
The Emerging Role of Metabolism in Brain-Heart Axis: New Challenge for the Therapy and Prevention of Alzheimer Disease. May Thioredoxin Interacting Protein (TXNIP) Play a Role?

{kind=link}
{kind=link}
Abstract
:1. Introduction
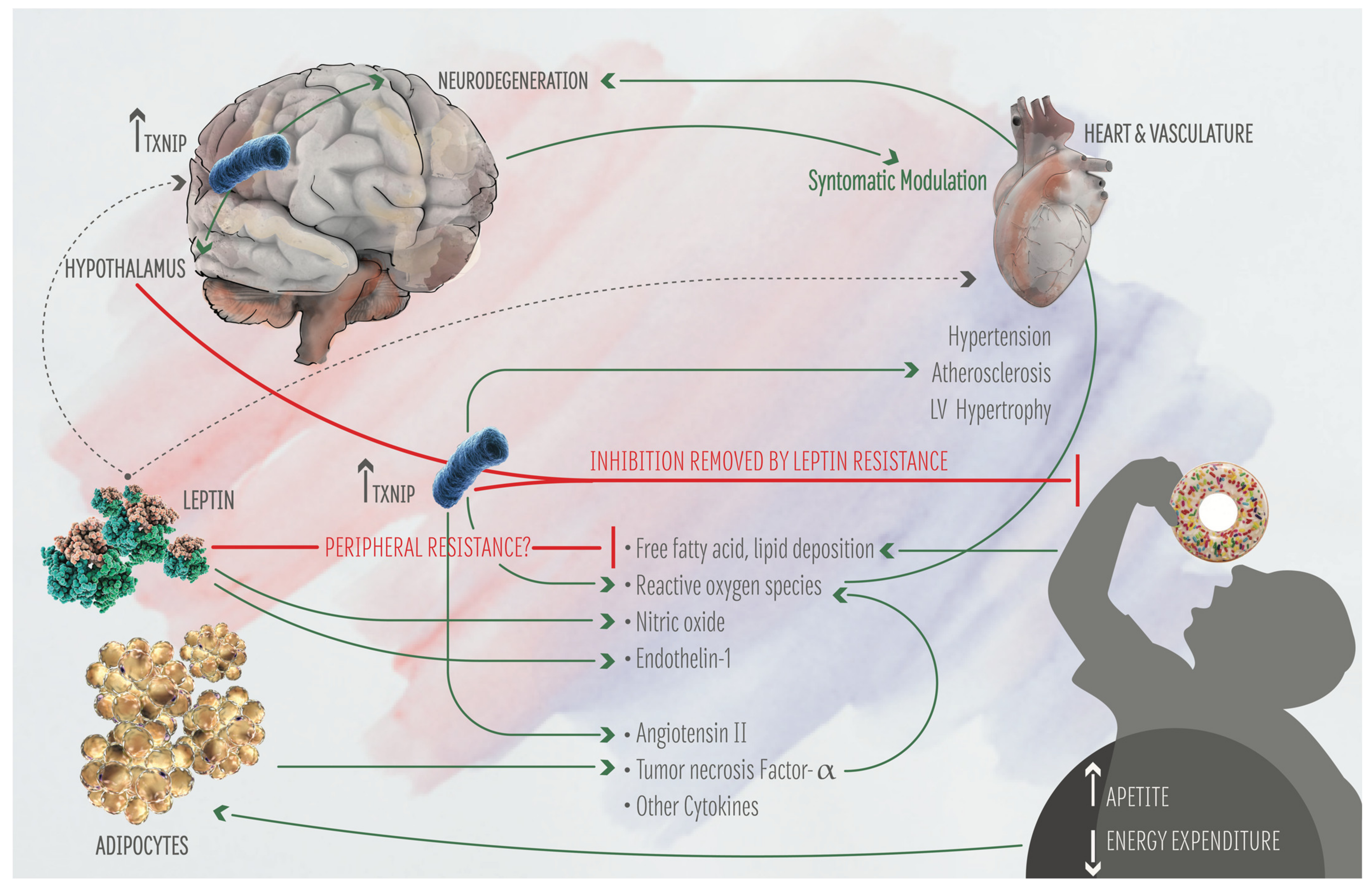
2. TXNIP: Its Function on Metabolism and Brain-Heart Axis
3. The Heart-Brain Axis and the Bi-Directional Connection with AD Role of Txnip
4. The Central Autonomic Network (CAN): Effect on AD
5. Metabolic AD Risk Factors and Central Autonomic Network: A Bi-Directional Regulation
6. The Renin-Angiotensin System: Role in AD and AD Risk Factors
7. The Natriuretic Peptides and Endothelins Counteract the Renin-Angiotensin Pathway: A Role in AD?
8. Role of the Gut Microbiota in the Brain-Heart Axis: Effect of Metabolism
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer Disease |
| TXNIP | Thioredoxin Interacting protein |
| Aβ | amyloid beta |
| APP | amyloid precursor protein |
| PSEN1 and PSEN2 | presenilin 1 and 2 |
| LOAD | late onset Alzheimer Disease |
| FAD | familiar Alzheimer Disease |
| NMDA | N-methyl-D-aspartate |
| ChEIs | inhibitors of the cholinesterase |
| ADMA | Asymmentric DiMethyl Arginine |
| SVD | small vessels disease |
| RAGE | Receptor for Advanced Glycation Endproducts |
| BBB | Blood Brain Barrier |
| Trx | Thioredoxin |
| ROS | reactive oxygen species |
| TBP-2 | Thioredoxin Binding protein 2 |
| VDUP-1 | Vitamin D Upregulated Protein 1 |
| AGEs | Advanced Glycation Endproducts |
| HMGB1 | high mobiliy group B1 |
| NLRP3 | NOD-like receptor family, pyrin domain containing 3 |
| PD | Parkinson Disease |
| HRV | heart rate variability |
| ApoE4 | apolipoprotein E4 |
| CAN | central autonomic network |
| ANS | autonomic nervous system |
| aMCI | amnestic mild cognitive impairment |
| HR | heart rate |
| ob | obese gene |
| db | diabetes gene |
| ObR | leptin receptor |
| BMI | body mass index |
| AGT | angiotensinogen |
| ANGII | angiotensin II |
| ACE1 | angiotensin converting enzyme 1 |
| ATRs | angiotensin receptors |
| PRR | pro-renin receptor |
| NPs | natriuretic peptides |
| ANP | atrial natriuretic peptide |
| BNP | brain natriuretic peptide |
| CNP | C-type natriuretic peptide |
| NPRA | Natriuretic Receptor A |
| NPRB | Natriuretic Receptor B |
| NPRC | Natriuretic Receptor C |
| NT-proBNP | N-terminal pro-brain natriuretic peptide |
| ET-1, 2, 3 | Endothelin 1, 2, 3 |
| PKC | Protein Kinase C |
| CNS | central nervous system |
| GM | gut microbiome |
| PAMPs | pathogen-associated molecular patterns |
| LPS | lipopolysaccharides |
| HFD | high-fat diet |
| MS | metabolic syndrome |
| GLP-1 | glucagon-like peptide 1 |
| GLP-1R | GLP-1 receptor |
References
- Prince, M.; Ali, G.C.; Guerchet, M.; Prina, A.M.; Albanese, E.; Wu, Y.T. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers Res. Ther. 2016, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimenova, A.A.; Raj, T.; Goate, A.M. Untangling Genetic Risk for Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 300–310. [Google Scholar] [CrossRef]
- Perrone, L.; Sbai, O.; Nawroth, P.P.; Bierhaus, A. The Complexity of Sporadic Alzheimer’s Disease Pathogenesis: The Role of RAGE as Therapeutic Target to Promote Neuroprotection by Inhibiting Neurovascular Dysfunction. Int. J. Alzheimers Dis. 2012, 2012, 734956–734966. [Google Scholar] [CrossRef] [Green Version]
- Steinbrook, R. The Accelerated Approval of Aducanumab for Treatment of Patients with Alzheimer Disease. JAMA Intern. Med. 2021, 181, 1281. [Google Scholar] [CrossRef]
- Crosson, F.J.; Covinsky, K.; Redberg, R.F. Medicare and the Shocking US Food and Drug Administration Approval of Aducanumab: Crisis or Opportunity? JAMA Intern. Med. 2021, 181, 1278–1280. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yao, L.; Liu, J.; Jiang, Y.; Ma, G.; Chen, Z.; Zhao, B.; Li, K.; (GERAD1) Consortium. Cardiovascular disease contributes to Alzheimer’s disease: Evidence from large-scale genome-wide association studies. Neurobiol. Aging 2014, 35, 786–792. [Google Scholar] [CrossRef]
