
Co-Stimulatory Molecules during Immune Control of Epstein Barr Virus Infection


Department of Viral Immunobiology, Institute of Experimental Immunology, University of Zürich, 8057 Zurich, Switzerland
Biomolecules 2022, 12(1), 38; https://doi.org/10.3390/biom12010038
Submission received: 6 December 2021
/
Revised: 21 December 2021
/
Accepted: 22 December 2021
/
Published: 28 December 2021
(This article belongs to the Special Issue Immunity and Pathogenesis of Epstein-Barr Virus Infection)
{kind=link}
{kind=link}
Abstract
:The Epstein Barr virus (EBV) is one of the prominent human tumor viruses, and it is efficiently immune-controlled in most virus carriers. Cytotoxic lymphocytes strongly expand during symptomatic primary EBV infection and in preclinical in vivo models of this tumor virus infection. In these models and patients with primary immunodeficiencies, antibody blockade or deficiencies in certain molecular pathways lead to EBV-associated pathologies. In addition to T, NK, and NKT cell development, as well as their cytotoxic machinery, a set of co-stimulatory and co-inhibitory molecules was found to be required for EBV-specific immune control. The role of CD27/CD70, 4-1BB, SLAMs, NKG2D, CD16A/CD2, CTLA-4, and PD-1 will be discussed in this review. Some of these have just been recently identified as crucial for EBV-specific immune control, and for others, their important functions during protection were characterized in in vivo models of EBV infection and its immune control. These insights into the phenotype of cytotoxic lymphocytes that mediate the near-perfect immune control of EBV-associated malignancies might also guide immunotherapies against other tumors in the future.
Keywords:
T cells; natural killer cells; cytotoxic lymphocytes; CD27; NKG2D; 4-1BB; PD-1; CTLA-4; 2B4; CD21. Introduction on EBV Infection and Its Malignancies
The Epstein Barr virus (EBV) is a class I WHO carcinogen [1]. At the same time, it is present as a persistent infection in more than 90% of the adult human population [2,3]. Therefore, it is not only one of the most growth-transforming pathogens, readily immortalizing human B cells to lymphoblastoid cell lines (LCLs) in vitro, but also one of the most widely distributed viruses in humans, to which most likely the human immune system had to adapt to prevent EBV-associated malignancies prior to reproductive age. EBV is thought to be primarily transmitted via saliva, e.g., during kissing. This leads to two thirds of European and US populations being infected before the age of two years, and African populations being nearly uniformly infected at this age [4]. EBV crosses the oropharyngeal mucosa most likely by transcytosis [5,6] to infect human B cells in submucosal secondary lymphoid tissues, such as the tonsils [2]. It then drives these infected B cells into proliferation and differentiation through the germinal center reaction to gain access to long-lived memory B cells for persistence. From this memory B-cell compartment, it can reactivate to produce infectious viral particles most likely after B-cell receptor engagement of the respective memory B cells. The immediate early transcription factor BZLF1 of EBV drives this lytic reactivation. The resulting viral particles can then infect epithelial cells from the basolateral side, presumably for another round of lytic replication before being shed into the saliva for transmission. B-cell proliferation and differentiation is supported by so-called latency gene transcription programs. In naïve B cells, latency III is found with the expression of all six nuclear antigens (EBNA1, 2, 3A-C, and -LP), two latent membrane proteins (LMP1 and 2), two EBV-encoded small RNAs (EBER1 and 2), and more than 40 miRNAs of the BHRF1 and BART clusters [7,8,9]. In germinal center B cells then, only EBNA1, LMP1, LMP2, EBER, and BART miRNA expression can be found [8]. This is called the latency IIa program. Finally, in memory B cells, only non-translated EBER and BART RNAs are expressed (latency 0) with transient expression of EBNA1 during homeostatic proliferation (latency I) [10,11]. Accordingly, these latent gene products fulfill important functions along this B-cell differentiation trajectory. EBNA2 drives early proliferation and EBNA3A/3C prevent apoptosis during this early proliferation phase [3]. LMP1 and 2 provide proliferation and survival signals for germinal center B cells [12]. EBNA1 maintains the viral DNA in all proliferating cells [13]. These latent EBV gene product functions are also abused by EBV-associated malignancies [14]. Post-transplant lymphoproliferative disease (PTLD) in transplant patients and immunoblastic lymphomas in HIV-infected individuals often express latency III, while EBV-associated Hodgkin’s lymphoma expresses latency IIa and Burkitt’s lymphoma latency I. In addition, EBV is also present in around 10% of gastric carcinoma (latency I) and nasopharyngeal carcinoma (latency II) which constitute the bulk of EBV-associated tumors that make up 1–2% of all human cancers [15]. Thus, the latency programs that can be found in EBV-associated malignancies include expression of viral oncogenes that are present in healthy virus carriers, and the human immune responses needs to keep these constantly in check during viral persistent infection.
2. T Cells as the Cornerstone of EBV-Specific Immune Control
This immune control is primarily mediated by cytotoxic lymphocytes, including natural killer (NK), NKT, and γδ T cells, but primarily αβ T cells, especially CD8+ T cells [16,17,18,19]. Several lines of evidence point in this direction, namely expansion of these lymphocyte compartments during symptomatic primary infection, such as infectious mononucleosis (IM), their efficacy in controlling EBV infection in preclinical animal models, and their genetic, co-infection-induced or iatrogenic defects in patients that suffer from EBV-associated pathologies. During IM NK, NKT, γδ T, and CD8+ αβ, T cells expand [20,21,22]. Early differentiated NK cells primarily target lytic EBV infection [21,23]. NKT cells might preferentially target latency II infected B cells [24,25]. Furthermore, Vγ9Vδ2 T cells get primarily stimulated by latency I infected B cells [22,26]. αβ CD8+ T cells that expand during IM are primarily directed against lytic EBV antigens [27,28], but adoptive transfer of T-cell lines specific for EBNA1 or LMP1 and 2 can cure EBV-associated malignancies in patients [29,30,31,32,33]. Therefore, IM seems to drive the expansion of several innate and adaptive lymphocyte compartments, suggesting their involvement in the immune control of EBV.
Their contribution to EBV-specific immune control can be tested in preclinical in vivo models, such as non-human primates and mice with reconstituted human immune system components (humanized mice) [34,35]. Accordingly, NK cell depletion leads to higher viral loads and increased lymphomagenesis in EBV-infected humanized mice [36,37]. This only affects infection with wild-type, but not lytic infection deficient BZLF1 knock-out EBV [36]. Moreover, mixed HLA disparate human immune reconstitution improves EBV-specific immune control, presumably by limiting inhibitory NK cell receptor engagement by EBV-infected B cells of the co-reconstituted donor [37]. Moreover, adoptive transfer of CD8+ NKT cells that expand during EBV infection limits lymphomagenesis in humanized mice [25]. Similarly, expansion or adoptive transfer of Vγ9Vδ2 T cells in humanized mice limited PTLD-like disease [38,39]. In addition to these innate lymphocytes, αβ CD8+ T cells massively expand during EBV infection of humanized mice [40,41,42,43]. Furthermore, their antibody-mediated depletion increases viral loads and EBV-associated lymphomagenesis [40,43,44,45,46]. The role of CD4+ T cells in EBV-specific immune control in humanized mice is less clear. Their depletion by antibodies or HIV co-infection increases viral loads and lymphomagenesis [40,45]. Furthermore, immune-suppressive FK506 (tacrolimus) treatment mainly affects IL-2 production and activation of CD4+ T cells in humanized mice and also increases viral loads and lymphomagenesis [47]. Finally, adoptive transfer of cytotoxic EBV-specific CD4+ T cells can control LCL growth in humanized mice [48]. However, non-cytotoxic CD4+ T-cell functions can also support EBV-associated lymphoma growth and possibly latency I/II infection in humanized mice [49,50]. Thus, depending on their polarization, CD4+ T cells might play pro- or anti-viral roles during EBV infection.
