
Integrative Organelle-Based Functional Proteomics: In Silico Prediction of Impaired Functional Annotations in SACS KO Cell Model

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Subcellular Fractionation of Cells for Mitochondrial and Lysosome Enrichment
2.3. Proteomic Analysis
2.4. Bioinformatic Analysis and Categorization of Proteomic Data
2.5. Oxygen Consumption Rate (OCR) Measurement
2.6. Mitochondrial Oxidative Stress Measurement
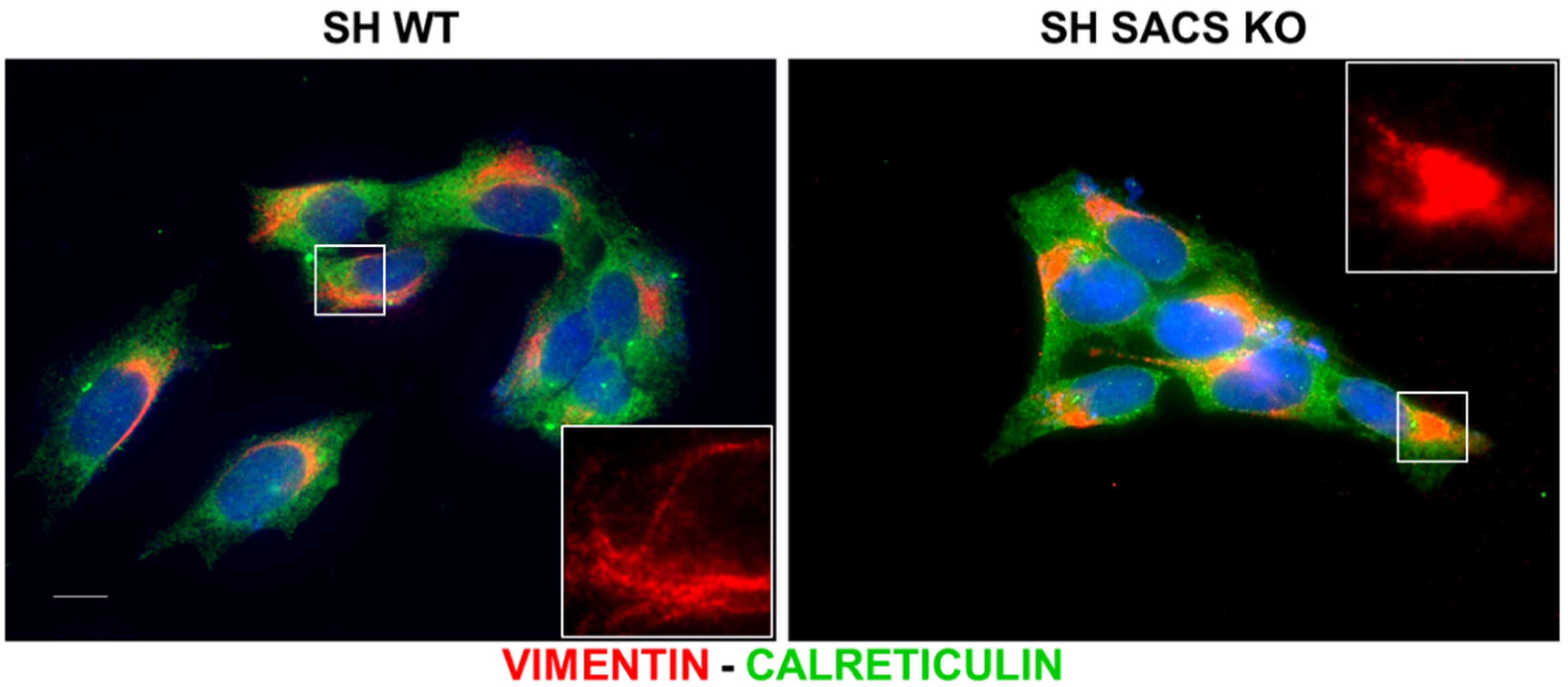
2.7. Immunofluorescence Analysis
2.8. Statistics
3. Results
3.1. Mitochondrial-Specific Proteome Profile
3.2. Lysosome-Specific Proteomic Profile
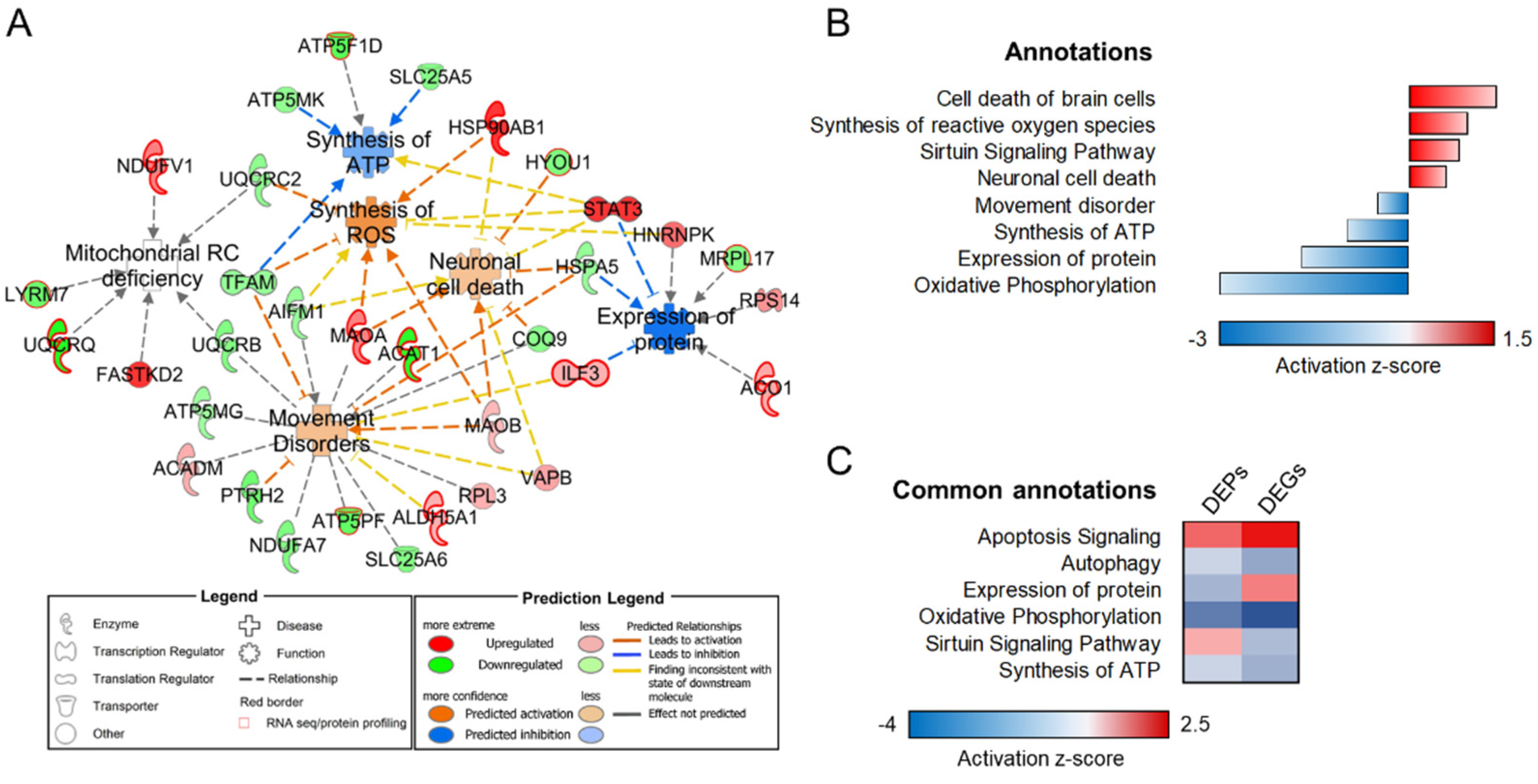
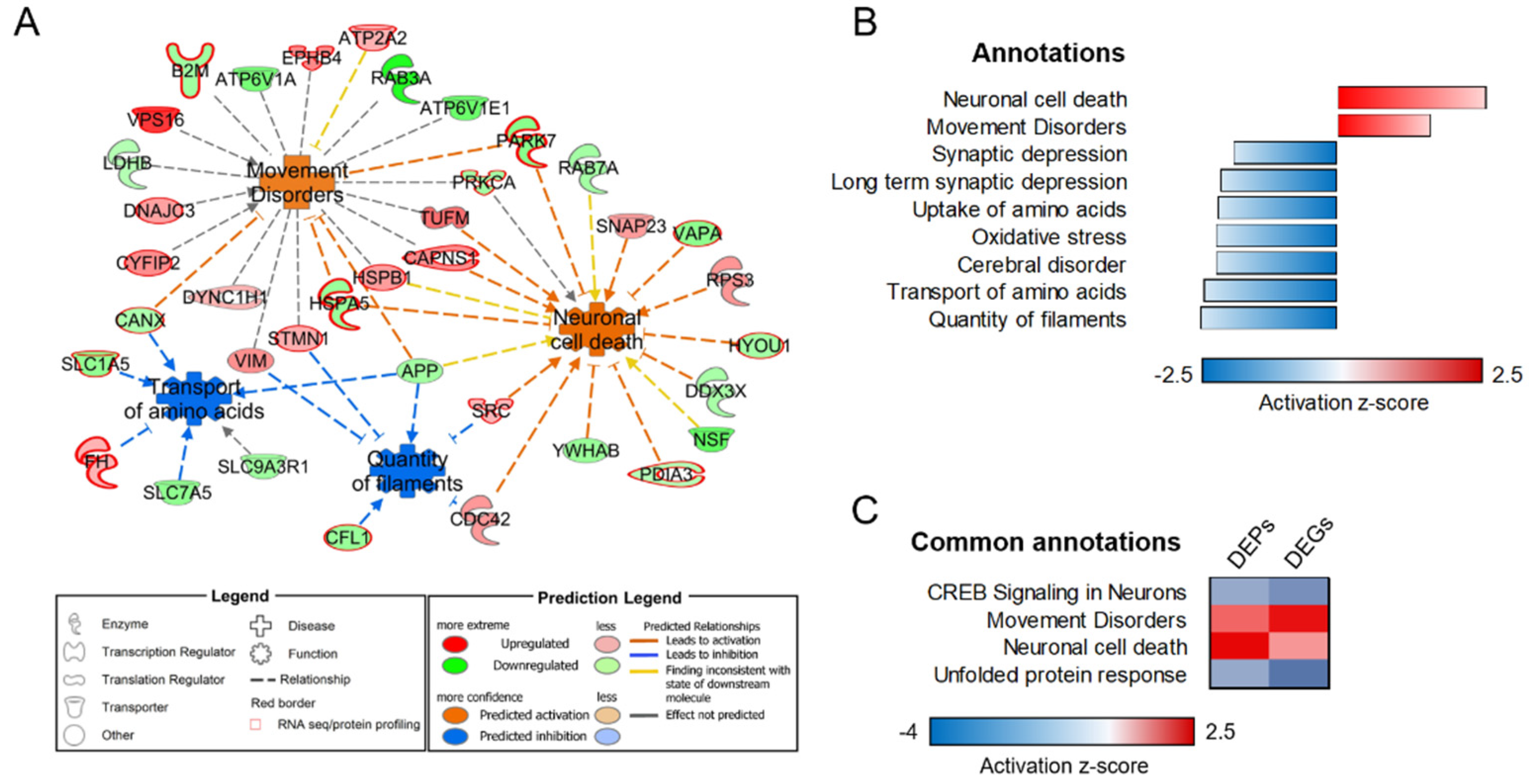
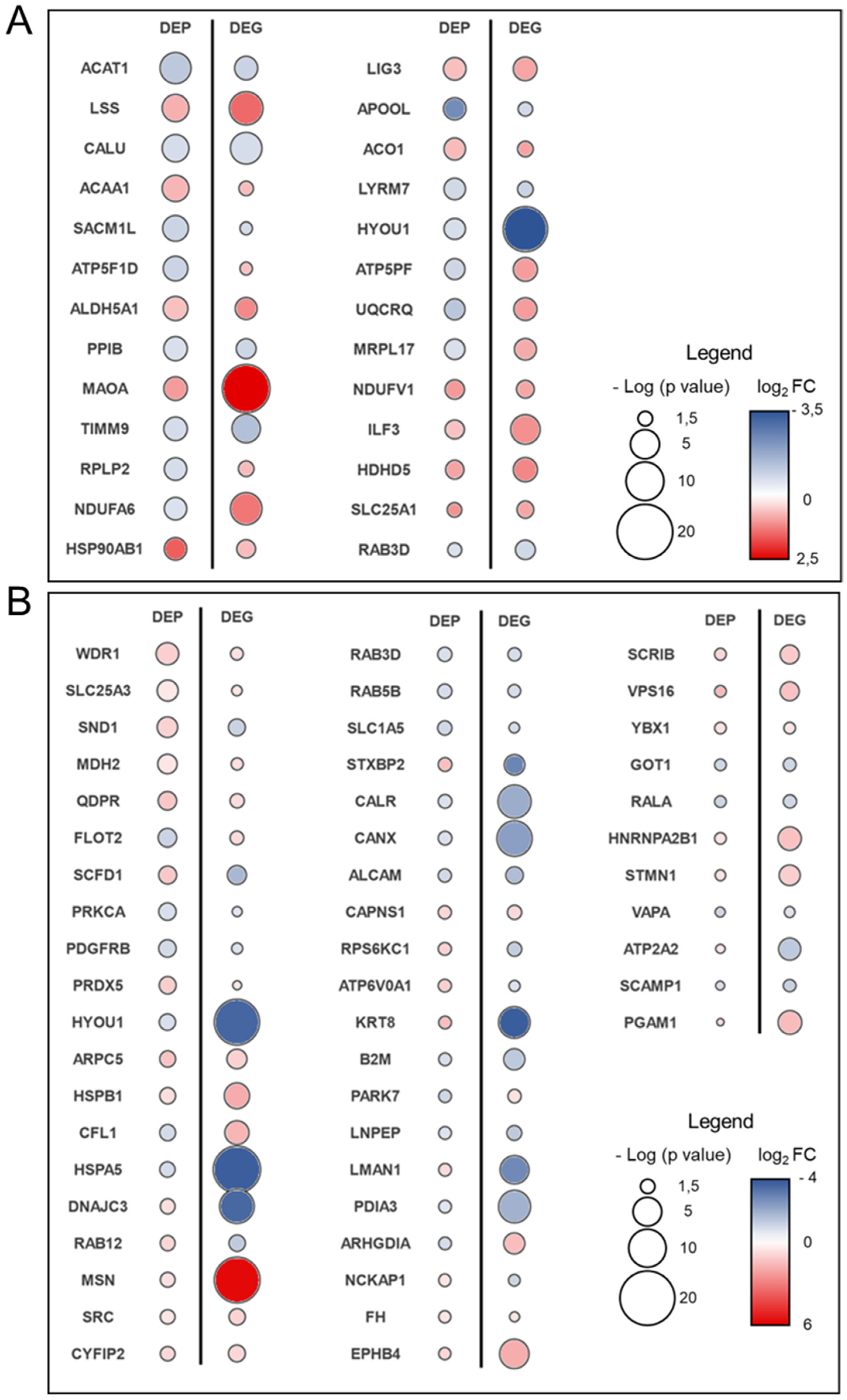
3.3. Transcriptome–Proteome Integrative Analysis
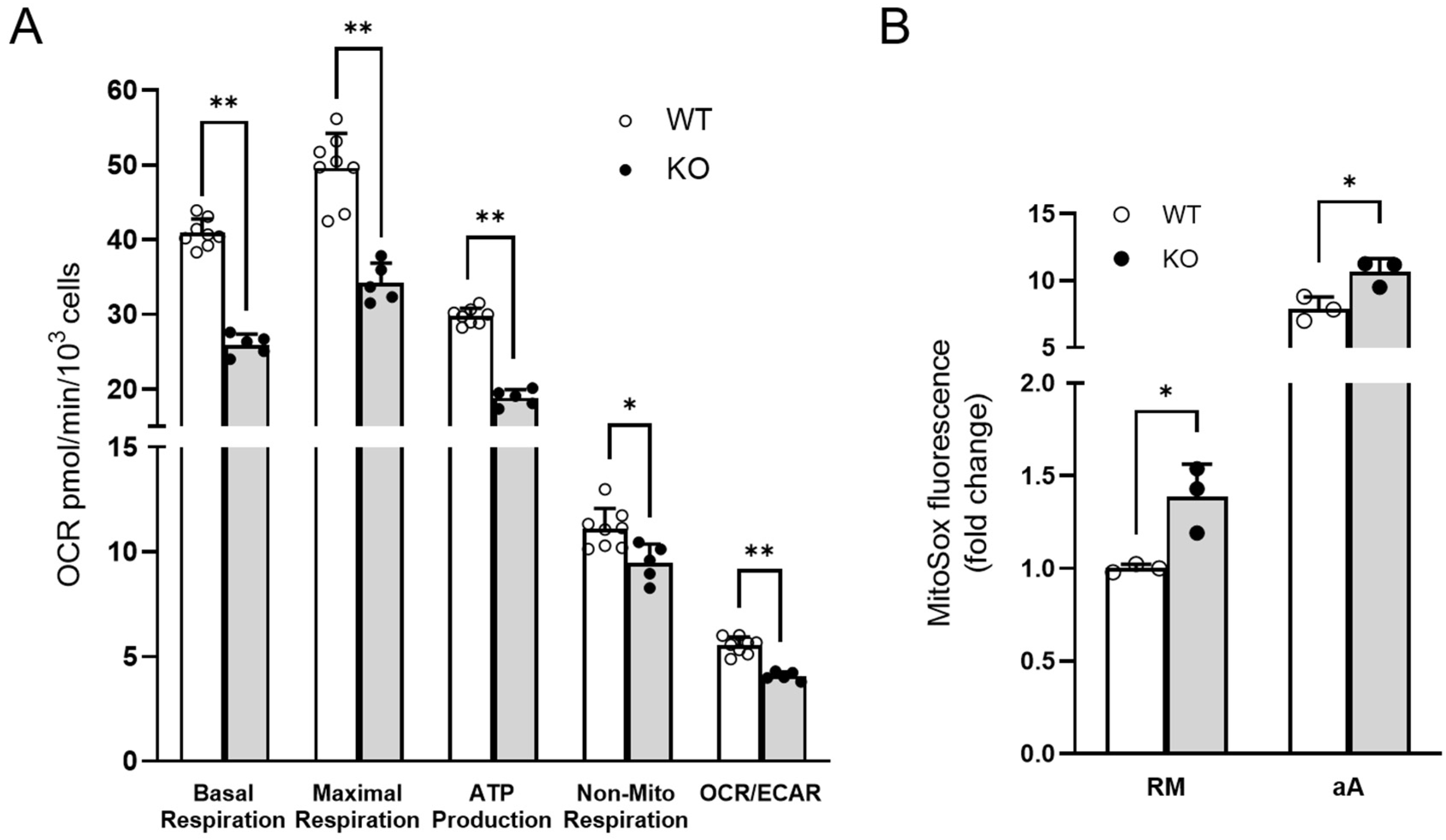
3.4. Experimental Validations to Corroborate In Silico Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engert, J.C.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.-P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000, 24, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Tessa, A.; Barca, A.; Fattori, F.; de Leva, M.F.; Terracciano, A.; Storelli, C.; Santorelli, F.M.; Verri, T. Comparative analysis and functional mapping of SACS mutations reveal novel insights into sacsin repeated architecture. Hum. Mutat. 2013, 34, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.F.; Siller, E.; Barral, J.M. The neurodegenerative-disease-related protein sacsin is a molecular chaperone. J. Mol. Biol. 2011, 411, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Lariviere, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.M.; Duchen, M.R.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, C.; Procaccini, C.; Meschini, M.C.; Cianflone, A.; Carbone, R.; Doccini, S.; Devos, D.; Nesti, C.; Vuillaume, I.; Pellegrino, M.; et al. Powerhouse failure and oxidative damage in autosomal recessive spastic ataxia of Charlevoix-Saguenay. J. Neurol. 2015, 262, 2755–2763. [Google Scholar] [CrossRef] [Green Version]
- Morani, F.; Doccini, S.; Sirica, R.; Paterno, M.; Pezzini, F.; Ricca, I.; Simonati, A.; Delledonne, M.; Santorelli, F.M. Functional Transcriptome Analysis in ARSACS KO Cell Model Reveals a Role of Sacsin in Autophagy. Sci. Rep. 2019, 9, 11878. [Google Scholar] [CrossRef]