- Ungvari, Z.; Toth, P.; Tarantini, S.; Prodan, C.I.; Sorond, F.; Merkely, B.; Csiszar, A. Hypertension-induced cognitive impairment: From pathophysiology to public health. Nat. Rev. Nephrol. 2021, 14, 1–16. [Google Scholar]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s Disease: Mechanisms and Nutritional Aspects. Clin. Nutr. Res. 2018, 7, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Apo, E.; Mondragón-Maya, A.; Ferrari-Díaz, M.; Silva-Pereyra, J. Structural Brain Changes Associated with Overweight and Obesity. J. Obes. 2021, 2021, 6613385. [Google Scholar] [CrossRef]
- Wanamaker, B.L.; Swiger, K.J.; Blumenthal, R.S.; Martin, S.S. Cholesterol, statins, and dementia: What the cardiologist should know. Clin. Cardiol. 2015, 38, 243–250. [Google Scholar] [CrossRef]
- Janes, F.; Cifù, A.; Pessa, M.E.; Domenis, R.; Gigli, G.L.; Sanvilli, N.; Nilo, A.; Garbo, R.; Curcio, F.; Giacomello, R.; et al. ADMA as a possible marker of endothelial damage. A study in young asymptomatic patients with cerebral small vessel disease. Sci. Rep. 2019, 9, 14207. [Google Scholar] [CrossRef] [PubMed]
- Arlt, S.; Schulze, F.; Eichenlaub, M.; Maas, R.; Lehmbeck, J.T.; Schwedhelm, E.; Jahn, H.; Böger, R.H. Asymmetrical dimethylarginine is increased in plasma and decreased in cerebrospinal fluid of patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2008, 26, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Malden, D.E.; Mangoni, A.A.; Woodman, R.J.; Thies, F.; McNeil, C.; Murray, A.D.; Soiza, R.L. Circulating asymmetric dimethylarginine and cognitive decline: A 4-year follow-up study of the 1936 Aberdeen Birth Cohort. Int. J. Geriatr. Psychiatry 2020, 35, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.L.; Xiang, Y.; Jin, W.S.; Wang, J.; Shen, L.L.; Huang, Z.L.; Zhang, K.; Liu, Y.H.; Zeng, F.; Liu, J.H.; et al. Blood-derived amyloid-beta protein induces Alzheimer’s disease pathologies. Mol. Psychiatry 2018, 23, 1948–1956. [Google Scholar] [CrossRef] [PubMed]
- Tublin, J.M.; Adelstein, J.M.; Del Monte, F.; Combs, C.K.; Wold, L.E. Getting to the Heart of Alzheimer Disease. Circ. Res. 2019, 124, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Troncone, L.; Luciani, M.; Coggins, M.; Wilker, E.H.; Ho, C.Y.; Codispoti, K.E.; Frosch, M.P.; Kayed, R.; Del Monte, F. Abeta Amyloid Pathology Affects the Hearts of Patients With Alzheimer’s Disease: Mind the Heart. J. Am. Coll. Cardiol. 2016, 68, 2395–2407. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Tao, X.; Ma, X.; Zhao, R.; Cao, Z. Cognitive Dysfunction after Heart Disease: A Manifestation of the Heart-Brain Axis. Oxid. Med. Cell. Longev. 2021, 2021, 4899688. [Google Scholar] [CrossRef] [PubMed]
- Qayyum, N.; Haseeb, M.; Kim, M.S.; Choi, S. Role of Thioredoxin-Interacting Protein in Diseases and Its Therapeutic Outlook. Int. J. Mol. Sci. 2021, 22, 2754. [Google Scholar] [CrossRef]
- Kim, S.Y.; Suh, H.W.; Chung, J.W.; Yoon, S.R.; Choi, I. Diverse functions of VDUP1 in cell proliferation, differentiation, and diseases. Cell. Mol. Immunol. 2007, 4, 345–351. [Google Scholar] [PubMed]
- Perrone, L.; Devi, T.S.; Hosoya, K.C.; Terasaki, T.; Singh, L.P. Thioredoxin Interacting Protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Devi, T.S.; Hosoya, K.C.; Terasaki, T.; Singh, L.P. Inhibition of TXNIP Expression In Vivo Blocks Early Pathologies of Diabetic Retinopathy. Cell Death Dis. 2010, 1, e65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingues, A.; Jolibois, J.; Marquet de Rougé, P.; Nivet-Antoine, V. The Emerging Role of TXNIP in Ischemic and Cardiovascular Diseases; A Novel Marker and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 1693. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Sbai, O.; Devi, T.S.; Melone, M.A.B.; Feron, F.; Khrestchatisky, M.; Singh, L.P.; Perrone, L. RAGE-TXNIP axis is required for S100B-promoted Schwann cell migration, fibronectin expression and cytokine secretion. J. Cell Sci. 2010, 123, 4332–4339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubaki, H.; Tooyama, I.; Walker, D.G. Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9357. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Bharti, V.; Zhou, H.; Hoi, V.; Tan, H.; Wu, Z.; Nagakannan, P.; Eftekharpour, E.; Wang, J.F. Upregulation of Thioredoxin-Interacting Protein in Brain of Amyloid-β Protein Precursor/Presenilin 1 Transgenic Mice and Amyloid-β Treated Neuronal Cells. J. Alzheimers Dis. 2019, 72, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Hokama, M.; Oka, S.; Leon, J.; Ninomiya, T.; Honda, H.; Sasaki, K.; Iwaki, T.; Ohara, T.; Sasaki, T.; LaFerla, F.M.; et al. Altered Expression of Diabetes-Related Genes in Alzheimer’s Disease Brains: The Hisayama Study. Cereb. Cortex. 2014, 24, 2476–2488. [Google Scholar] [CrossRef]
- Melone, M.A.B.; Dato, C.; Paladino, S.; Coppola, C.; Trebini, C.; Giordana, M.; Perrone, L. Verapamil Inhibits Ser202/Thr205 Phosphorylation of Tau by Blocking TXNIP/ROS/p38 MAPK Pathway. Pharm. Res. 2018, 35, 44. [Google Scholar] [CrossRef]
- Matrone, C.; Djelloul, M.; Taglialatela, G.; Perrone, L. Inflammatory risk factors and pathologies promoting Alzheimer’s disease progression: Is RAGE the key? Histol. Histopathol. 2015, 30, 125–139. [Google Scholar]
- Abate, G.; Uberti, D.L.; Djelloul, M.; Sbai, O.; Ieraci, A.; Perrone, L. RAGE-TXNIP axis drives inflammation in Alzheimer’s by targeting Aβ to mitochondria in microglia. Preprint 2020. [Google Scholar] [CrossRef]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jin, F.; Cao, G.; Mei, R.; Wang, Y.; Long, P.; Wang, X.; Ge, W. ApoE4 May be a Promising Target for Treatment of Coronary Heart Disease and Alzheimer’s Disease. Curr. Drug Targets 2018, 19, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.A.; Huan, T.; Ligthart, S.; Gondalia, R.; Jhun, M.A.; Brody, J.A.; Irvin, M.R.; Marioni, R.; Shen, J.; Tsai, P.C.; et al. DNA Methylation Analysis Identifies Loci for Blood Pressure Regulation. Am. J. Hum. Genet. 2017, 101, 888–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Huang, J.; Yang, X.; Sun, X.; Xu, Q.; Wang, B.; Zhong, P.; Wei, Z. Altered Expression of TXNIP in the peripheral leukocytes of patients with coronary atherosclerotic heart disease. Medicine 2017, 96, e9108. [Google Scholar] [CrossRef] [PubMed]
- Palma, J.A.; Benarroch, E.E. Neural control of the heart. Recent concepts and clinical correlations. Neurology 2014, 8, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Van der Wall, E.E.; van Gilst, W.H. Neurocardiology: Close interaction between heart and brain. Neth. Heart J. 2013, 21, 51–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thayer, J.F.; Hansen, A.L.; Saus-Rose, E.; Johnsen, B.H. Heart rate variability, prefrontal neural function, and cognitive performance: The neurovisceral integration perspective on self-regulation, adaptation, and health. Ann. Behav. Med. 2009, 37, 141–153. [Google Scholar] [CrossRef]
- Tahsili-Fahadan, P.; Geocadin, R.G. Heart–brain axis: Effects of neurologic injury on cardiovascular function. Circ. Res. 2017, 120, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, P.; Ciulla, M.M.; De Asmundis, C.; Magrini, F.; Brugada, P. The prognostic value of heart rate variability in the elderly, changing the perspective: From sympathovagal balance to chaos theory. Pacing Clin. Electrophysiol. 2012, 35, 622–638. [Google Scholar] [CrossRef]