However, their depletion by HIV co-infection also increases EBV-associated lymphomagenesis in patients [51]; even so, MHC class II deficient patients with a compromised CD4+ T-cell compartment usually have no problems with persistent EBV infection [52]. Indeed, the notion that lymphocyte cytotoxicity-supporting functions of CD4+ T cells are important for EBV-specific immune control, while other helper cytokines might rather drive immune pathology or even lymphomagenesis, is supported by patients with primary immunodeficiencies due to mutations in the cytotoxic machinery [53,54,55]. These often suffer from cytokine-driven hemophagocytic lymphohistiocytosis (HLH), which is driven by poorly controlled EBV infection in the absence of cytotoxicity. The importance of cytotoxic lymphocytes in the immune control of EBV is also supported by other primary immunodeficiencies that predispose for EBV-associated pathologies. These include factors for their development (GATA2, MCM4) and others that are required for the expansion of these lymphocytes (XIAP, STK4, CTPS1) [16,17,18,19]. Moreover, the importance of T cells is underscored by the susceptibility of individuals with mutations in molecules of T-cell receptor signaling (ITK, RASGRP1, ZAP70, PI3K). However, while these factors are generally required for human T-cell development and function, mutations in co-stimulatory molecules and their mechanistic studies provide insights into the particular interactions of T cells with EBV-infected cells and/or antigen-presenting cells that are required for the control of this ubiquitous human tumor virus. The discussion of these interactions is the main focus of this review.
3. Co-Stimulatory CD27/CD70 Signaling
Homozygous mutations in the tumor necrosis factor (TNF) receptor superfamily member CD27 or its ligand CD70, which have so far been recorded in 49 patients from 29 families, predispose affected individuals to EBV-induced lymphoproliferations, often developing into lymphomas such as Hodgkin’s lymphoma [19,56]. Most of these patients benefit from B-cell depletion by antibody therapy or bone marrow transplantation as curative treatments [56]. Several, but not all, of their EBV-specific T-cell responses are reduced and demonstrate some deficiencies in killing EBV-transformed B cells [56,57,58]. It was postulated that this functional deficiency could result from diminished expansion and differentiation of EBV-specific T cells in the absence of CD27 that would result in their decreased surface expression of co-stimulatory molecules, such as 2B4 and NKG2D, which are directly involved in recognizing EBV-infected B cells [19].
The CD27 interaction with CD70 is in addition to the CD28 interaction with CD80 and CD86, considered to be one of the key co-stimulations during the initiation of T-cell responses [59,60]. Therefore, it is surprising that CD27 deficiency predominantly predisposes for EBV-associated pathologies and not for susceptibilities to most other infectious diseases, and that CD27 or CD70 deficient patients still have some EBV-specific T-cell responses. Along these lines, it was recently found in humanized mice that antibody-mediated blockade of CD27 primarily limits expansion of lytic EBV antigen-specific CD8+ T-cell responses with intact expansion of latent EBV antigen-specific CD8+ T-cell responses [61]. In addition, CD27 blockade also inhibited cytotoxicity of lytic EBV antigen-specific CD8+ T-cell clones against EBV-transformed autologous LCLs (Figure 1), and CD70 is upregulated upon B-cell transformation by EBV. Accordingly, CD27 blockade increased viral load and EBV-infected B-cell lymphoproliferations during wild-type but not BZLF1 deficient lytic replication incompetent EBV infection. Therefore, CD27 might play a role during expansion and EBV-transformed B-cell recognition for a subset of EBV-specific T-cell responses that are required for EBV-specific immune control. This might explain the emergence of EBV-associated pathologies despite the presence of some EBV-specific T-cell responses in patients with homozygous CD27 or CD70 deficiencies.
4. Co-Stimulation by 4-1BB
Another TNF receptor superfamily member that is required for EBV-specific immune control is CD137/4-1BB [62,63,64,65]. Less than 10 patients have been so far diagnosed with this deficiency and suffer from lymphoproliferations and lymphomas. However, one of these EBV infections spreads to T cells (Figure 1), possibly assisted by a concomitant loss of function mutation in the p110δ subunit of phosphatidylinositol 3-kinase (PIK3CD) [64]. Furthermore, in two studies, siblings with the same recessive loss-of-function mutations in 4-1BB were identified that did not present with any clinical symptoms [63,64]. Therefore, 4-1BB does not seem to be as essential for EBV-specific immune control as CD27. Nevertheless, patients with 4-1BB mutations that suffer from EBV-driven lymphoproliferations can be successfully treated with B-cell depleting therapy [62,63,65] if infection has not spread to T cells [64] and EBV-associated lymphomas have not lost the target antigen CD20 [65]. Alternatively, bone marrow transplantation offers a curative treatment [62,65].
Similar to CD27 deficiency, T-cell expansion and cytotoxicity of EBV-infected cells is compromised by 4-1BB deficiency (Figure 1) [62,63]. This can also be mimicked by antibody-mediated 4-1BB blocking in vitro [62]. Accordingly, 4-1BBL is upregulated by EBV transformation of B cells [66]. The lower penetrance of EBV-associated pathologies in individuals with homozygous 4-1BB mutations, as demonstrated by healthy siblings of the patients that carry the same mutations, suggests that compensatory co-stimulation might rescue EBV-specific immune control in these siblings. Along these lines, CD28 stimulation can compensate for 4-1BB deficiency for T-cell proliferation [63]. Thus, 4-1BB seems to support T-cell expansion and cytotoxicity against EBV-infected cells but is not as strictly required for these functions as CD27 during EBV-specific immune control.
5. Co-Stimulatory Molecules of the SLAM Receptor Family
Loss of function mutations in the associated protein of signaling lymphocyte activation molecules (SLAM), called SAP and encoded by the SH2D1A gene, cause x-linked lymphoproliferative disease type 1 (XLP1) [67,68,69,70,71]. The majority of the affected individuals develop viremia and HLH after primary EBV infection, and up to 50% develop lymphomas that are associated with this γ-herpesvirus, while other viral infections, including α- and β-herpesviruses, are controlled by these patients [72,73,74]. Bone marrow transplantation and antibody-mediated B-cell depletion are successful treatments in XLP1 patients [73,75,76,77]. Thus, SAP deficiency renders affected individuals exquisitely sensitive to EBV-associated pathologies and thereby implicates SLAM receptors in EBV-specific immune control.
SAP converts SLAM signaling from inhibitory to activating [78,79]. Accordingly, cytotoxic lymphocytes such as CD8+ T cells and NK cells cannot efficiently kill EBV-transformed LCLs without SAP (Figure 1) [80,81]. In addition, NKT cells are lacking in XLP1 patients [82]. The inability of cytotoxic XLP1 lymphocytes to target EBV-transformed B cells is at least in part due to the inability of the two SLAM receptors 2B4 and NTB-A to activate CD8+ T cells and NK cells in this interaction without SAP [83,84]. These two receptors are in particular required for cytotoxic immune synapse formation with B cells, but not other antigen-presenting cells, such as dendritic cells (DCs) and fibroblasts [80,85,86]. The 2B4 ligand CD48, especially, is also strongly upregulated upon EBV infection of B cells [66]. This strict requirement for SAP in cytotoxic lymphocyte recognition of EBV-transformed B cells results in EBV-specific CD8+ T cells of female carriers of SAP mutations expressing uniformly wild-type SAP, while CD8+ T cells against human cytomegalovirus or influenza virus express mutant or wild-type SAP [86]. Furthermore, EBV-specific CD8+ T cells with revertant SAP mutations can be found in XLP1 patients [87]. The lack of eliminating EBV-transformed B cells in the absence SAP can at least in part be mimicked by antibody blocking of 2B4 [44]. This leads to higher viral loads and lymphoma formation in humanized mice, while CD8+ T cell expansion upon EBV infection is not altered by 2B4 blockade in this model. Furthermore, simultaneous antibody-mediated depletion of CD8+ T cells leads to loss of EBV-specific immune control that cannot be worsened by 2B4 blockade. This suggests that 2B4 is mainly required on CD8+ T cells for eliminating EBV-infected B cells. Thus, the SLAM receptors, especially 2B4, seem to be mainly required for lymphocyte cytotoxicity against EBV-infected B cells but much less for EBV-specific T-cell expansion.