- Gentil, B.J.; Lai, G.-T.; Menade, M.; Larivière, R.; Minotti, S.; Gehring, K.; Chapple, J.-P.; Brais, B.; Durham, H.D. Sacsin, mutated in the ataxia ARSACS, regulates intermediate filament assembly and dynamics. FASEB J. 2019, 33, 2982–2994. [Google Scholar] [CrossRef] [Green Version]
- Duncan, E.J.; Larivière, R.; Bradshaw, T.Y.; Longo, F.; Sgarioto, N.; Hayes, M.J.; Romano, L.E.L.; Nethisinghe, S.; Giunti, P.; Bruntraeger, M.B.; et al. Altered organization of the intermediate filament cytoskeleton and relocalization of proteostasis modulators in cells lacking the ataxia protein sacsin. Hum. Mol. Genet. 2017, 26, 3130–3143. [Google Scholar] [CrossRef] [Green Version]
- Ady, V.; Toscano-Márquez, B.; Nath, M.; Chang, P.K.; Hui, J.; Cook, A.; Charron, F.; Larivière, R.; Brais, B.; McKinney, R.A.; et al. Altered synaptic and firing properties of cerebellar Purkinje cells in a mouse model of ARSACS. J. Physiol. 2018, 596, 4253–4267. [Google Scholar] [CrossRef]
- Takiyama, Y. Sacsinopathies: Sacsin-related ataxia. Cerebellum 2007, 6, 353–359. [Google Scholar] [CrossRef]
- Vermeer, S.; Meijer, R.P.P.; Pijl, B.J.; Timmermans, J.; Cruysberg, J.R.M.; Bos, M.M.; Schelhaas, H.J.; van de Warrenburg, B.P.C.; Knoers, N.V.A.M.; Scheffer, H.; et al. ARSACS in the Dutch population: A frequent cause of early-onset cerebellar ataxia. Neurogenetics 2008, 9, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synofzik, M.; Soehn, A.S.; Gburek-Augustat, J.; Schicks, J.; Karle, K.N.; Schüle, R.; Haack, T.B.; Schöning, M.; Biskup, S.; Rudnik-Schöneborn, S.; et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): Expanding the genetic, clinical and imaging spectrum. Orphanet J. Rare Dis. 2013, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Varman, M.; Yoganathan, S.; Subhash, P.K.; Mani, S. Teaching NeuroImages: Autosomal recessive spastic ataxia of Charlevoix-Saguenay: Typical MRI findings. Neurology 2018, 90, e1271–e1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagaria, J.; Bagyinszky, E.; An, S.S.A. Genetics of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) and Role of Sacsin in Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 552. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Götzl, J.K.; Lang, C.M.; Haass, C.; Capell, A. Impaired protein degradation in FTLD and related disorders. Ageing Res. Rev. 2016, 32, 122–139. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Haslett, L.J. The lysosomal storage disease continuum with ageing-related neurodegenerative disease. Ageing Res. Rev. 2016, 32, 104–121. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.-S.; Lee, J.-H. Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy 2008, 4, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Doccini, S.; Morani, F.; Nesti, C.; Pezzini, F.; Calza, G.; Soliymani, R.; Signore, G.; Rocchiccioli, S.; Kanninen, K.M.; Huuskonen, M.T.; et al. Proteomic and functional analyses in disease models reveal CLN5 protein involvement in mitochondrial dysfunction. Cell Death Discov. 2020, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Lie, P.P.Y.; Nixon, R.A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Overly, C.C.; Lee, K.D.; Berthiaume, E.; Hollenbeck, P.J. Quantitative measurement of intraorganelle pH in the endosomal-lysosomal pathway in neurons by using ratiometric imaging with pyranine. Proc. Natl. Acad. Sci. USA 1995, 92, 3156–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overly, C.C.; Hollenbeck, P.J. Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J. Neurosci. 1996, 16, 6056–6064. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Shastry, P.; Basu, A.; Rajadhyaksha, M.S. Neuroblastoma cell lines—A versatile in vitro model in neurobiology. Int. J. Neurosci. 2001, 108, 109–126. [Google Scholar] [CrossRef]
- Scifo, E.; Szwajda, A.; Soliymani, R.; Pezzini, F.; Bianchi, M.; Dapkunas, A.; Debski, J.; Uusi-Rauva, K.; Dadlez, M.; Gingras, A.C.; et al. Proteomic analysis of the palmitoyl protein thioesterase 1 interactome in SH-SY5Y human neuroblastoma cells. J. Proteom. 2015, 123, 42–53. [Google Scholar] [CrossRef]
- Laakkonen, E.K.; Soliymani, R.; Karvinen, S.; Kaprio, J.; Kujala, U.M.; Baumann, M.; Sipilä, S.; Kovanen, V.; Lalowski, M. Estrogenic regulation of skeletal muscle proteome: A study of premenopausal women and postmenopausal MZ cotwins discordant for hormonal therapy. Aging Cell 2017, 16, 1276–1287. [Google Scholar] [CrossRef]
- Zhang, J.; Nuebel, E.; Wisidagama, D.R.R.; Setoguchi, K.; Hong, J.S.; Van Horn, C.M.; Imam, S.S.; Vergnes, L.; Malone, C.S.; Koehler, C.M.; et al. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat. Protoc. 2012, 7, 1068–1085. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.A.; Fisher-Wellman, K.H.; Neufer, P.D. From OCR and ECAR to energy: Perspectives on the design and interpretation of bioenergetics studies. J. Biol. Chem. 2021, 297, 101140. [Google Scholar] [CrossRef]