- Al Hazzouri, A.Z.; Haan, M.N.; Deng, Y.; Neuhaus, J.; Yaffe, K. Reduced heart rate variability is associated with worse cognitive performance in elderly Mexican Americans. Hypertension 2014, 63, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, V.P.; Ramalho Oliveira, B.R.; Tavares Mello, R.G.; Moraes, H.; Deslandes, A.C.; Laks, J. Heart Rate Variability Indexes in Dementia: A Systematic Review with a Quantitative Analysis. Curr. Alzheimer Res. 2018, 15, 80–88. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Z.; Li, S. Can multi-modal neuroimaging evidence from hippocampus provide biomarkers for the progression of amnestic mild cognitive impairment? Neurosci. Bull. 2015, 31, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayram, E.; Caldwell, J.Z.K.; Banks, S.J. Current understanding of magnetic resonance imaging biomarkers and memory in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, D.A.; Mraovitch, S.; Granata, A.R.; Anwar, M.; Reis, D.J. A role of insular cortex in cardiovascular function. J. Comp. Neurol. 1987, 257, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.; Jennings, J.R.; Gianaros, P.J.; Thayer, J.F.; Manuck, S.B. Resting high-frequency heart rate variability is related to resting brain perfusion. Psychophysiology 2015, 52, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmerly, D.S. A review of human neuroimaging investigations involved with central autonomic regulation of baroreflexmediated cardiovascular control. Auton. Neurosci. 2017, 207, 10–21. [Google Scholar] [CrossRef]
- Valenza, G.; Passamonti, L.; Duggento, A.; Toschi, N.; Barbieri, R. Uncovering complex central autonomic networks at rest: A functional magnetic resonance imaging study on complex cardiovascular oscillations. J. R. Soc. Interface 2020, 17, 20190878. [Google Scholar] [CrossRef] [Green Version]
- Frontoni, S.; Bracaglia, D.; Gigli, F. Relationship between autonomic dysfunction, insulin resistance and hypertension, in diabetes. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Enriori, P.J.; Sinnayah, P.; Simonds, S.E.; Garcia Rudaz, C.; Cowley, M.A. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. J. Neurosci. 2011, 31, 12189–12197. [Google Scholar] [CrossRef] [Green Version]
- Guarino, D.; Nannipieri, M.; Iervasi, G.; Taddei, S.; Bruno, R.M. The Role of the Autonomic Nervous System in the Pathophysiology of Obesity. Front. Physiol. 2017, 8, 665. [Google Scholar] [CrossRef] [Green Version]
- Landsberg, L. Insulin-mediated sympathetic stimulation: Role in the pathogenesis of obesity-related hypertension (or, how insulin affects blood pressure, and why). J. Hypertens. 2001, 19, 523–528. [Google Scholar] [CrossRef]
- Battault, S.; Meziat, C.; Nascimento, A.; Braud, L.; Gayrard, S.; Legros, C.; De Nardi, F.; Drai, J.; Cazorla, O.; Thireau, J.; et al. Vascular endothelial function masks increased sympathetic vasopressor activity in rats with metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H497–H507. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.E.; Omae, S.; Pereira, A.; Rodrigues, M.V.; Miyakawa, A.A.; Campos, L.C.; Santos, P.C.; Dallan, L.A.; Martinez, T.L.; Santos, R.D.; et al. Thioredoxin interacting protein genetic variation is associated with diabetes and hypertension in the Brazilian general population. Atherosclerosis 2012, 221, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Alvim, R.O.; Santos, P.C.; Ferreira, N.E.; Mill, J.G.; Krieger, J.E.; Pereira, A.C. Thioredoxin interacting protein (TXNIP) rs7212 polymorphism is associated with arterial stiffness in the Brazilian general population. J. Hum. Hypertens. 2012, 26, 340–342. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.B.; Han, Y.D.; Zhang, S.; Cui, N.H.; Liu, Z.J.; Huang, Z.L.; Li, C.; Zheng, F. Associations of polymorphisms in TXNIP and gene-environment interactions with the risk of coronary artery disease in a Chinese Han population. J. Cell Mol. Med. 2016, 20, 2362–2373. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, E. TXNIP/TBP-2, A Master Regulator for Glucose Homeostasis. Antioxidants 2020, 9, 765. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-Dependent Degradation of TXNIP upon Energy Stress Leads to Enhanced Glucose Uptake via GLUT1. Mol. Cell. 2013, 49, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, R.; Beerens, S.; Adan, R.A.H. Role of leptin in energy expenditure: The hypothalamic perspective. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 312, R938–R947. [Google Scholar] [CrossRef] [Green Version]