6. Stimulatory NKG2D Engagement
Another receptor that is engaged to kill EBV-infected B cells is NKG2D [23,88]. NKG2D ligands seem to be mainly upregulated during the transition from latently EBV-infected B cells to viral lytic replication (Figure 1) [23]. This is also consistent with preferential recognition of B cells with lytic EBV infection by NK cells which uniformly express NKG2D [21,23,36]. Accordingly, NK cells limit wild-type EBV infection and associated lymphomagenesis in humanized mice, but do not influence infection with BZLF1-deficient lytic EBV infection incompetent virus [36]. In the primary X-linked immunodeficiency with magnesium defect, the Epstein-Barr virus infection, and neoplasia (XMEN), magnesium transporter 1 (MAGT1) is mutated and the diminished intracellular magnesium levels compromise glycosylation and surface expression of NKG2D [88]. Restoring NKG2D surface expression by magnesium supplementation was able to increase NK cell cytotoxicity against EBV-transformed LCLs. Thus, NKG2D, in addition to SLAM receptors, seems to be required for the direct recognition of EBV-infected B cells. However, NKG2D-mediated recognition might primarily be important for the immune control of lytic EBV infection.
7. Modifying CD2 Co-Stimulation by CD16
The low affinity IgG receptor CD16 (FcγRIII) was found to be mutated or deleted in a few patients that suffer from EBV-associated pathologies [89,90,91]. In a family with three siblings that all lacked CD16A on NK cells but retained CD16B on monocytes, elevated EBV viral loads were associated with CD16A deficiency [91]. In one of these, persistent EBV viremia with clinical symptoms was associated with T-cell infection by EBV (Figure 1). CD16A with one altered amino acid was also the cause of poorly controlled herpesvirus infections in the originally described two patients [89,90]. How the low affinity FcγRIIIA is required for EBV-specific immune control is unclear, however, because antibody responses do not seem to be required to keep EBV in check [18]. It has been proposed that it either influences the function of the adhesion molecule CD2 that binds to CD58 on target cells [90], or maturation of NK cells to high CD2 expression [91]. KSHV co-infection also diminishes CD2 expressing NK cells and increases EBV-associated lymphomagenesis in humanized mice [92,93]. In one of the original studies that identified poorly controlled EBV infection in patients with CD16A mutations, direct association of CD16A with CD2 was demonstrated, thereby modifying NK cell cytotoxicity (Figure 1) [90]. Furthermore, CD16A mutant NK cells were less capable of forming this association with CD2 and compromised in their CD2-dependent cytotoxicity. CD58, the ligand of CD2, is transiently upregulated in the first five days after EBV infection of B cells [66]. This period is dominated by EBNA2-driven proliferation of infected B cells without LMP expression, the so-called latency IIb program [94]. Thus, mutant CD16A or CD16A deficiency is associated with loss of EBV-specific immune control but possibly not by modifying IgG opsonized target cell recognition, and rather by loss of cytotoxic immune synapse formation due to decreased CD2 levels or function.
8. The Co-Inhibitory Receptors PD-1 and CTLA-4
Not only the loss of co-stimulatory receptors, but also the loss of co-inhibitory receptors was found to cause EBV pathologies. Among these, CTLA-4 deficiency, often even haploinsufficiency, is associated with increased viral loads in more than half of the individuals, as well as EBV-associated lymphoma and gastric carcinoma formation [95,96,97]. Diffuse large B-cell lymphomas, Burkitt lymphomas, and Hodgkin lymphomas have been observed in CTLA-4-deficient patients [96]. By now, more than 150 CTLA-4-insufficient patients have been described, and they present usually with decreased T-cell but increased regulatory T-cell numbers (Figure 2) [95]. Accordingly, both effector T-cell exhaustion and/or increased T-cell regulation could account for the lack of EBV-specific immune control. The increased T-cell lymphoproliferation causing activation induced cell death, which might be the cause of T-cell lymphopenia in these patients, can be treated with recombinant CTLA-4-Fc reagents, such as Abatacept and Belatacept [95,97,98]. However, EBV reactivation needs to be monitored during such treatments. Thus, the role of CTLA-4 during EBV-specific immune control is incompletely understood.
Another inhibitory co-receptor on T cells, PD-1, was also explored during EBV infection in humanized mice. Surprisingly, increased viral loads and EBV-associated lymphomagenesis was observed during PD-1-specific antibody blockade (Figure 2) [42,99]. Only in an EBV-infected cord blood transfer model into immune compromised mice did PD-1 blockade alone or together with CTLA-4 blockade decrease EBV-associated lymphomagenesis [100]. In humanized mice, the loss of EBV-specific immune control during PD-1 blockade was associated with elevated levels of the immune-suppressive cytokine IL-10, which is also produced by a subset of regulatory T cells [42,99]. Both PD-1 ligands, PD-L1 and PD-L2, are upregulated on B cells after EBV infection [66]. Along these lines, some cancer patients that receive therapeutic PD-1-targeted immune check-point blockade develop neurological symptoms. In some of these, EBV viral loads were elevated, and both EBV-infected B cells as well as EBV-specific T cells were suggested to home into the central nervous system (CNS), in part being responsible for the observed neurological side effects [101]. Such respective clonal B- and T-cell expansions, pointing to EBV-infected B- and EBV-specific T-cell homing to the CNS, were, however, primarily reported in one patient after immune checkpoint blockade. Therefore, it is tempting to speculate that both CTLA-4 haploinsufficiency and PD-1 blockade might increase regulatory T-cell function during EBV infection, resulting in virus-associated pathologies.
9. Conclusions and Outlook
The summarized studies above highlight molecular requirements for the near-perfect immune control of EBV and its associated malignancies by cytotoxic lymphocytes. Similarly, robust cytotoxic T and innate lymphocyte responses would also be desirable to target other infectious diseases or tumors. Thus, identifying the means to elicit such robust cell-mediated immune responses by vaccination or adoptive lymphocyte transfer would be highly desirable. Maybe also for this goal, EBV infection can provide us with some interesting insights.
As discussed, EBV has a near-exclusive tropism for human B cells. Indeed, it remains so far unclear under which physiological circumstances the virus infects other cells, such as epithelial and T cells from which EBV-associated pathologies emerge [14]. This particular tropism of EBV for B cells indicates primarily cross-presentation by DCs from EBV-infected B cells [102] and/or T-cell stimulation by EBV-infected B cells [103] as the source of EBV-specific T-cell responses. Along these lines, EBV-based and DC-targeted vaccine formulations elicited only low T-cell responses in humanized mice [104,105], while EBV-based and B-cell-targeted vaccination induced protective T-cell responses in the same model [106]. Furthermore, B cells activated by transgenic LMP1 expression were able to elicit tumor-associated, antigen-specific CD4+ and CD8+ T cells in a mouse model [107]. Therefore, the molecular requirements of this B-cell-elicited T-cell stimulation should be characterized in the future. These might provide complementary pathways to DC-mediated T-cell stimulation in eliciting cytotoxic cellular immune control. Thus, multiple antigen-presenting cell populations, including both DCs and B cells, could be targeted to elicit robust T-cell-mediated immune control, not only of EBV infection and associated malignancies, but also for tumor immunology in general.