- Luo, C.; Zhao, S.; Dai, W.; Zheng, N.; Wang, J. Proteomic Analysis of Lysosomal Membrane Proteins in Bovine Mammary Epithelial Cells Illuminates Potential Novel Lysosome Functions in Lactation. J. Agric. Food Chem. 2018, 66, 13041–13049. [Google Scholar] [CrossRef]
- The Human Lysosome Gene Database—hLGDBhLGDB V.1.2 hLGDB v.1.2. (2013)—University of Perugia, Dept. of Experimental Medicine and Biochemical Sciences. Available online: http://lysosome.unipg.it/ (accessed on 8 June 2022).
- Brozzi, A.; Urbanelli, L.; Luc Germain, P.; Magini, A.; Emiliani, C. hLGDB: A database of human lysosomal genes and their regulation. Database 2013, 2013, bat024. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Doccini, S.; Marchese, M.; Morani, F.; Gammaldi, N.; Mero, S.; Pezzini, F.; Soliymani, R.; Santi, M.; Signore, G.; Ogi, A.; et al. Lysosomal Proteomics Links Disturbances in Lipid Homeostasis and Sphingolipid Metabolism to CLN5 Disease. Cells 2022, 11, 1840. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Schilder, B.M.; Navarro, E.; Raj, T. Multi-omic insights into Parkinson’s Disease: From genetic associations to functional mechanisms. Neurobiol. Dis. 2022, 163, 105580. [Google Scholar] [CrossRef]
- Wang, M.; Li, A.; Sekiya, M.; Beckmann, N.D.; Quan, X.; Schrode, N.; Fernando, M.B.; Yu, A.; Zhu, L.; Cao, J.; et al. Transformative Network Modeling of Multi-omics Data Reveals Detailed Circuits, Key Regulators, and Potential Therapeutics for Alzheimer’s Disease. Neuron 2021, 109, 257–272.e14. [Google Scholar] [CrossRef]
- Ruffini, N.; Klingenberg, S.; Schweiger, S.; Gerber, S. Common Factors in Neurodegeneration: A Meta-Study Revealing Shared Patterns on a Multi-Omics Scale. Cells 2020, 9, 2642. [Google Scholar] [CrossRef]
- Nativio, R.; Lan, Y.; Donahue, G.; Sidoli, S.; Berson, A.; Srinivasan, A.R.; Shcherbakova, O.; Amlie-Wolf, A.; Nie, J.; Cui, X.; et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat. Genet. 2020, 52, 1024–1035. [Google Scholar] [CrossRef]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef]
- Makioka, K.; Yamazaki, T.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Involvement of endoplasmic reticulum stress defined by activated unfolded protein response in multiple system atrophy. J. Neurol. Sci. 2010, 297, 60–65. [Google Scholar] [CrossRef]