- Elmquist, J.K.; Elias, C.F.; Saper, C.B. From lesions to leptin: Hypothalamic control of food intake and body weight. Neuron 1999, 22, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Bell, B.B.; Rahmouni, K. Leptin as a Mediator of Obesity-Induced Hypertension. Curr. Obes. Rep. 2016, 5, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.A.; do Carmo, J.M.; Hall, J.E. CNS Regulation of Glucose Homeostasis: Role of the Leptin-Melanocortin System. Curr. Diab. Rep. 2020, 26, 29. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.; Pan, C.; Contreras, R.E.; Wiedemann, T.; Morgan, D.A.; Skowronski, A.A.; Lefort, S.; Murat, C.D.; Le Thuc, O.; Legutko, B.; et al. Obesity-associated hyperleptinemia alters the gliovascular interface of the hypothalamus to promote hypertension. Cell Metab. 2021, 33, 1155–1170. [Google Scholar] [CrossRef] [PubMed]
- Meakin, P.J.; Jalicy, S.M.; Montagut, G.; Allsop, D.J.P.; Cavellini, D.L.; Irvine, S.W.; McGinley, C.; Liddell, M.K.; McNeilly, A.D.; Parmionova, K.; et al. Bace1-dependent amyloid processing regulates hypothalamic leptin sensitivity in obese mice. Sci. Rep. 2018, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooqi, I.S.; O’Rahilly, S. 20 years of leptin: Human disorders of leptin action. J. Endocrinol. 2014, 223, T63–T70. [Google Scholar] [CrossRef]
- Shanley, L.J.; Irving, A.J.; Harvey, J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 2001, 21, RC186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.L.; Aou, S.; Oomura, Y.; Hori, N.; Fukunaga, K.; Hori, T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 2002, 113, 607–615. [Google Scholar] [CrossRef]
- Ahima, R.S.; Bjorbaek, C.; Osei, S.; Flier, J.S. Regulation of neuronal and glial proteins by leptin: Implications for brain development. Endocrinology 1999, 140, 2755–2762. [Google Scholar] [CrossRef]
- Perrone, L.; Grant, W.B. Observational and ecological studies of dietary advanced glycation end products in national diets and Alzheimer’s disease incidence and prevalence. J. Alzheimers Dis. 2015, 45, 965–979. [Google Scholar] [CrossRef]
- Power, D.A.; Noel, J.; Collins, R.; O’Neill, D. Circulating leptin levels and weight loss in Alzheimer’s disease patients. Dement. Geriatr. Cogn. Disord. 2001, 12, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Lieb, W.; Beiser, A.S.; Vasan, R.S.; Tan, Z.S.; Au, R.; Harris, T.B.; Roubenoff, R.; Auerbach, S.; DeCarli, C.; Wolf, P.A.; et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA 2009, 302, 2565–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J. 20 years of leptin: Leptin at 20, An overview. J. Endocrinol. 2014, 223, T1–T8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guérin, O.; Andrieu, S.; Schneider, S.M.; Cortes, F.; Cantet, C.; Gillette-Guyonnet, S.; Vellas, B. Characteristics of Alzheimer’s disease patients with a rapid weight loss during a six-year follow-up. Clin. Nutr. 2009, 28, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Rabin, J.S.; Shirzadi, Z.; Swardfager, W.; MacIntosh, B.J.; Schultz, A.; Yang, H.S.; Buckley, R.F.; Gatchel, J.R.; Kirn, D.; Pruzin, J.J.; et al. Amyloid-beta burden predicts prospective decline in body mass index in clinically normal adults. Neurobiol. Aging 2020, 93, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Schmitz, S.; Hansson, O.; Walsh, C.; Fitzpatrick, A.; Bennett, D.; Minthon, L.; Trojanowski, J.Q.; Shaw, L.M.; Faluyi, Y.O.; et al. Body mass index is associated with biological CSF markers of core brain pathology of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1599–1608. [Google Scholar] [CrossRef] [Green Version]