Funding
Research in the author’s laboratory is funded by Cancer Research Switzerland, Grant number KFS-4962-02-2020; KFSP-PrecisionMS and HMZ ImmunoTargET of the University of Zurich; the Cancer Research Center Zurich; the Sobek Foundation; the Swiss Vaccine Research Institute; the Swiss MS Society; Roche; Novartis; Innosuisse; and the Swiss National Science Foundation, Grant number 310030_204470/1, 310030L_197952/1 and CRSII5_180323. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens--part b: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Münz, C. Latency and lytic replication in the oncogenesis of the Epstein Barr virus. Nat. Rev. Micobiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.J. Epstein-Barr virus and cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Dunmire, S.K.; Verghese, P.S.; Balfour, H.H., Jr. Primary Epstein-Barr virus infection. J. Clin. Virol. 2018, 102, 84–92. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Herrera, R.; Palefsky, J.M. Epstein-Barr virus transcytosis through polarized oral epithelial cells. J. Virol. 2013, 87, 8179–8194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münz, C. Immune escape by non-coding RNAs of the Epstein Barr virus. Front. Microbiol. 2021, 12, 657387. [Google Scholar] [CrossRef]
- Babcock, J.G.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.M.; Babcock, G.J.; Thorley-Lawson, D.A. Cells expressing the Epstein-Barr virus growth program are present in and restricted to the naive B-cell subset of healthy tonsils. J. Virol. 2000, 74, 9964–9971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the Burkitt’s lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Frappier, L. EBNA1. Curr. Top. Microbiol. Immunol. 2015, 391, 3–34. [Google Scholar] [PubMed]
- Shannon-Lowe, C.; Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr virus: An important vaccine target for cancer prevention. Sci. Transl. Med. 2011, 3, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damania, B.; Münz, C. Immunodeficiencies that predispose to pathologies by human oncogenic gamma-herpesviruses. FEMS Microbiol. Rev. 2019, 43, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.; Latour, S. Immunity to EBV as revealed by immunedeficiencies. Curr. Opin. Immunol. 2021, 72, 107–115. [Google Scholar] [CrossRef]
- Latour, S.; Fischer, A. Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: Lessons from genetic diseases. Immunol. Rev. 2019, 291, 174–189. [Google Scholar] [CrossRef]
- Tangye, S.G.; Latour, S. Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood 2020, 135, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Dunmire, S.K.; Grimm, J.M.; Schmeling, D.O.; Balfour, H.H., Jr.; Hogquist, K.A. The incubation period of primary Epstein-Barr virus infection: Viral dynamics and immunologic events. PLoS Pathog. 2015, 11, e1005286. [Google Scholar] [CrossRef]
- Azzi, T.; Lunemann, A.; Murer, A.; Ueda, S.; Beziat, V.; Malmberg, K.J.; Staubli, G.; Gysin, C.; Berger, C.; Münz, C.; et al. Role for early-differentiated Natural Killer cells in infectious mononucleosis. Blood 2014, 124, 2533–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djaoud, Z.; Guethlein, L.A.; Horowitz, A.; Azzi, T.; Nemat-Gorgani, N.; Olive, D.; Nadal, D.; Norman, P.J.; Münz, C.; Parham, P. Two alternate strategies for innate immunity to Epstein-Barr virus: One using NK cells and the other NK cells and gammadelta T cells. J. Exp. Med. 2017, 214, 1827–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The switch from latent to productive infection in Epstein-Barr virus-infected B cells is associated with sensitization to NK cell killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, B.K.; Tsai, K.; Allan, L.L.; Zheng, D.J.; Nie, J.C.; Biggs, C.M.; Hasan, M.R.; Kozak, F.K.; van den Elzen, P.; Priatel, J.J.; et al. Innate immune control of EBV-infected B cells by invariant natural killer T cells. Blood 2013, 122, 2600–2608. [Google Scholar] [CrossRef] [Green Version]
- Yuling, H.; Ruijing, X.; Li, L.; Xiang, J.; Rui, Z.; Yujuan, W.; Lijun, Z.; Chunxian, D.; Xinti, T.; Wei, X.; et al. EBV-induced human CD8+ NKT cells suppress tumorigenesis by EBV-associated malignancies. Cancer Res. 2009, 69, 7935–7944. [Google Scholar] [CrossRef] [Green Version]
- Djaoud, Z.; Parham, P. Dimorphism in the TCRgamma-chain repertoire defines 2 types of human immunity to Epstein-Barr virus. Blood Adv. 2020, 4, 1198–1205. [Google Scholar] [CrossRef]
- Callan, M.F.; Steven, N.; Krausa, P.; Wilson, J.D.; Moss, P.A.; Gillespie, G.M.; Bell, J.I.; Rickinson, A.B.; McMichael, A.J. Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat. Med. 1996, 2, 906–911. [Google Scholar] [CrossRef]
- Callan, M.F.; Tan, L.; Annels, N.; Ogg, G.S.; Wilson, J.D.; O’Callaghan, C.A.; Steven, N.; McMichael, A.J.; Rickinson, A.B. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus in vivo. J. Exp. Med. 1998, 187, 1395–1402. [Google Scholar] [CrossRef] [Green Version]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef]
- Bollard, C.M.; Tripic, T.; Cruz, C.R.; Dotti, G.; Gottschalk, S.; Torrano, V.; Dakhova, O.; Carrum, G.; Ramos, C.A.; Liu, H.; et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed Hodgkin lymphoma. J. Clin. Oncol. 2018, 36, 1128–1139. [Google Scholar] [CrossRef]
- Grant, M.L.; Bollard, C.M. Cell therapies for hematological malignancies: Don’t forget non-gene-modified T cells! Blood Rev. 2018, 32, 203–224. [Google Scholar] [CrossRef]
- Icheva, V.; Kayser, S.; Wolff, D.; Tuve, S.; Kyzirakos, C.; Bethge, W.; Greil, J.; Albert, M.H.; Schwinger, W.; Nathrath, M.; et al. Adoptive transfer of Epstein-Barr virus (EBV) nuclear antigen 1-specific T cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J. Clin. Oncol. 2013, 31, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Tsang, J.; Beagley, L.; Chua, D.; Lee, V.; Li, V.; Moss, D.J.; Coman, W.; Chan, K.H.; Nicholls, J.; et al. Effective treatment of metastatic forms of Epstein-Barr virus-associated nasopharyngeal carcinoma with a novel adenovirus-based adoptive immunotherapy. Cancer Res. 2012, 72, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D.; Caduff, N.; Murer, A.; Engelmann, C.; Deng, Y.; Zdimerova, H.; Zens, K.; Chijioke, O.; Münz, C. Infection and immune control of human oncogenic gamma-herpesviruses in humanized mice. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhe, J.; Wang, F. Non-human primate lymphocryptoviruses: Past, present, and future. Curr. Top. Microbiol. Immunol. 2015, 391, 385–405. [Google Scholar] [PubMed]
