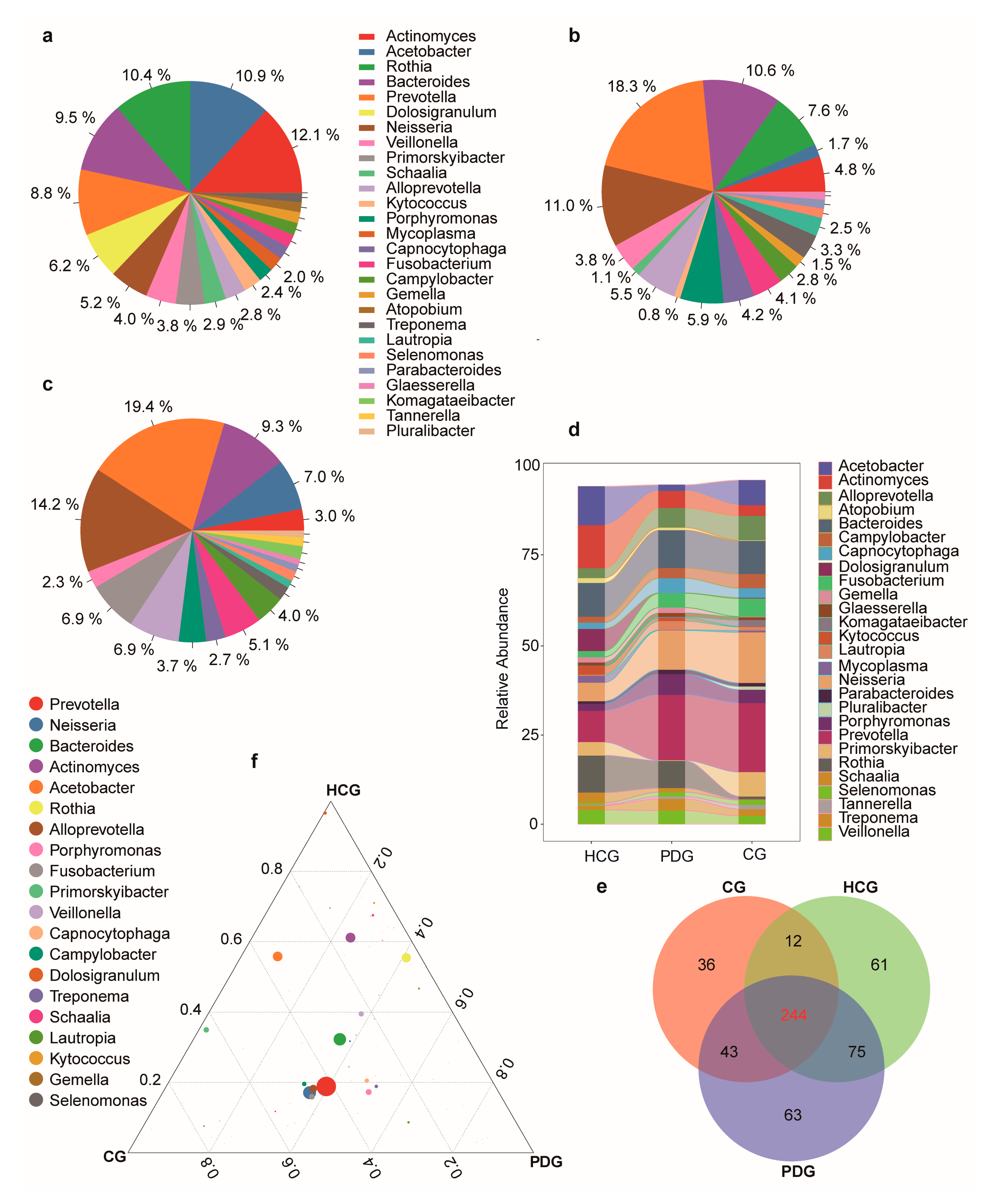
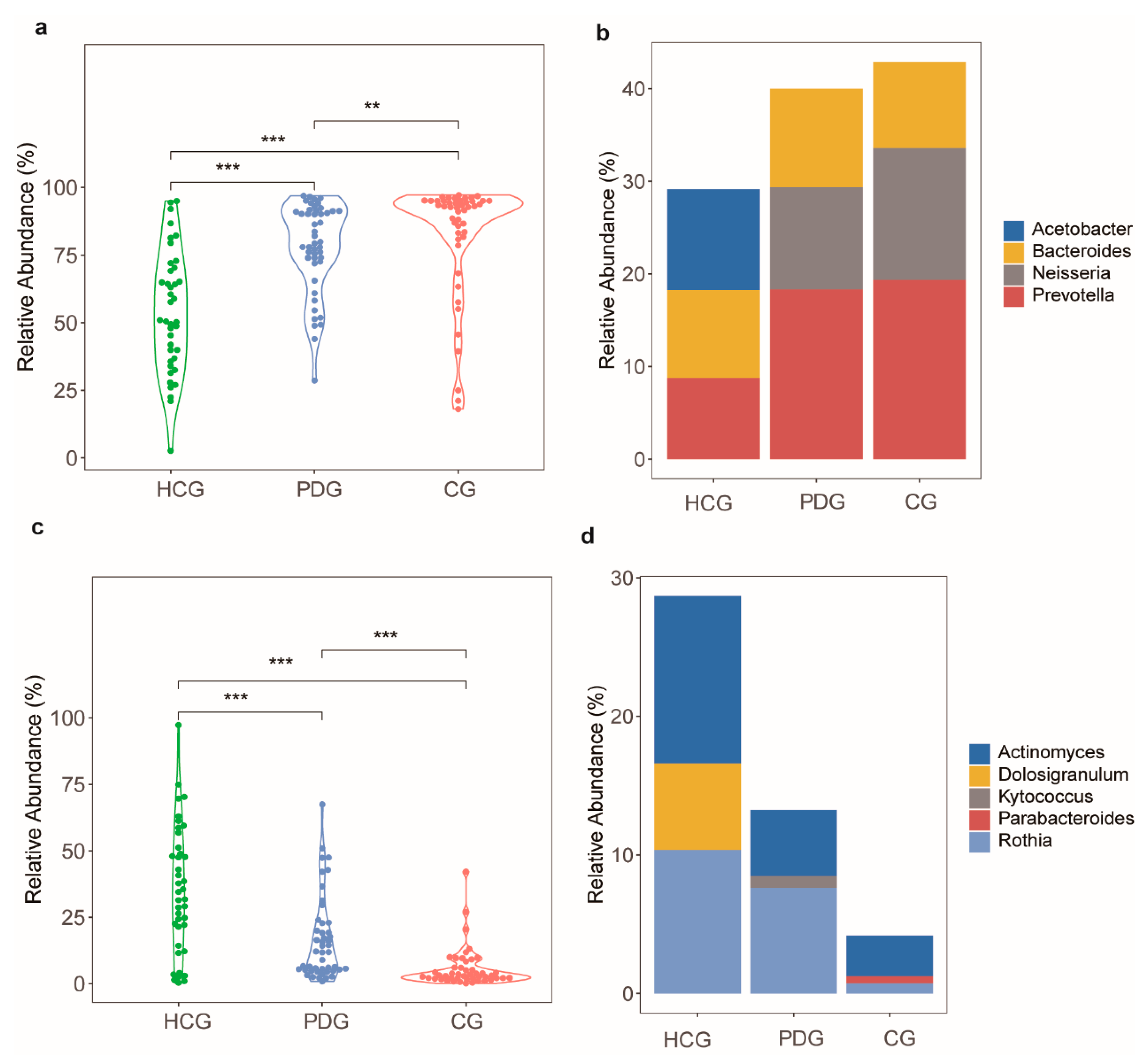
3.2. Relative Abundance Alterations in Oropharyngeal Bacteria during the Infection and Clearance Processes of SARS-CoV-2
3.3. Relative Abundance Alterations in Oropharyngeal Fungi during the Infection and Clearance Processes of SARS-CoV-2
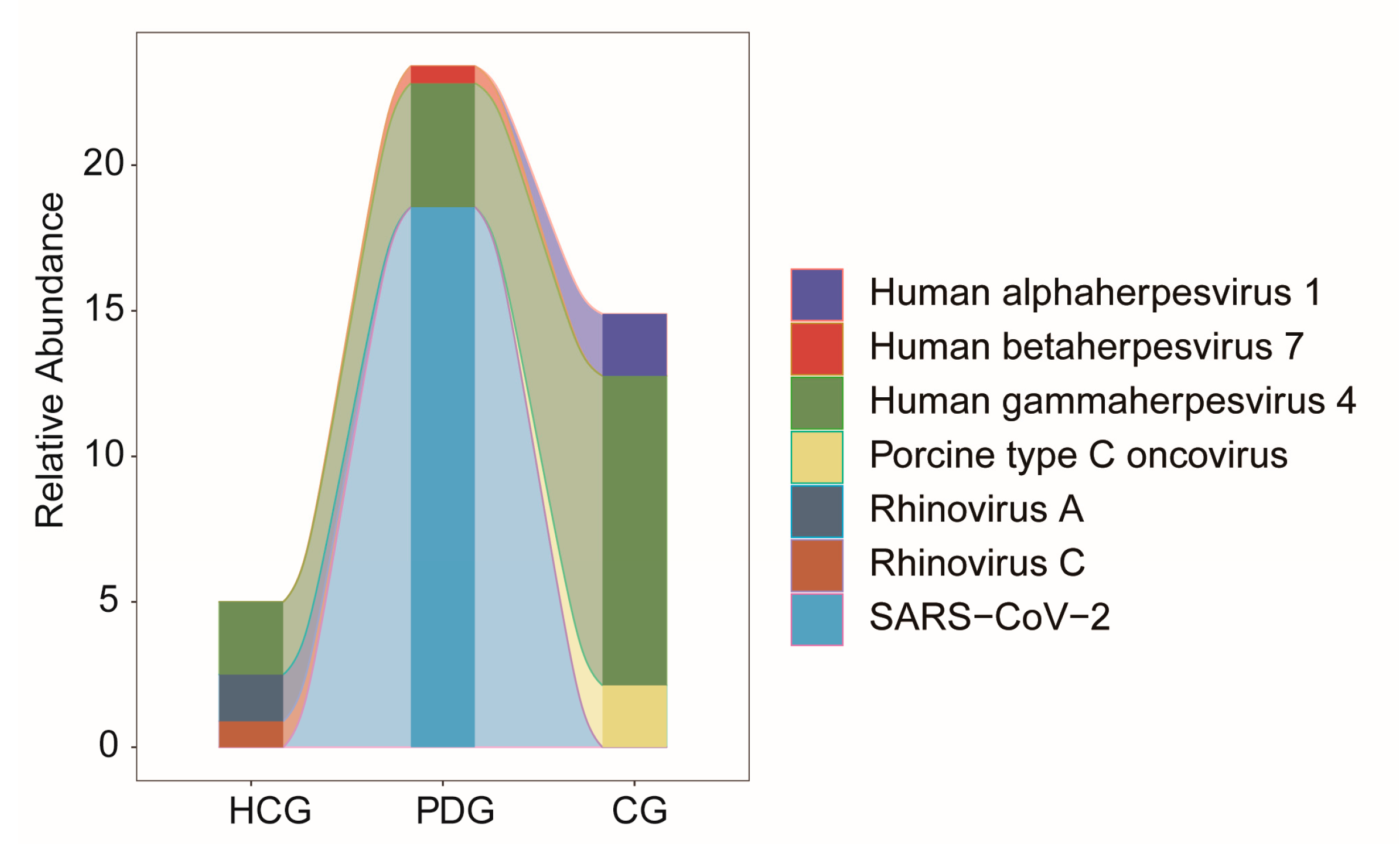
3.4. Relative Abundance Alterations in Oropharyngeal Viruses during the Infection and Clearance Processes of SARS-CoV-2
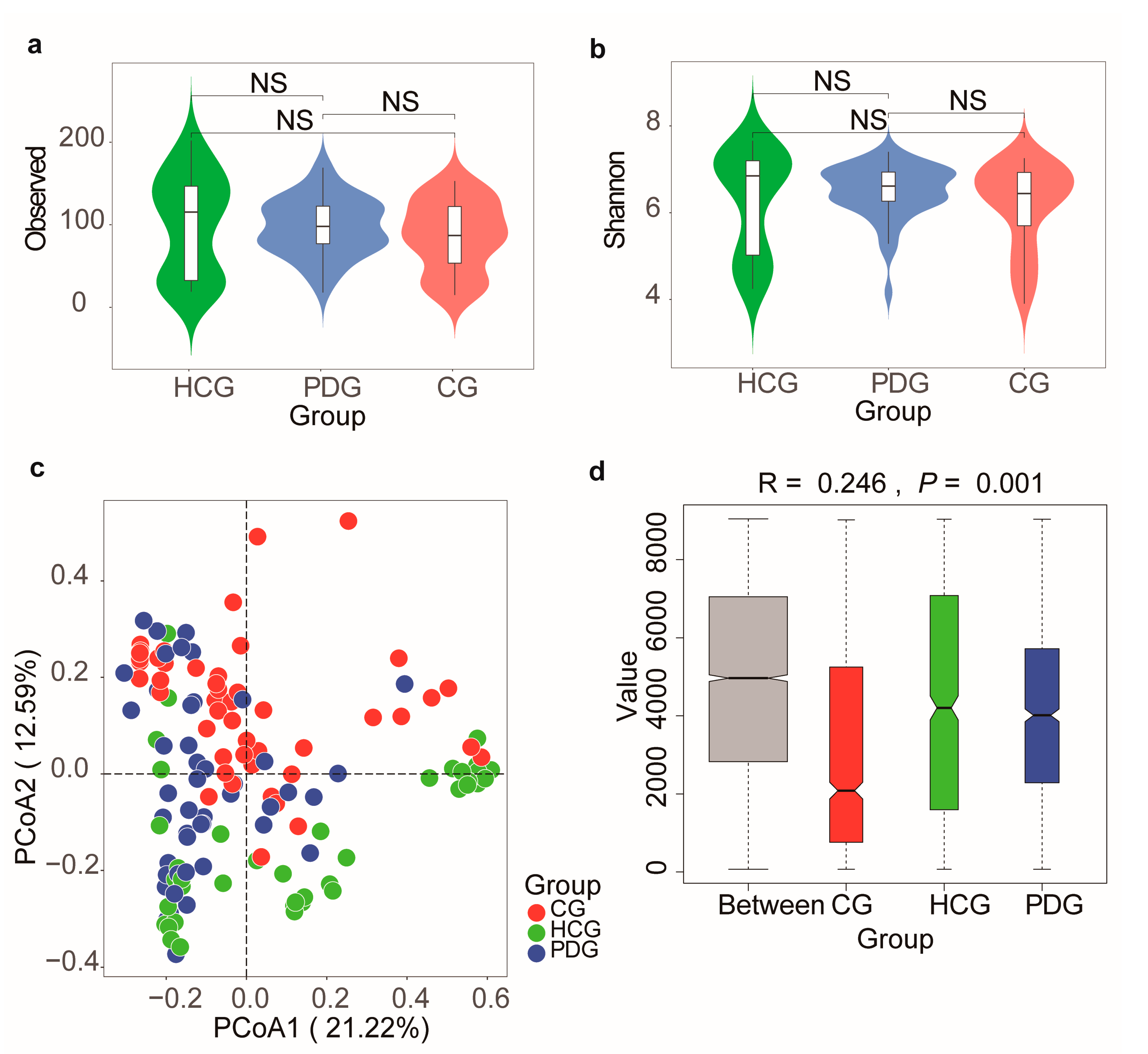
3.5. Diversity Alterations in Oropharyngeal Microbiome during the Infection and Clearance Processes of SARS-CoV-2
3.6. Potential Function Alterations in Oropharyngeal Microbiome during the Infection and Clearance Processes of SARS-CoV-2
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thierry, A.R. Host/genetic factors associated with COVID-19 call for precision medicine. Precis. Clin. Med. 2020, 3, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.-Y.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Zuo, T.; Zhan, H.; Zhang, F.; Liu, Q.; Tso, E.Y.; Lui, G.C.; Chen, N.; Li, A.; Lu, W.; Chan, F.K.; et al. Alterations in fecal fungal microbiome of patients with COVID-19 during time of hospitalization until discharge. Gastroenterology 2020, 159, 1302–1310. [Google Scholar] [CrossRef]