- Cova, I.; Clerici, F.; Rossi, A.; Cucumo, V.; Ghiretti, R.; Maggiore, L.; Pomati, S.; Galimberti, D.; Scarpini, E.; Mariani, C.; et al. Weight Loss Predicts Progression of Mild Cognitive Impairment to Alzheimer’s Disease. PLoS ONE 2016, 11, e0151710. [Google Scholar] [CrossRef] [Green Version]
- Robison, L.S.; Gannon, O.J.; Thomas, M.A.; Salinero, A.E.; Abi-Ghanem, C.; Poitelon, Y.; Belin, S.; Zuloaga, K.L. Role of sex and high-fat diet in metabolic and hypothalamic disturbances in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 285. [Google Scholar] [CrossRef]
- López-Gambero, A.J.; Rosell-Valle, C.; Medina-Vera, D.; Navarro, J.A.; Vargas, A.; Rivera, P.; Sanjuan, C.; Rodríguez de Fonseca, F.; Suárez, J. A Negative Energy Balance Is Associated with Metabolic Dysfunctions in the Hypothalamus of a Humanized Preclinical Model of Alzheimer’s Disease, the 5XFAD Mouse. Int. J. Mol. Sci. 2021, 22, 5365. [Google Scholar] [CrossRef]
- Johnson, D.K.; Wilkins, C.H.; Morris, J.C. Accelerated weight loss may precede diagnosis in Alzheimer disease. Arch. Neurol. 2006, 63, 1312–1317. [Google Scholar] [CrossRef]
- McGregor, G.; Harvey, J. Food for thought: Leptin regulation of hippocampal function and its role in Alzheimer’s disease. Neuropharmacology. Neuropharmacology 2018, 136, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Iadecola, C. Metabolic and non-cognitive manifestations of Alzheimer’s disease: The hypothalamus as both culprit and target of pathology. Cell Metab. 2015, 22, 761–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, J.R.; Lyra, E.; Silva, N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Katashima, C.K.; Razolli, D.; Carvalho, B.; Frazao, R.; et al. Alzheimer-associated Aβ oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Blouet, C.; Schwartz, G.J. Nutrient-sensing hypothalamic TXNIP links nutrient excess to energy imbalance in mice. J. Neurosci. 2011, 31, 6019–6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blouet, C.; Liu, S.M.; Jo, Y.H.; Chua, S.; Schwartz, G.J. TXNIP in Agrp neurons regulates adiposity, energy expenditure, and central leptin sensitivity. J. Neurosci. 2012, 32, 9870–9877. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, P.; Sigmund, C.D. How Is the Brain Renin-Angiotensin System Regulated? Hypertension 2017, 70, 10–18. [Google Scholar] [CrossRef]
- Sinn, P.L.; Sigmund, C.D. Identification of three human renin mRNA isoforms from alternative tissue-specific transcriptional initiation. Physiol. Genomics. 2000, 3, 25–31. [Google Scholar] [CrossRef]
- Phillips, M.I.; de Oliveira, E.M. Brain renin angiotensin in disease. J. Mol. Med. 2008, 86, 715–722. [Google Scholar] [CrossRef]
- Rao, Y.; Chen, J.; Guo, Y.; Ji, T.; Xie, P. Rivaroxaban ameliorates angiotensin II-induced cardiac remodeling by attenuating TXNIP/Trx2 interaction in KKAy mice. Thromb. Res. 2021, 193, 45–52. [Google Scholar] [CrossRef]
- Cheung, B.M.; Li, C. Diabetes and hypertension: Is there a common metabolic pathway? Curr. Atheroscler. Rep. 2012, 14, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denver, P.; McClean, P. Distinguishing normal brain aging from the development of Alzheimer’s disease: Inflammation, insulin signaling and cognition. Neural. Regen. Res. 2018, 13, 1719. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Shi, J.; Zhang, Y.; Wang, B.; Liu, W.; Chen, Z.; Tong, Q. Central angiotensin II stimulation promotes β amyloid production in Sprague Dawley rats. PLoS ONE 2011, 6, e16037. [Google Scholar] [CrossRef] [Green Version]
- AbdAlla, S.; Lother, H.; el Missiry, A.; Langer, A.; Sergeev, P.; el Faramawy, Y.; Quitterer, U. Angiotensin II AT2 receptor oligomers mediate G-protein dysfunction in an animal model of Alzheimer disease. J. Biol. Chem. 2009, 284, 6554–6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosarderelioglu, C.; Nidadavolu, L.S.; George, C.J.; Oh, E.S.; Bennett, D.A.; Walston, J.D.; Abadir, P.M. Brain Renin-Angiotensin System at the Intersect of Physical and Cognitive Frailty. Front. Neurosci. 2020, 14, 586314. [Google Scholar] [CrossRef] [PubMed]