- Lindquist, S.L.; Kelly, J.W. Chemical and Biological Approaches for Adapting Proteostasis to Ameliorate Protein Misfolding and Aggregation Diseases-Progress and Prognosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004507. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remondelli, P.; Renna, M. The Endoplasmic Reticulum Unfolded Protein Response in Neurodegenerative Disorders and Its Potential Therapeutic Significance. Front. Mol. Neurosci. 2017, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.H.; Zlokovic, B.; Fernández, J.A. Activated protein C: Potential therapy for severe sepsis, thrombosis, and stroke. Semin. Hematol. 2002, 39, 197–205. [Google Scholar] [CrossRef]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, M.; Xue, J.; Chong, Y. Calreticulin protects the HT22 hippocampal neurons from injury induced by hypoxia. Mol. Cell. Toxicol. 2018, 14, 79–86. [Google Scholar] [CrossRef]
- Fricker, M.; Oliva-Martín, M.J.; Brown, G.C. Primary phagocytosis of viable neurons by microglia activated with LPS or Aβ is dependent on calreticulin/LRP phagocytic signalling. J. Neuroinflammation 2012, 9, 196. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Cao, Y.; Gao, J. Serum calreticulin is a negative biomarker in patients with Alzheimer’s disease. Int. J. Mol. Sci. 2014, 15, 21740–21753. [Google Scholar] [CrossRef] [Green Version]
- Kuang, X.-L.; Liu, F.; Chen, H.; Li, Y.; Liu, Y.; Xiao, J.; Shan, G.; Li, M.; Snider, B.J.; Qu, J.; et al. Reductions of the components of the calreticulin/calnexin quality-control system by proteasome inhibitors and their relevance in a rodent model of Parkinson’s disease. J. Neurosci. Res. 2014, 92, 1319–1329. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.M.; Park, S.H.; Chung, K.C.; Oh, Y.J. Proteomic analysis reveals upregulation of calreticulin in murine dopaminergic neuronal cells after treatment with 6-hydroxydopamine. Neurosci. Lett. 2003, 352, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Corson, T.W.; Woo, K.K.; Li, P.P.; Warsh, J.J. Cell-type specific regulation of calreticulin and Bcl-2 expression by mood stabilizer drugs. Eur. Neuropsychopharmacol. 2004, 14, 143–150. [Google Scholar] [CrossRef]