- Chen, Y.; Gu, S.; Chen, Y.; Lu, H.; Shi, D.; Guo, J.; Wu, W.-R.; Yang, Y.; Li, Y.; Xu, K.-J.; et al. Six-month follow-up of gut microbiota richness in patients with COVID-19. Gut 2022, 71, 222–225. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Xu, M.; Yi, P.; Liu, J.; Tao, J.J.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, I.; Chung, M.; Angel, L.; Tsay, J.-C.J.; Wu, B.G.; Yeung, S.T.; Krolikowski, K.; Li, Y.; Duerr, R.; Schluger, R.; et al. Microbial signatures in the lower airways of mechanically ventilated COVID-19 patients associated with poor clinical outcome. Nat. Microbiol. 2021, 6, 1245–1258. [Google Scholar] [CrossRef]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Westwood, D.; MacFadden, D.R.; Soucy, J.-P.R.; Daneman, N. Bacterial co-infection and secondary infection in patients with COVID-19: A living rapid review and meta-analysis. Clin. Microbiol. Infect. 2020, 26, 1622–1629. [Google Scholar] [CrossRef]
- Bao, L.; Zhang, C.; Dong, J.; Zhao, L.; Li, Y.; Sun, J. Oral Microbiome and SARS-CoV-2: Beware of Lung Co-infection. Front. Microbiol. 2020, 11, 1840. [Google Scholar] [CrossRef]
- Vaillancourt, M.; Jorth, P. The unrecognized threat of secondary bacterial infections with COVID-19. Mbio 2020, 11, e01806-20. [Google Scholar] [CrossRef]
- Kumpitsch, C.; Koskinen, K.; Schöpf, V.; Moissl-Eichinger, C. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 2019, 17, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Zhang, F.; Zhou, F.; Li, H.; Ge, W.; Gan, R.; Nie, H.; Li, B.; Wang, Y.; Wu, M.; et al. Metagenomic analysis reveals oropharyngeal microbiota alterations in patients with COVID-19. Signal Transduct. Target. Ther. 2021, 6, 191. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-f.; Li, A.; Zhang, T.; Ren, Z.-g.; He, K.-x.; Zhang, H.; Luo, Q.-x.; Zhou, K.; Chen, C.-l.; Chen, X.-l.; et al. Disordered oropharyngeal microbial communities in H7N9 patients with or without secondary bacterial lung infection. Emerg. Microbes Infect. 2017, 6, e112. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic diversity of severe acute respiratory Syndrome–Coronavirus 2 in patients with coronavirus disease 2019. Clin. Infect. Dis. 2020, 71, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Liu, J.; Zhao, X.; Liu, C.; Wang, W.; Wang, D.; Xu, W.; Zhang, C.; Yu, J.; Jiang, B.; et al. Clinical characteristics of imported cases of coronavirus disease 2019 (COVID-19) in Jiangsu province: A multicenter descriptive study. Clin. Infect. Dis. 2020, 71, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tan, L.; Wang, X.; Liu, W.; Lu, Y.; Cheng, L.; Sun, Z. Comparison of nasopharyngeal and oropharyngeal swabs for SARS-CoV-2 detection in 353 patients received tests with both specimens simultaneously. Int. J. Infect. Dis. 2020, 94, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. FASTP: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Clarke, E.L.; Taylor, L.J.; Zhao, C.; Connell, A.; Lee, J.-J.; Fett, B.; Bushman, F.D.; Bittinger, K. Sunbeam: An extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome 2019, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Shaaban, H.; Westfall, D.A.; Mohammad, R.; Danko, D.; Bezdan, D.; Afshinnekoo, E.; Segata, N.; Mason, C.E. The