
Monoamine Oxidase Inhibition by Major Tanshinones from Salvia miltiorrhiza and Selective Muscarinic Acetylcholine M4 Receptor Antagonism by Tanshinone I

 ,
,  , ,
, ,
Abstract
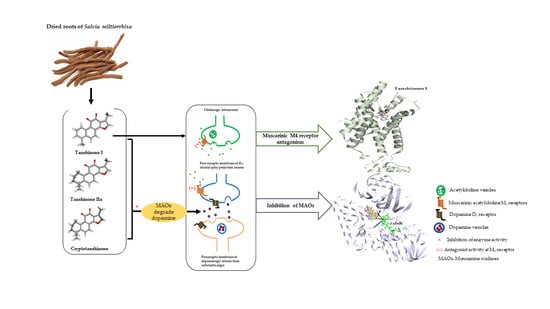
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Human Monoamine Oxidase Inhibition Assay
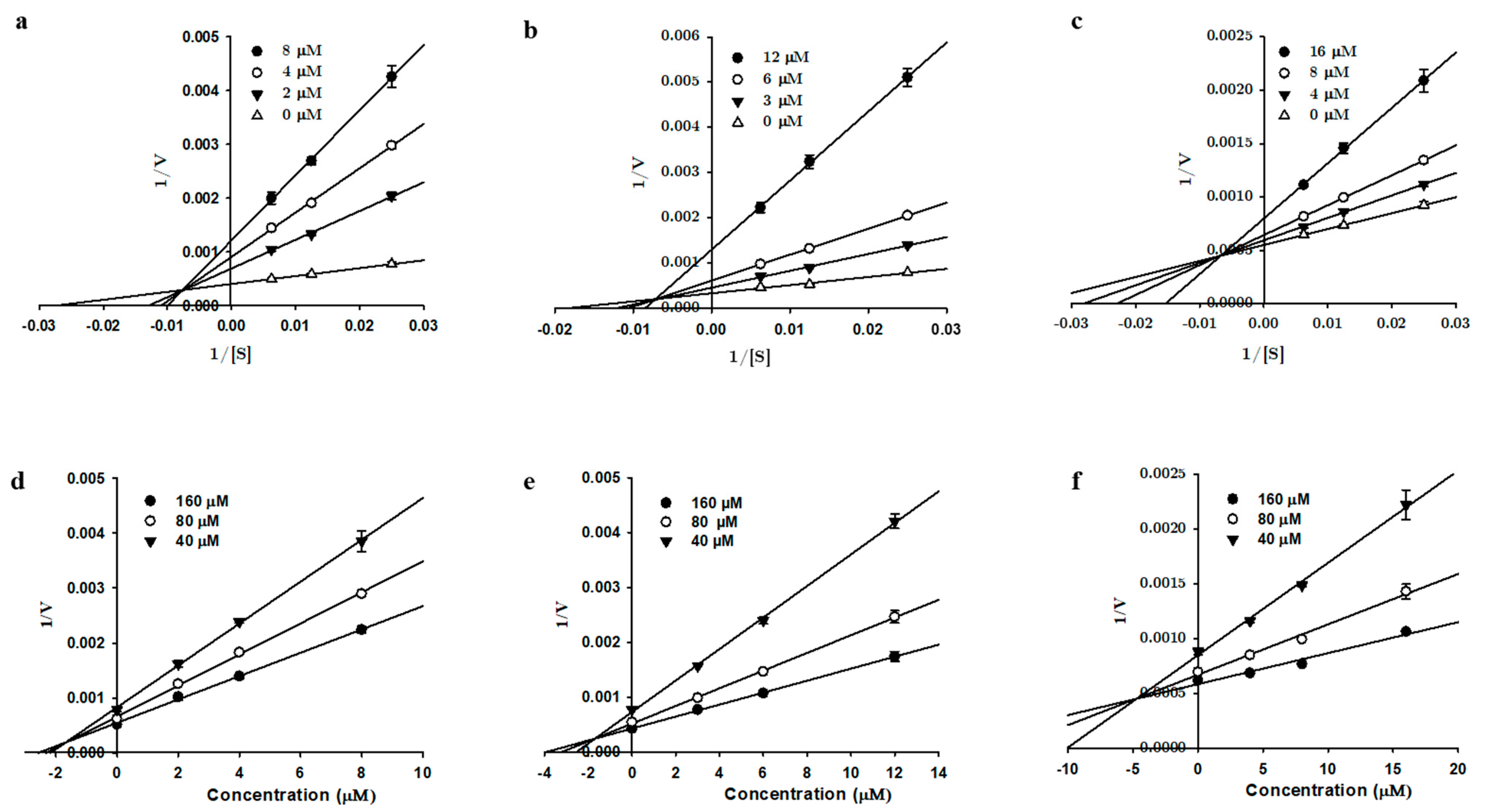
2.3. hMAO Enzyme Kinetics Experiment
2.4. Prediction of Protein Targets