- Farokhashtiani, T.; Mirabzadeh, A.; Olad Nabi, M.; Magham, Z.G.; Khorshid, H.R.K.; Najmabadi, H.; Ohadi, M. Reversion of the human calreticulin gene promoter to the ancestral type as a result of a novel psychosis-associated mutation. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Ohadi, M.; Mirabzadeh, A.; Esmaeilzadeh-Gharehdaghi, E.; Rezazadeh, M.; Hosseinkhanni, S.; Oladnabi, M.; Firouzabadi, S.G.; Darvish, H. Novel evidence of the involvement of calreticulin in major psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 37, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Cunnea, P.; Mháille, A.N.; McQuaid, S.; Farrell, M.; McMahon, J.; FitzGerald, U. Expression profiles of endoplasmic reticulum stress-related molecules in demyelinating lesions and multiple sclerosis. Mult. Scler. 2011, 17, 808–818. [Google Scholar] [CrossRef]
- Ní Fhlathartaigh, M.; McMahon, J.; Reynolds, R.; Connolly, D.; Higgins, E.; Counihan, T.; FitzGerald, U. Calreticulin and other components of endoplasmic reticulum stress in rat and human inflammatory demyelination. Acta Neuropathol. Commun. 2013, 1, 37. [Google Scholar] [CrossRef] [Green Version]
- Edenharter, O.; Schneuwly, S.; Navarro, J.A. Mitofusin-Dependent ER Stress Triggers Glial Dysfunction and Nervous System Degeneration in a Drosophila Model of Friedreich’s Ataxia. Front. Mol. Neurosci. 2018, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Hong, J.-S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Shastri, N.; Cardinaud, S.; Schwab, S.R.; Serwold, T.; Kunisawa, J. All the peptides that fit: The beginning, the middle, and the end of the MHC class I antigen-processing pathway. Immunol. Rev. 2005, 207, 31–41. [Google Scholar] [CrossRef]
- Warre-Cornish, K.; Perfect, L.; Nagy, R.; Duarte, R.R.R.; Reid, M.J.; Raval, P.; Mueller, A.; Evans, A.L.; Couch, A.; Ghevaert, C.; et al. Interferon-γ signaling in human iPSC–derived neurons recapitulates neurodevelopmental disorder phenotypes. Sci. Adv. 2020, 6, eaay9506. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; He, Y.; Park, J.-S.; Bieri, G.; Snethlage, C.E.; Lin, K.; Gontier, G.; Wabl, R.; Plambeck, K.E.; Udeochu, J.; et al. β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 2015, 21, 932–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morani, F.; Doccini, S.; Chiorino, G.; Fattori, F.; Galatolo, D.; Sciarrillo, E.; Gemignani, F.; Züchner, S.; Bertini, E.S.; Santorelli, F.M. Functional Network Profiles in ARSACS Disclosed by Aptamer-Based Proteomic Technology. Front. Neurol. 2020, 11, 603774. [Google Scholar] [CrossRef] [PubMed]