- Hodes, A.; Lichtstein, D. Natriuretic hormones in brain function. Front. Endocrinol. 2014, 5, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabayan, B.; van Buchem, M.A.; Sigurdsson, S.; Zhang, Q.; Meirelles, O.; Harris, T.B.; Gudnason, V.; Arai, A.E.; Launer, L.J. Cardiac and Carotid Markers Link with Accelerated Brain Atrophy: The AGES-Reykjavik Study (Age, Gene/Environment Susceptibility-Reykjavik). Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2246–2251. [Google Scholar] [CrossRef] [Green Version]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimer’s Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, J.A.; Moffitt, P.; Perez-Moreno, A.C.; Walters, M.R.; Broomfield, N.M.; McMurray, J.J.V.; Quinn, T.J. Cognitive Impairment and Heart Failure: Systematic Review and Meta-Analysis. J. Card. Fail. 2017, 23, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Rukavina Mikusic, N.L.; Kouyoumdzian, N.M.; Puyó, A.M.; Fernández, B.E.; Choi, M.R. Role of natriuretic peptides in the cardiovascular-adipose communication: A tale of two organs. Pflugers Arch. 2021, 1–15. [Google Scholar]
- Fouda, A.Y.; Fagan, S.C.; Ergul, A. Brain Vasculature and Cognition. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Ritthaler, T.; Scholz, H.; Ackermann, M.; Riegger, G.; Kurtz, A.; Krämer, B.K. Effects of endothelins on renin secretion from isolated mouse renal juxtaglomerular cells. Am. J. Physiol. 1995, 268, F39–F45. [Google Scholar] [CrossRef]
- Pohjolainen, L.; Easton, J.; Solanki, R.; Ruskoaho, H.; Talman, V. Pharmacological Protein Kinase C Modulators Reveal a Pro-hypertrophic Role for Novel Protein Kinase C Isoforms in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Pharmacol. 2021, 11, 553852. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Behl, T.; Kumar, A.; Sehgal, A.; Singh, S.; Sharma, N.; Hatia, S.; Al-Harrasi, A.; Bungau, S. Targeting Endothelin in Alzheimer’s Disease: A Promising Therapeutic Approach. Biomed. Res. Int. 2021, 2021, 7396580. [Google Scholar] [CrossRef]
- Wang, N.Y.; Li, J.N.; Liu, W.L.; Huang, Q.; Li, W.X.; Tan, Y.H.; Liu, F.; Song, Z.H.; Wang, M.Y.; Xie, N.; et al. Ferulic Acid Ameliorates Alzheimer’s Disease-like Pathology and Repairs Cognitive Decline by Preventing Capillary Hypofunction in APP/PS1 Mice. Neurotherapeutics 2021, 18, 1064–1080. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, D.J. Dysregulation of Endothelin-1, Implications for Health Disparities in Alzheimer’s Disease. J. Pers. Med. 2020, 10, 199. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Correction to: Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 119. [Google Scholar] [CrossRef]
- Sommer, F.; Bäckhed, F. The gut microbiota–masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Bazin, T.; Pellissier, S. The vagus nerve at the interface of the microbiota-gut-brain axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrenovich, M.; Sankar Chittoor Mana, T.; Rai, H.; Shola, D.; Christopher, S.; McCloskey, B.; Levison, B.S. Recent findings within the microbiota- gut-brain-endocrine metabolic interactome. Pathol. Lab. Med. Int. 2017, 9, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Brierley, S.M.; Linden, D.R. Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 611–627. [Google Scholar] [CrossRef]
- Jonsson, A.L.; Bäckhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Cani, P.D. Diabetes, obesity and gut microbiota. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Cantone, E.; Cassarano, S.; Tuccinardi, D.; Barrea, L.; Savastano, S.; Colao, A. Gut microbiota: A new path to treat obesity. Int. J. Obes. Suppl. 2019, 9, 10–19. [Google Scholar] [CrossRef]