- Chijioke, O.; Muller, A.; Feederle, R.; Barros, M.H.; Krieg, C.; Emmel, V.; Marcenaro, E.; Leung, C.S.; Antsiferova, O.; Landtwing, V.; et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep. 2013, 5, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Landtwing, V.; Raykova, A.; Pezzino, G.; Beziat, V.; Marcenaro, E.; Graf, C.; Moretta, A.; Capaul, R.; Zbinden, A.; Ferlazzo, G.; et al. Cognate HLA absence in trans diminishes human nk cell education. J. Clin. Investig. 2016, 126, 3772–3782. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Liu, Y.; Zheng, J.; Liu, M.; Lv, A.; Gao, Y.; Hu, H.; Lam, K.T.; Chan, G.C.; Yang, Y.; et al. Targeted activation of human Vgamma9Vdelta2-T cells controls Epstein-Barr virus-induced B cell lymphoproliferative disease. Cancer Cell 2014, 26, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Zumwalde, N.A.; Sharma, A.; Xu, X.; Ma, S.; Schneider, C.L.; Romero-Masters, J.C.; Hudson, A.W.; Gendron-Fitzpatrick, A.; Kenney, S.C.; Gumperz, J.E. Adoptively transferred Vgamma9Vdelta2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis model. JCI Insight 2017, 2, e93179. [Google Scholar] [CrossRef] [Green Version]
- Strowig, T.; Gurer, C.; Ploss, A.; Liu, Y.F.; Arrey, F.; Sashihara, J.; Koo, G.; Rice, C.M.; Young, J.W.; Chadburn, A.; et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J. Exp. Med. 2009, 206, 1423–1434. [Google Scholar] [CrossRef] [Green Version]
- Antsiferova, O.; Müller, A.; Rämer, P.; Chijioke, O.; Chatterjee, B.; Raykova, A.; Planas, R.; Sospedra, M.; Shumilov, A.; Tsai, M.H.; et al. Adoptive transfer of EBV specific CD8+ T cell clones can transiently control EBV infection in humanized mice. PLoS Pathog. 2014, 10, e1004333. [Google Scholar] [CrossRef]
- Chatterjee, B.; Deng, Y.; Holler, A.; Nunez, N.; Azzi, T.; Vanoaica, L.D.; Müller, A.; Zdimerova, H.; Antsiferova, O.; Zbinden, A.; et al. CD8+ T cells retain protective functions despite sustained inhibitory receptor expression during Epstein-Barr virus infection in vivo. PLoS Pathog. 2019, 15, e1007748. [Google Scholar] [CrossRef] [Green Version]
- Murer, A.; Ruhl, J.; Zbinden, A.; Capaul, R.; Hammerschmidt, W.; Chijioke, O.; Münz, C. MicroRNAs of Epstein-Barr virus attenuate T-cell-mediated immune control in vivo. mBio 2019, 10, e01941-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chijioke, O.; Marcenaro, E.; Moretta, A.; Capaul, R.; Münz, C. The SAP-dependent 2B4 receptor mediates CD8+ T cell dependent immune control of Epstein Barr virus infection in mice with reconstituted human immune system components. J. Infect. Dis. 2015, 212, 803–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, D.; Myburgh, R.; Caduff, N.; Spohn, M.; Kok, Y.L.; Keller, C.W.; Murer, A.; Chatterjee, B.; Rühl, J.; Engelmann, C.; et al. EBV renders B cells susceptible to HIV-1 in humanized mice. Life Sci. Alliance 2020, 3, e202000640. [Google Scholar] [CrossRef]
- Yajima, M.; Imadome, K.; Nakagawa, A.; Watanabe, S.; Terashima, K.; Nakamura, H.; Ito, M.; Shimizu, N.; Yamamoto, N.; Fujiwara, S. T cell-mediated control of Epstein-Barr virus infection in humanized mice. J. Infect. Dis. 2009, 200, 1611–1615. [Google Scholar] [CrossRef] [Green Version]
- Caduff, N.; McHugh, D.; Murer, A.; Ramer, P.; Raykova, A.; Landtwing, V.; Rieble, L.; Keller, C.W.; Prummer, M.; Hoffmann, L.; et al. Immunosuppressive FK506 treatment leads to more frequent EBV-associated lymphoproliferative disease in humanized mice. PLoS Pathog. 2020, 16, e1008477. [Google Scholar]
- Linnerbauer, S.; Behrends, U.; Adhikary, D.; Witter, K.; Bornkamm, G.W.; Mautner, J. Virus and autoantigen-specific CD4+ T cells are key effectors in a scid mouse model of EBV-associated post-transplant lymphoproliferative disorders. PLoS Pathog. 2014, 10, e1004068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.D.; Xu, X.; Plowshay, J.; Ranheim, E.A.; Burlingham, W.J.; Jensen, J.L.; Asimakopoulos, F.; Tang, W.; Gulley, M.L.; Cesarman, E.; et al. LMP1-deficient Epstein-Barr virus mutant requires T cells for lymphomagenesis. J. Clin. Investig. 2015, 125, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Heuts, F.; Rottenberg, M.E.; Salamon, D.; Rasul, E.; Adori, M.; Klein, G.; Klein, E.; Nagy, N. T cells modulate Epstein-Barr virus latency phenotypes during infection of humanized mice. J. Virol. 2014, 88, 3235–3245. [Google Scholar] [CrossRef] [Green Version]
- Totonchy, J.; Cesarman, E. Does persistent HIV replication explain continued lymphoma incidence in the era of effective antiretroviral therapy? Curr. Opin. Virol. 2016, 20, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hawary, R.E.; Mauracher, A.A.; Meshaal, S.S.; Eldash, A.; Abd Elaziz, D.S.; Alkady, R.; Lotfy, S.; Opitz, L.; Galal, N.M.; Boutros, J.A.; et al. MHC-II deficiency among egyptians: Novel mutations and unique phenotypes. J. Allergy Clin. Immunol. Pract. 2019, 7, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Ali, M.A.; Patera, A.C.; Catalfamo, M.; Jaffe, E.S.; Kimura, H.; Dale, J.K.; Straus, S.E.; Cohen, J.I. Chronic active Epstein-Barr virus infection associated with mutations in perforin that impair its maturation. Blood 2004, 103, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Rohr, J.; Beutel, K.; Maul-Pavicic, A.; Vraetz, T.; Thiel, J.; Warnatz, K.; Bondzio, I.; Gross-Wieltsch, U.; Schundeln, M.; Schutz, B.; et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica 2010, 95, 2080–2087. [Google Scholar] [CrossRef]
- Cohen, J.I.; Niemela, J.E.; Stoddard, J.L.; Pittaluga, S.; Heslop, H.; Jaffe, E.S.; Dowdell, K. Late-onset severe chronic active ebv in a patient for five years with mutations in STXBP2 (MUNC18-2) and PRF1 (perforin 1). J. Clin. Immunol. 2015, 35, 445–448. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Kostel Bal, S.; Edwards, E.S.J.; Pillay, B.; Jimenez-Heredia, R.; Rao, G.; Erol Cipe, F.; Salzer, E.; Zoghi, S.; Abolhassani, H.; et al. Extended clinical and immunological phenotype and transplant outcome in CD27 and CD70 deficiency. Blood 2020, 136, 2638–2655. [Google Scholar] [CrossRef]
- Izawa, K.; Martin, E.; Soudais, C.; Bruneau, J.; Boutboul, D.; Rodriguez, R.; Lenoir, C.; Hislop, A.D.; Besson, C.; Touzot, F.; et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J. Exp. Med. 2017, 214, 73–89. [Google Scholar] [CrossRef]
- Abolhassani, H.; Edwards, E.S.; Ikinciogullari, A.; Jing, H.; Borte, S.; Buggert, M.; Du, L.; Matsuda-Lennikov, M.; Romano, R.; Caridha, R.; et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J. Exp. Med. 2017, 214, 91–106. [Google Scholar] [CrossRef]
- Watts, T.H. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005, 23, 23–68. [Google Scholar] [CrossRef]
- Croft, M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat. Rev. Immunol. 2003, 3, 609–620. [Google Scholar] [CrossRef]
- Deng, Y.; Chatterjee, B.; Zens, K.; Zdimerova, H.; Müller, A.; Schuhmachers, P.; Ligeon, L.-A.; Bongiovanni, A.; Capaul, R.; Zbinden, A.; et al. CD27 is required for protective lytic EBV antigen specific CD8+ 1 T cell expansion. Blood 2021, 137, 3225–3236. [Google Scholar] [CrossRef] [PubMed]