- Paulin, D.; Lilienbaum, A.; Kardjian, S.; Agbulut, O.; Li, Z. Vimentin: Regulation and pathogenesis. Biochimie 2022, 197, 96–112. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Ravichandran, R.; Bansal, S.; Bremner, R.M.; Smith, M.A.; Mohanakumar, T. Tissue-associated self-antigens containing exosomes: Role in allograft rejection. Hum. Immunol. 2018, 79, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Wehmeyer, J.C.; Schmidt, V.; Lichtenberg, A.; Akhyari, P. Rapid Fluorescence—Based Characterization of Single Extracellular Vesicles in Human Blood with Nanoparticle-tracking Analysis. J. Vis. Exp. 2019, 143, e58731. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.; Mokhdomi, T.A.; Chikan, N.A.; Amin, A.; Qazi, H.; Wani, S.H.; Wafai, A.H.; Tyub, S.; Mustafa, F.; Mir, M.S.; et al. Affinity proteomics led identification of vimentin as a potential biomarker in colon cancers: Insights from serological screening and computational modelling. Mol. Biosyst. 2015, 11, 159–169. [Google Scholar] [CrossRef]
- Yang, H.; Wang, L.; Zang, C.; Wang, Y.; Shang, J.; Zhang, Z.; Liu, H.; Bao, X.; Wang, X.; Zhang, D. Src Inhibition Attenuates Neuroinflammation and Protects Dopaminergic Neurons in Parkinson’s Disease Models. Front. Neurosci. 2020, 14, 45. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.S.; Meera, P.; Altindag, B.; Chopra, R.; Perkins, E.M.; Paul, S.; Scoles, D.R.; Tarapore, E.; Magri, J.; Huang, H.; et al. MTSS1/Src family kinase dysregulation underlies multiple inherited ataxias. Proc. Natl. Acad. Sci. USA 2018, 115, E12407–E12416. [Google Scholar] [CrossRef] [Green Version]
- Liu, D. Repurposing cancer drugs to treat neurological diseases—Src inhibitors as examples. Neural Regen. Res. 2017, 12, 910. [Google Scholar] [CrossRef]
- Gage, M.C.; Thippeswamy, T. Inhibitors of Src Family Kinases, Inducible Nitric Oxide Synthase, and NADPH Oxidase as Potential CNS Drug Targets for Neurological Diseases. CNS Drugs 2021, 35, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Palha, J.A.; Ballinari, D.; Amboldi, N.; Cardoso, I.; Fernandes, R.; Bellotti, V.; Merlini, G.; Saraiva, M.J. 4′-Iodo-4′-Deoxydoxorubicin Disrupts the Fibrillar Structure of Transthyretin Amyloid. Am. J. Pathol. 2000, 156, 1919–1925. [Google Scholar] [CrossRef]
- Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Cheson, B.D.; Barlogie, B.; Kyle, R.A.; Palladini, G.; Geyer, S.M.; Merlini, G. A multicenter phase II trial of 4′-iodo-4′-deoxydoxorubicin (IDOX) in primary amyloidosis (AL). Amyloid 2002, 9, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Ascari, E.; Amboldi, N.; Bellotti, V.; Arbustini, E.; Perfetti, V.; Ferrari, M.; Zorzoli, I.; Marinone, M.G.; Garini, P. Interaction of the anthracycline 4’-iodo-4’-deoxydoxorubicin with amyloid fibrils: Inhibition of amyloidogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 2959–2963. [Google Scholar] [CrossRef] [Green Version]