- Cani, P.D.; Van Hul, M.; Lefort, C.; Depommier, C.; Rastelli, M.; Everard, A. Microbial regulation of organismal energy homeostasis. Nat. Metab. 2019, 1, 34. [Google Scholar] [CrossRef]
- Geurts, L.; Neyrinck, A.M.; Delzenne, N.M.; Knauf, C.; Cani, P.D. Gut microbiota controls adipose tissue expansion, gut barrier and glucose metabolism: Novel insights into molecular targets and interventions using prebiotics. Benef. Microbes. 2014, 5, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front. Neurosci. 2018, 12, 930. [Google Scholar] [CrossRef]
- Heiss, C.N.; Mannerås-Holm, L.; Lee, Y.S.; Serrano-Lobo, J.; Håkansson Gladh, A.; Seeley, R.J.; Drucker, D.J.; Bäckhed, F.; Olofsson, L.E. The gut microbiota regulates hypothalamic inflammation and leptin sensitivity in Western diet-fed mice via a GLP-1R-dependent mechanism. Cell Rep. 2021, 35, 109163. [Google Scholar] [CrossRef]
- Peng, J.; Xiao, X.; Hu, M.; Zhang, X. Interaction between gut microbiome and cardiovascular disease. Life Sci. 2018, 214, 153–157. [Google Scholar] [CrossRef]
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-linked pathophysiological alterations in the gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef]
- Tanaka, M.; Itoh, H. Hypertension as a metabolic disorder and the novel role of the gut. Curr. Hypertens. Rep. 2019, 21, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryniarski, M.A.; Hamarneh, F.; Yacoub, R. The role of chronic kidney disease-associated dysbiosis in cardiovascular disease. Exp. Biol. Med. 2019, 244, 514–525. [Google Scholar] [CrossRef]
- Oliveira Andrade, J.M.; de Farias Lelis, D.; Mafra, V.; Cota, J. The Angiotensin Converting Enzyme 2 (ACE2), Gut Microbiota, and Cardiovascular Health. Protein Pept. Lett. 2017, 24, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Dewan, S.; Zheng, S.; Xia, S.; Bill, K. Senescent remodeling of the immune system and its contribution to the predisposition of the elderly to infections. Chin. Med. J. 2012, 125, 3325–3331. [Google Scholar] [PubMed]
- Snarr, B.D.; Qureshi, S.T.; Sheppard, D.C. Immune Recognition of Fungal Polysaccharides. J. Fungi. 2017, 3, 47. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, F.; Xin, R.; Cui, D.; He, J.; Zhang, S.X.; Sun, Y. The gut microbiota attenuate neuroinflammation in manganese exposure by inhibiting cerebral NLRP3 inflammasome. Biomed. Pharmacother. 2020, 129, 110449. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Choi, H.; Hyun, D.W.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef]
- Mezö, C.; Dokalis, N.; Mossad, O.; Staszewski, O.; Neuber, J.; Yilmaz, B.; Schnepf, D.; de Agüero, M.G.; Ganal-Vonarburg, S.C.; Macpherson, A.J.; et al. Different effects of constitutive and induced microbiota modulation on microglia in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef] [PubMed]
- Joachim, C.L.; Mori, H.; Selkoe, D.J. Amyloid beta-protein deposition in tissues other than brain in Alzheimer’s disease. Nature 1989, 341, 226–230. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrone, L.; Valente, M. The Emerging Role of Metabolism in Brain-Heart Axis: New Challenge for the Therapy and Prevention of Alzheimer Disease. May Thioredoxin Interacting Protein (TXNIP) Play a Role? Biomolecules 2021, 11, 1652. https://doi.org/10.3390/biom11111652
Perrone L, Valente M. The Emerging Role of Metabolism in Brain-Heart Axis: New Challenge for the Therapy and Prevention of Alzheimer Disease. May Thioredoxin Interacting Protein (TXNIP) Play a Role? Biomolecules. 2021; 11(11):1652. https://doi.org/10.3390/biom11111652
Chicago/Turabian StylePerrone, Lorena, and Mariarosaria Valente. 2021. "The Emerging Role of Metabolism in Brain-Heart Axis: New Challenge for the Therapy and Prevention of Alzheimer Disease. May Thioredoxin Interacting Protein (TXNIP) Play a Role?" Biomolecules 11, no. 11: 1652. https://doi.org/10.3390/biom11111652