- Alosaimi, M.F.; Hoenig, M.; Jaber, F.; Platt, C.D.; Jones, J.; Wallace, J.; Debatin, K.M.; Schulz, A.; Jacobsen, E.; Moller, P.; et al. Immunodeficiency and EBV-induced lymphoproliferation caused by 4-1BB deficiency. J. Allergy Clin. Immunol. 2019, 144, 574–583.e5. [Google Scholar] [CrossRef] [Green Version]
- Somekh, I.; Thian, M.; Medgyesi, D.; Gulez, N.; Magg, T.; Gallon Duque, A.; Stauber, T.; Lev, A.; Genel, F.; Unal, E.; et al. CD137 deficiency causes immune dysregulation with predisposition to lymphomagenesis. Blood 2019, 134, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Fournier, B.; Cordeiro, D.J.; Winter, S.; Izawa, K.; Martin, E.; Boutboul, D.; Lenoir, C.; Fraitag, S.; Kracker, S.; et al. Concomitant PIK3CD and TNFRSF9 deficiencies cause chronic active Epstein-Barr virus infection of T cells. J. Exp. Med. 2019, 216, 2800–2818. [Google Scholar] [CrossRef]
- Wildermann, C.; Alosaimi, M.; Liebenehm, S.; Jacobsen, E.M.; Barth, T.F.E.; Moller, P.; Debatin, K.M.; Schulz, A.; Sirin, M.; Abosoudah, I.F.; et al. Successful hematopoietic stem cell transplantation in a 4-1BB deficient patient with EBV-induced lymphoproliferation. Clin. Immunol. 2021, 222, 108639. [Google Scholar] [CrossRef] [PubMed]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [Green Version]
- Bar, R.S.; DeLor, C.J.; Clausen, K.P.; Hurtubise, P.; Henle, W.; Hewetson, J.F. Fatal infectious mononucleosis in a family. N. Engl. J. Med. 1974, 290, 363–367. [Google Scholar] [CrossRef]
- Purtilo, D.T.; Cassel, C.K.; Yang, J.P.; Harper, R. X-linked recessive progressive combined variable immunodeficiency (Duncan’s disease). Lancet 1975, 1, 935–940. [Google Scholar] [CrossRef]
- Provisor, A.J.; Iacuone, J.J.; Chilcote, R.R.; Neiburger, R.G.; Crussi, F.G. Acquired agammaglobulinemia after a life-threatening illness with clinical and laboratory features of infectious mononucleosis in three related male children. N. Engl. J. Med. 1975, 293, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Coffey, A.J.; Brooksbank, R.A.; Brandau, O.; Oohashi, T.; Howell, G.R.; Bye, J.M.; Cahn, A.P.; Durham, J.; Heath, P.; Wray, P.; et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat. Genet. 1998, 20, 129–135. [Google Scholar] [CrossRef]
- Nichols, K.E.; Harkin, D.P.; Levitz, S.; Krainer, M.; Kolquist, K.A.; Genovese, C.; Bernard, A.; Ferguson, M.; Zuo, L.; Snyder, E.; et al. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc. Natl. Acad. Sci. USA 1998, 95, 13765–13770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumegi, J.; Huang, D.; Lanyi, A.; Davis, J.D.; Seemayer, T.A.; Maeda, A.; Klein, G.; Seri, M.; Wakiguchi, H.; Purtilo, D.T.; et al. Correlation of mutations of the SH2D1A gene and Epstein-Barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood 2000, 96, 3118–3125. [Google Scholar] [PubMed]
- Booth, C.; Gilmour, K.C.; Veys, P.; Gennery, A.R.; Slatter, M.A.; Chapel, H.; Heath, P.T.; Steward, C.G.; Smith, O.; O’Meara, A.; et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: A multicenter study on the manifestations, management and outcome of the disease. Blood 2011, 117, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachlopnik Schmid, J.; Canioni, D.; Moshous, D.; Touzot, F.; Mahlaoui, N.; Hauck, F.; Kanegane, H.; Lopez-Granados, E.; Mejstrikova, E.; Pellier, I.; et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood 2011, 117, 1522–1529. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; Tsai, D.E.; Hodinka, R.L.; Silverman, L.B.; Malbran, A.; Wasik, M.A.; Nichols, K.E. Treatment of primary Epstein-Barr virus infection in patients with X-linked lymphoproliferative disease using B-cell-directed therapy. Blood 2005, 105, 994–996. [Google Scholar] [CrossRef] [Green Version]
- Sharapova, S.O.; Fedorova, A.S.; Pashchenko, O.E.; Vahliarskaya, S.S.; Guryanova, I.E.; Migas, A.A.; Kondratenko, I.V.; Aleinikova, O.V. Novel mutations in SH2D1A gene in X-linked lymphoproliferative syndrome, diagnosed after B-cell non-Hodgkin lymphoma. J. Pediatr. Hematol. Oncol. 2017, 39, e203–e206. [Google Scholar] [CrossRef] [PubMed]
- Shamriz, O.; Vilk, S.R.; Wolf, D.G.; Ta-Shma, A.; Averbuch, D.; Weintraub, M.; Stepensky, P. Hematopoietic stem cell transplantation conditioning with use of rituximab in EBV related lymphoproliferative disorders. Clin. Immunol. 2014, 151, 79–83. [Google Scholar] [CrossRef]
- Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L. SLAM family receptors and SAP adaptors in immunity. Annu. Rev. Immunol. 2011, 29, 665–705. [Google Scholar] [CrossRef]
- Latour, S.; Gish, G.; Helgason, C.D.; Humphries, R.K.; Pawson, T.; Veillette, A. Regulation of SLAM-mediated signal transduction by SAP, the X-linked lymphoproliferative gene product. Nat. Immunol. 2001, 2, 681–690. [Google Scholar] [CrossRef]
- Dupre, L.; Andolfi, G.; Tangye, S.G.; Clementi, R.; Locatelli, F.; Arico, M.; Aiuti, A.; Roncarolo, M.G. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood 2005, 105, 4383–4389. [Google Scholar] [CrossRef]
- Hislop, A.D.; Palendira, U.; Leese, A.M.; Arkwright, P.D.; Rohrlich, P.S.; Tangye, S.G.; Gaspar, H.B.; Lankester, A.C.; Moretta, A.; Rickinson, A.B. Impaired Epstein-Barr virus-specific CD8+ T-cell function in X-linked lymphoproliferative disease is restricted to SLAM family-positive B-cell targets. Blood 2010, 116, 3249–3257. [Google Scholar] [CrossRef] [Green Version]
- Nichols, K.E.; Hom, J.; Gong, S.Y.; Ganguly, A.; Ma, C.S.; Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L.; Koretzky, G.A.; Stein, P.L. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat. Med. 2005, 11, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Bottino, C.; Falco, M.; Parolini, S.; Marcenaro, E.; Augugliaro, R.; Sivori, S.; Landi, E.; Biassoni, R.; Notarangelo, L.D.; Moretta, L.; et al. NTB-A, a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J. Exp. Med. 2001, 194, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Parolini, S.; Bottino, C.; Falco, M.; Augugliaro, R.; Giliani, S.; Franceschini, R.; Ochs, H.D.; Wolf, H.; Bonnefoy, J.Y.; Biassoni, R.; et al. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J. Exp. Med. 2000, 192, 337–346. [Google Scholar] [CrossRef]
- Zhao, F.; Cannons, J.L.; Dutta, M.; Griffiths, G.M.; Schwartzberg, P.L. Positive and negative signaling through SLAM receptors regulate synapse organization and thresholds of cytolysis. Immunity 2012, 36, 1003–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palendira, U.; Low, C.; Chan, A.; Hislop, A.D.; Ho, E.; Phan, T.G.; Deenick, E.; Cook, M.C.; Riminton, D.S.; Choo, S.; et al. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol. 2011, 9, e1001187. [Google Scholar] [CrossRef]