microbe directory: An annotated, searchable inventory of microbes’ characteristics. Gates Open Res. 2018, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.; Mackenzie, B.W.; Waldvogel-Thurlow, S.; Middleditch, M.; Jullig, M.; Zoing, M.; Taylor, M.W.; Douglas, R.G. Differentially regulated host proteins associated with chronic rhinosinusitis are correlated with the sinonasal microbiome. Front. Cell. Infect. Microbiol. 2017, 7, 504. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J.; Walsh, D.C. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 2013, 83, 557–574. [Google Scholar] [CrossRef]
- Lemon, K.P.; Klepac-Ceraj, V.; Schiffer, H.K.; Brodie, E.L.; Lynch, S.V.; Kolter, R. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. Mbio 2010, 1, e00129-10. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Hu, X.; Li, Z.; Tu, C.; Wang, Y.; Pang, P.; Zhang, H.; Zheng, X.; Liang, Y.; Shan, H.; et al. Effect of SARS-CoV-2 infection on the microbial composition of upper airway. Infect. Drug. Resist. 2020, 13, 2637–2640. [Google Scholar] [CrossRef]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef] [Green Version]
- Manna, S.; Baindara, P.; Mandal, S.M. Molecular pathogenesis of secondary bacterial infection associated to viral infections including SARS-CoV-2. J. Infect. Public Healthy 2020, 13, 1397–1404. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Chong, W.H.; Saha, B.K.; Ramani, A.; Chopra, A. State-of-the-art review of secondary pulmonary infections in patients with COVID-19 pneumonia. Infection 2021, 49, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Kurra, N.; Woodard, P.I.; Gandrakota, N.; Gandhi, H.; Polisetty, S.R.; Ang, S.P.; Patel, K.P.; Chitimalla, V.; Baig, M.M.A.; Samudrala, G. Opportunistic infections in COVID-19: A systematic review and meta-analysis. Cureus 2022, 14, e23687. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.H.; Annavajhala, M.K.; Chong, A.M.; Park, H.; Nobel, Y.R.; Soroush, A.; Blackett, J.W.; Krigel, A.; Phipps, M.M.; Freedberg, D.E.; et al. Oral microbiome alterations and SARS-CoV-2 Saliva viral load in patients with COVID-19. Microbiol. Spectr. 2021, 9, e0005521. [Google Scholar] [CrossRef]
- Gao, M.; Wang, H.; Luo, H.; Sun, Y.; Wang, L.; Ding, S.; Ren, H.; Gang, J.; Rao, B.; Liu, S.; et al. Characterization of the human oropharyngeal microbiomes in SARS-CoV-2 infection and recovery patients. Adv. Sci. 2021, 8, e2102785. [Google Scholar] [CrossRef]
- Dudoignon, E.; Caméléna, F.; Deniau, B.; Habay, A.; Coutrot, M.; Ressaire, Q.; Plaud, B.; Berçot, B.; Dépret, F. Bacterial pneumonia in COVID-19 critically Ill patients: A case series. Clin. Infect. Dis. 2021, 72, 905–906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Y.; Wu, J.; Li, Y.; Zhou, X.; Li, X.; Chen, H.; Guo, M.; Chen, S.; Sun, F.; et al. Risks and features of secondary infections in severe and critical ill COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 1958–1964. [Google Scholar] [CrossRef] [PubMed]