- Babic, I.; Nurmemmedov, E.; Yenugonda, V.M.; Juarez, T.; Nomura, N.; Pingle, S.C.; Glassy, M.C.; Kesari, S. Pritumumab, the first therapeutic antibody for glioma patients. Hum. Antibodies 2019, 26, 95–101. [Google Scholar] [CrossRef]
- Glassy, M.C.; Hagiwara, H. Summary analysis of the pre-clinical and clinical results of brain tumor patients treated with pritumumab. Hum. Antibodies 2009, 18, 127–137. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Family | Drug(s) | Mitochondrial | Lysosomal | Plasma/Serum | |||
|---|---|---|---|---|---|---|---|---|
| p-Value | Log2FC | p-Value | Log2FC | |||||
| CALR | P27797 | ER chaperone | 1.4 × 10−4 |  −0.45 −0.45 | 1.5 × 10−5 | −0.70 | x | |
| HSPA5 | P11021 | enzyme | SHetA2, PAT-SM6 | 1.2 × 10−5 | −0.55 | 6.3 × 10−7 | −0.82 | x |
| HYOU1 | Q9Y4L1 | other | 5.7 × 10−5 | −0.66 | 7.7 × 10−8 | −0.74 | x | |
| LDHB | P07195 | enzyme | 5.7 × 10−2 | −0.39 | 3.3 × 10−4 | −0.62 | x | |
| VIM | P08670 | other | pritumumab | 9.2 × 10−2 |  1.11 1.11 | 2.6 × 10−4 | 0.93 | x |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morani, F.; Doccini, S.; Galatolo, D.; Pezzini, F.; Soliymani, R.; Simonati, A.; Lalowski, M.M.; Gemignani, F.; Santorelli, F.M. Integrative Organelle-Based Functional Proteomics: In Silico Prediction of Impaired Functional Annotations in SACS KO Cell Model. Biomolecules 2022, 12, 1024. https://doi.org/10.3390/biom12081024
Morani F, Doccini S, Galatolo D, Pezzini F, Soliymani R, Simonati A, Lalowski MM, Gemignani F, Santorelli FM. Integrative Organelle-Based Functional Proteomics: In Silico Prediction of Impaired Functional Annotations in SACS KO Cell Model. Biomolecules. 2022; 12(8):1024. https://doi.org/10.3390/biom12081024
Chicago/Turabian StyleMorani, Federica, Stefano Doccini, Daniele Galatolo, Francesco Pezzini, Rabah Soliymani, Alessandro Simonati, Maciej M. Lalowski, Federica Gemignani, and Filippo M. Santorelli. 2022. "Integrative Organelle-Based Functional Proteomics: In Silico Prediction of Impaired Functional Annotations in SACS KO Cell Model" Biomolecules 12, no. 8: 1024. https://doi.org/10.3390/biom12081024