- Palendira, U.; Low, C.; Bell, A.I.; Ma, C.S.; Abbott, R.J.M.; Phan, T.G.; Riminton, D.S.; Choo, S.; Smart, J.M.; Lougaris, V.; et al. Expansion of somatically reverted memory CD8+ T cells in patients with X-linked lymphoproliferative disease caused by selective pressure from Epstein-Barr virus. J. Exp. Med. 2012, 209, 911–922. [Google Scholar] [CrossRef]
- Chaigne-Delalande, B.; Li, F.Y.; O’Connor, G.M.; Lukacs, M.J.; Jiang, P.; Zheng, L.; Shatzer, A.; Biancalana, M.; Pittaluga, S.; Matthews, H.F.; et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science 2013, 341, 186–191. [Google Scholar] [CrossRef] [Green Version]
- de Vries, E.; Koene, H.R.; Vossen, J.M.; Gratama, J.W.; von dem Borne, A.E.; Waaijer, J.L.; Haraldsson, A.; de Haas, M.; van Tol, M.J. Identification of an unusual Fc gamma receptor IIIA (CD16) on natural killer cells in a patient with recurrent infections. Blood 1996, 88, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Grier, J.T.; Forbes, L.R.; Monaco-Shawver, L.; Oshinsky, J.; Atkinson, T.P.; Moody, C.; Pandey, R.; Campbell, K.S.; Orange, J.S. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J. Clin. Investig. 2012, 122, 3769–3780. [Google Scholar] [CrossRef]
- Perez-Portilla, A.; Moraru, M.; Blazquez-Moreno, A.; Kolb, P.; Bravo Garcia-Morato, M.; Ranganath, T.; Esteso, G.; Gianelli, C.; Rodriguez-Pena, R.; Lozano-Rodriguez, R.; et al. Identification of the first cases of complete CD16A deficiency: Association with persistent EBV infection. J. Allergy Clin. Immunol. 2020, 145, 1288–1292. [Google Scholar] [CrossRef]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Rämer, P.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.T.; Spohn, M.; et al. Persistent KSHV infection increases EBV-associated tumor formation in vivo via enhanced EBV lytic gene expression. Cell Host Microbe 2017, 22, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caduff, N.; McHugh, D.; Rieble, L.; Forconi, C.S.; Ong’echa, J.M.; Oluoch, P.O.; Raykova, A.; Murer, A.; Böni, M.; Zuppiger, L.; et al. KSHV infection drives poorly cytotoxic CD56 negative natural killer cell differentiation in vivo upon KSHV/EBV dual infection. Cell Rep. 2021, 35, 109056. [Google Scholar] [CrossRef] [PubMed]
- Messinger, J.E.; Dai, J.; Stanland, L.J.; Price, A.M.; Luftig, M.A. Identification of host biomarkers of Epstein-Barr virus latency IIb and latency III. mBio 2019, 10, e01006-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, C.; Gabrysch, A.; Olbrich, P.; Patino, V.; Warnatz, K.; Wolff, D.; Hoshino, A.; Kobayashi, M.; Imai, K.; Takagi, M.; et al. Phenotype, penetrance, and treatment of 133 CTLA-4-insufficient individuals. J. Allergy Clin. Immunol. 2018, 142, 1932–1946. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Tanita, K.; Kanda, K.; Imadome, K.I.; Shikama, Y.; Yasumi, T.; Imai, K.; Takagi, M.; Morio, T.; Kanegane, H. High frequencies of asymptomatic Epstein-Barr virus viremia in affected and unaffected individuals with CTLA4 mutations. Clin. Immunol. 2018, 195, 45–48. [Google Scholar] [CrossRef]
- Egg, D.; Schwab, C.; Gabrysch, A.; Arkwright, P.D.; Cheesman, E.; Giulino-Roth, L.; Neth, O.; Snapper, S.; Okada, S.; Moutschen, M.; et al. Increased risk for malignancies in 131 affected CTLA4 mutation carriers. Front. Immunol. 2018, 9, 2012. [Google Scholar] [CrossRef]
- Egg, D.; Rump, I.C.; Mitsuiki, N.; Rojas-Restrepo, J.; Maccari, M.E.; Schwab, C.; Gabrysch, A.; Warnatz, K.; Goldacker, S.; Patino, V.; et al. Therapeutic options for CTLA-4 insufficiency. J. Allergy Clin. Immunol. 2021; in press. [Google Scholar] [CrossRef]
- Volk, V.; Theobald, S.J.; Danisch, S.; Khailaie, S.; Kalbarczyk, M.; Schneider, A.; Bialek-Waldmann, J.; Kronke, N.; Deng, Y.; Eiz-Vesper, B.; et al. PD-1 blockade aggravates Epstein-Barr virus+ post-transplant lymphoproliferative disorder in humanized mice resulting in central nervous system involvement and CD4+ T cell dysregulations. Front. Oncol. 2020, 10, 614876. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Xu, X.; Jones, R.; Delecluse, H.J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 blockade inhibits Epstein-Barr virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; McDonnell, W.J.; Gonzalez-Ericsson, P.I.; Al-Rohil, R.N.; Mobley, B.C.; Salem, J.E.; Wang, D.Y.; Sanchez, V.; Wang, Y.; Chastain, C.A.; et al. A case report of clonal EBV-like memory CD4+ T cell activation in fatal checkpoint inhibitor-induced encephalitis. Nat. Med. 2019, 25, 1243–1250. [Google Scholar] [CrossRef]
- Bickham, K.; Goodman, K.; Paludan, C.; Nikiforow, S.; Tsang, M.L.; Steinman, R.M.; Münz, C. Dendritic cells initiate immune control of Epstein-Barr virus transformation of B lymphocytes in vitro. J. Exp. Med. 2003, 198, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Maurer, M.A.; Meixlsperger, S.; Lippmann, A.; Cheong, C.; Zuo, J.; Haigh, T.A.; Taylor, G.S.; Münz, C. Robust T-cell stimulation by Epstein-Barr virus-transformed B cells after antigen targeting to DEC-205. Blood 2013, 121, 1584–1594. [Google Scholar] [CrossRef] [Green Version]
- Gurer, C.; Strowig, T.; Brilot, F.; Pack, M.; Trumpfheller, C.; Arrey, F.; Park, C.G.; Steinman, R.M.; Münz, C. Targeting the nuclear antigen 1 of Epstein-Barr virus to the human endocytic receptor DEC-205 stimulates protective T-cell responses. Blood 2008, 112, 1231–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meixlsperger, S.; Leung, C.S.; Ramer, P.C.; Pack, M.; Vanoaica, L.D.; Breton, G.; Pascolo, S.; Salazar, A.M.; Dzionek, A.; Schmitz, J.; et al. CD141+ dendritic cells produce prominent amounts of IFN-alpha after dsRNA recognition and can be targeted via DEC-205 in humanized mice. Blood 2013, 121, 5034–5044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zyl, D.G.; Tsai, M.H.; Shumilov, A.; Schneidt, V.; Poirey, R.; Schlehe, B.; Fluhr, H.; Mautner, J.; Delecluse, H.J. Immunogenic particles with a broad antigenic spectrum stimulate cytolytic T cells and offer increased protection against EBV infection ex vivo and in mice. PLoS Pathog. 2018, 14, e1007464. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Wang, Z.; Ke, Q.; Hong, M.; Paul, D.W., Jr.; Fernandes, S.M.; Hu, Z.; Stevens, J.; Guleria, I.; Kim, H.J.; et al. Mechanism of EBV inducing anti-tumour immunity and its therapeutic use. Nature 2021, 590, 157–162. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Co-stimulatory molecules that are involved in T and NK cell expansion as well as EBV-infected target cell recognition by these cytotoxic lymphocytes. The interaction of CD27 with CD70 as well as 4-1BB seems to be involved in T-cell expansion after EBV infection. SAP-associated SLAM receptors, such as 2B4, contribute to T-cell recognition of EBV-transformed B cells. CD27 and NKG2D are involved in T and NK cell recognition of lytically EBV-infected B cells, respectively. 4-1BB and CD2 (assisted by CD16A) might be involved in EBV-infected T-cell recognition.