- Al Hamdan, A.S.; Alghamdi, A.A.; Alyousif, G.F.; Hamza, F.A.; Shafey, M.M.; AlAmri, A.M.; Sunki, A.A. Evaluating the prevalence and the risk factors of gram-negative multi-drug resistant bacteria in Eastern Saudi Arabia. Infect. Drug. Resist. 2022, 15, 475–490. [Google Scholar] [CrossRef]
- Kariyawasam, R.M.; Julien, D.A.; Jelinski, D.C.; Larose, S.L.; Rennert-May, E.; Conly, J.M.; Dingle, T.C.; Chen, J.Z.; Tyrrell, G.J.; Ronksley, P.E.; et al. Antimicrobial resistance (AMR) in COVID-19 patients: A systematic review and meta-analysis (November 2019–June 2021). Antimicrob. Resist. Infect. Control. 2022, 11, 45. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Patients with COVID-19 | Healthy Controls |
|---|---|---|
| Number of subjects | 47 | 40 |
| Age-years (mean ± SD) | 44.7 ± 15.3 | 45.8 ± 16.4 |
| Gender | ||
| Male | 20/47(42.5%) | 20/40(50.0%) |
| Female | 27/47(57.5%) | 20/40(50.0%) |
| Disease severity category | ||
| Mild | 5/47 (10.6%) | NA |
| Moderate | 25/47 (53.2%) | NA |
| Severe | 10/47 (21.3%) | NA |
| Critical | 7/47 (14.9%) | NA |
| Symptoms at admission | ||
| Fever | 29/47 (61.7%) | NA |
| Cough | 29/47 (61.7%) | NA |
| Sputum | 19/47 (40.4%) | NA |
| Sore throat | 7/47 (14.9%) | NA |
| Shortness of breath | 17/47 (36.2%) | NA |
| Received antibiotics during first week of hospitalization by disease severity 1 | ||
| Mild | 1/47 (2.1%) | NA |
| Moderate | 3/47 (6.4%) | NA |
| Severe | 5/47 (10.6%) | NA |
| Critical | 7/47 (14.9%) | NA |
| Received antivirals during first week of hospitalization by disease severity 2 | ||