Figure 1.
Co-stimulatory molecules that are involved in T and NK cell expansion as well as EBV-infected target cell recognition by these cytotoxic lymphocytes. The interaction of CD27 with CD70 as well as 4-1BB seems to be involved in T-cell expansion after EBV infection. SAP-associated SLAM receptors, such as 2B4, contribute to T-cell recognition of EBV-transformed B cells. CD27 and NKG2D are involved in T and NK cell recognition of lytically EBV-infected B cells, respectively. 4-1BB and CD2 (assisted by CD16A) might be involved in EBV-infected T-cell recognition.

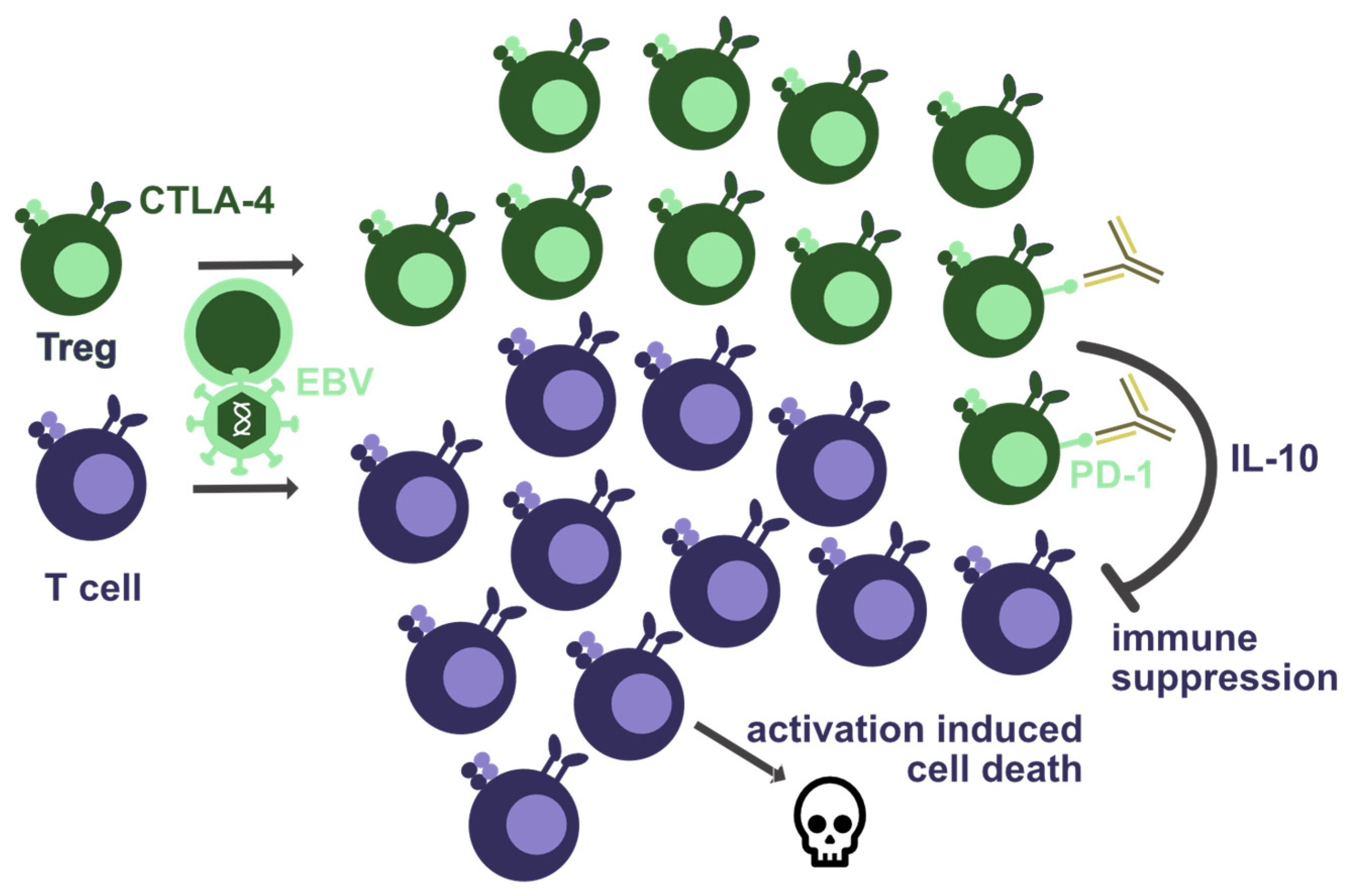
Figure 2.
Co-inhibitory receptors that are required for EBV-specific immune control. Haploinsufficiency of CTLA-4 seems to lead to T-cell lymphopenia and increased regulatory T-cell (Treg) numbers. This could result from activation-induced cell death due to T-cell hyperactivation, or from suppression by Tregs due to their diminished CTLA-4-mediated inhibition. Furthermore, PD-1 blockade by antibodies could further increase Treg-mediated suppression of EBV-specific immune control, e.g., via cytokines such as IL-10.
Figure 2.
Co-inhibitory receptors that are required for EBV-specific immune control. Haploinsufficiency of CTLA-4 seems to lead to T-cell lymphopenia and increased regulatory T-cell (Treg) numbers. This could result from activation-induced cell death due to T-cell hyperactivation, or from suppression by Tregs due to their diminished CTLA-4-mediated inhibition. Furthermore, PD-1 blockade by antibodies could further increase Treg-mediated suppression of EBV-specific immune control, e.g., via cytokines such as IL-10.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Münz, C. Co-Stimulatory Molecules during Immune Control of Epstein Barr Virus Infection. Biomolecules 2022, 12, 38. https://doi.org/10.3390/biom12010038
AMA Style
Münz C. Co-Stimulatory Molecules during Immune Control of Epstein Barr Virus Infection. Biomolecules. 2022; 12(1):38. https://doi.org/10.3390/biom12010038
Chicago/Turabian StyleMünz, Christian. 2022. "Co-Stimulatory Molecules during Immune Control of Epstein Barr Virus Infection" Biomolecules 12, no. 1: 38. https://doi.org/10.3390/biom12010038
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.