| Mild | 4/47 (8.5%) | NA |
| Moderate | 24/47 (51.1%) | NA |
| Severe | 10/47 (21.3%) | NA |
| Critical | 7/47 (14.9%) | NA |
| Comorbidities | ||
| Hypertension | 7/47 (14.9%) | 2/40 (5.0%) |
| Heart disease | 4/47 (8.5%) | 0 |
| Diabetes | 5/47 (10.6%) | 2/40 (5.0%) |
| Chronic obstructive pulmonary disease | 1/47 (2.1%) | 1/40 (2.5 %) |
| Chronic bronchitis | 2/47 (4.3%) | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Lu, S.; Wei, X.; Hu, Y.; Li, H.; Wang, J.; Lin, Y.; Li, M.; Wang, M.; Ma, J.; et al. Metatranscriptomic Analysis Reveals Disordered Alterations in Oropharyngeal Microbiome during the Infection and Clearance Processes of SARS-CoV-2: A Warning for Secondary Infections. Biomolecules 2023, 13, 6. https://doi.org/10.3390/biom13010006
Zhou Y, Lu S, Wei X, Hu Y, Li H, Wang J, Lin Y, Li M, Wang M, Ma J, et al. Metatranscriptomic Analysis Reveals Disordered Alterations in Oropharyngeal Microbiome during the Infection and Clearance Processes of SARS-CoV-2: A Warning for Secondary Infections. Biomolecules. 2023; 13(1):6. https://doi.org/10.3390/biom13010006
Chicago/Turabian StyleZhou, Yongzhao, Sifen Lu, Xiaozhen Wei, Ya Hu, Honghao Li, Jing Wang, Yifei Lin, Mengjiao Li, Minjin Wang, Jinmin Ma, and et al. 2023. "Metatranscriptomic Analysis Reveals Disordered Alterations in Oropharyngeal Microbiome during the Infection and Clearance Processes of SARS-CoV-2: A Warning for Secondary Infections" Biomolecules 13, no. 1: 6. https://doi.org/10.3390/biom13010006