2.5. In Vitro Functional GPCR Assay
2.5.1. Measurement of the cAMP Level
2.5.2. Measurement of the Intracellular Calcium Levels
2.6. Molecular Docking Simulation
2.7. Drug-Likeness and ADMET Prediction of Compound 1
3. Results
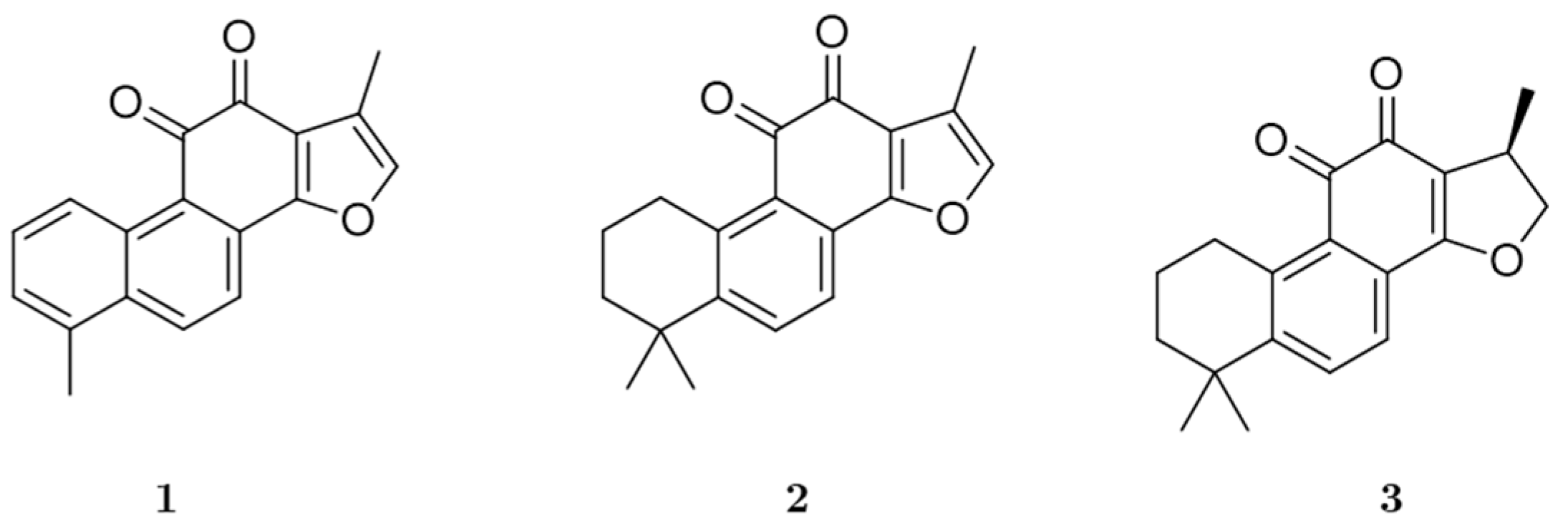
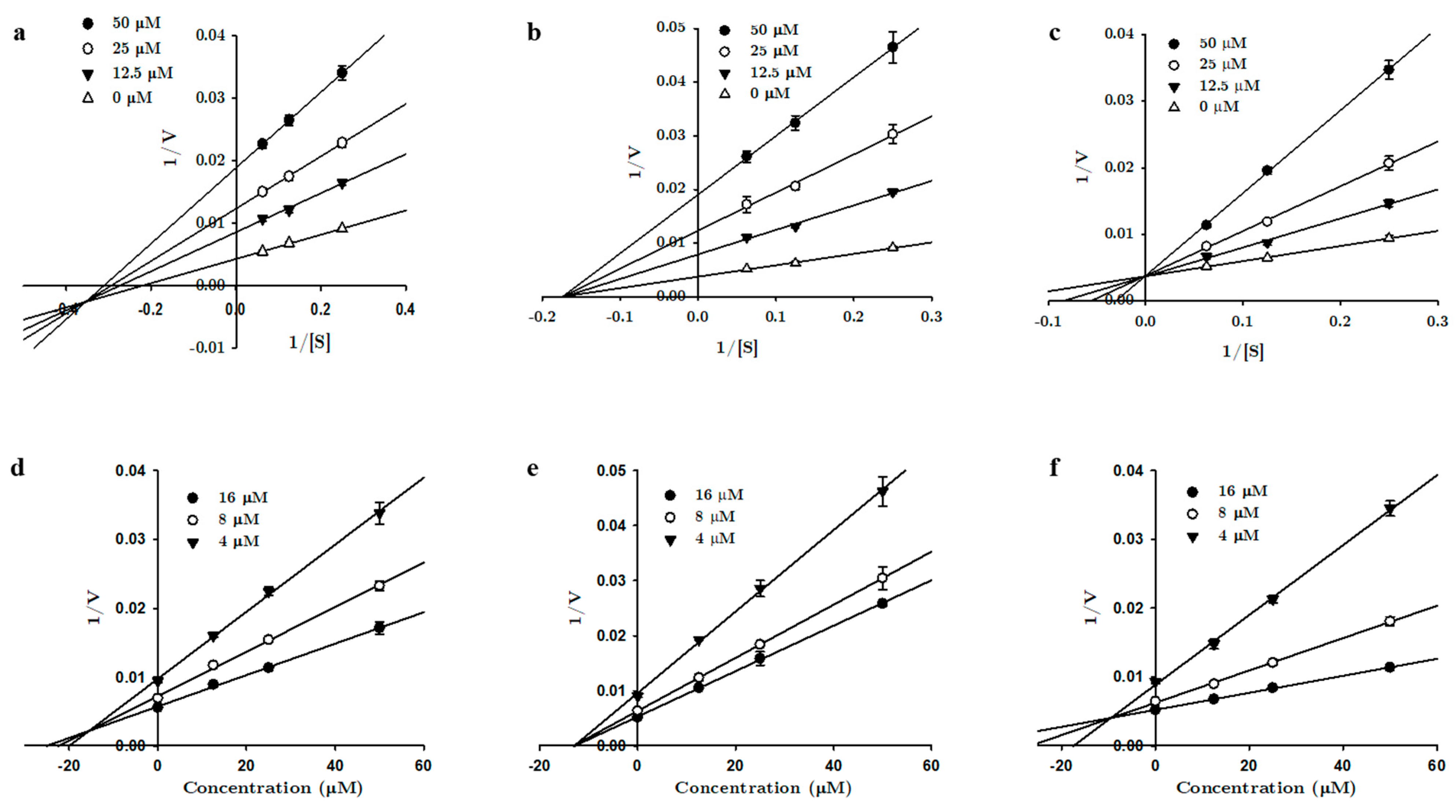
3.1. In Vitro Recombinant Human Monoamine Oxidase Inhibition by 1, 2, and 3
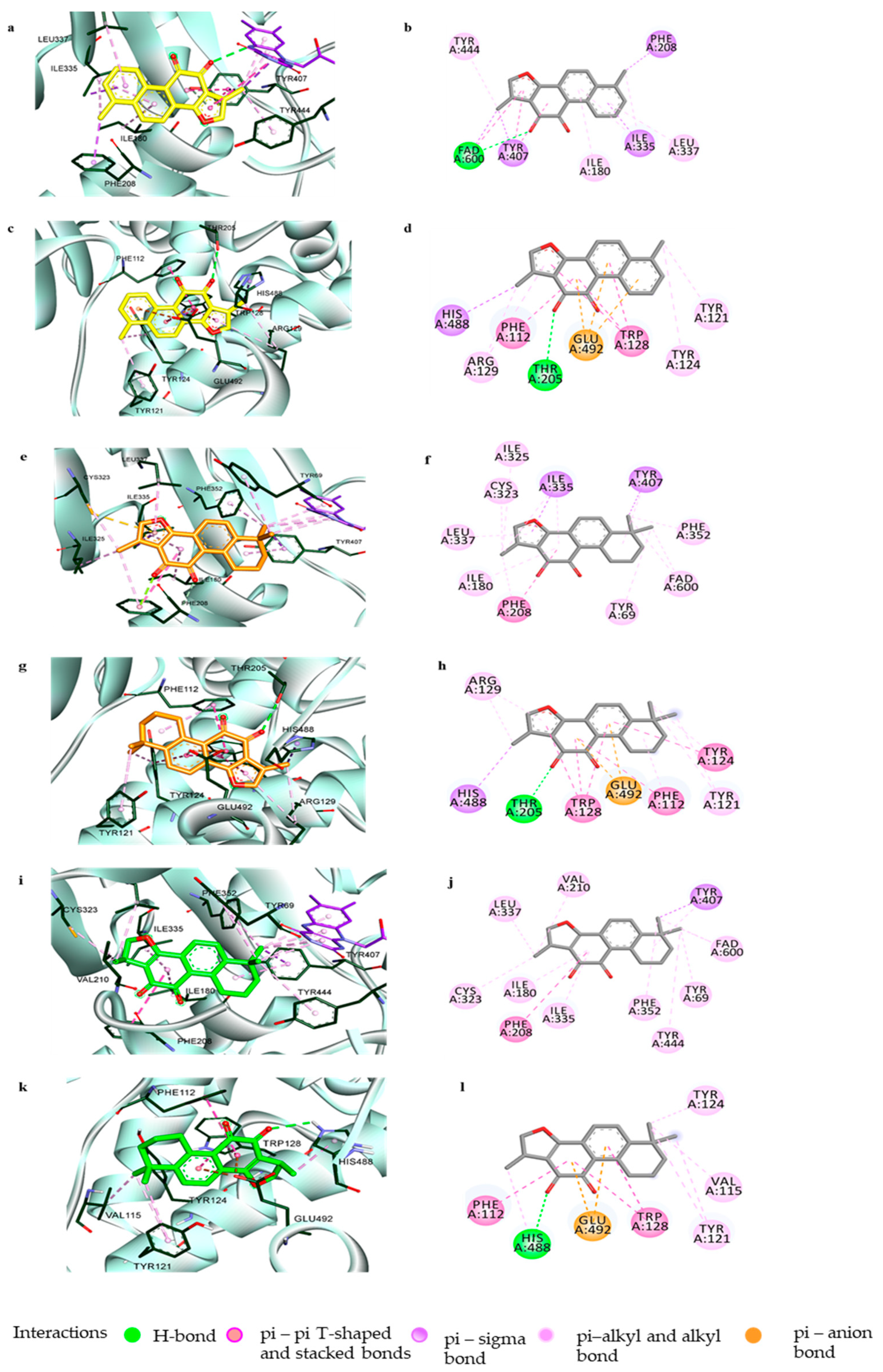
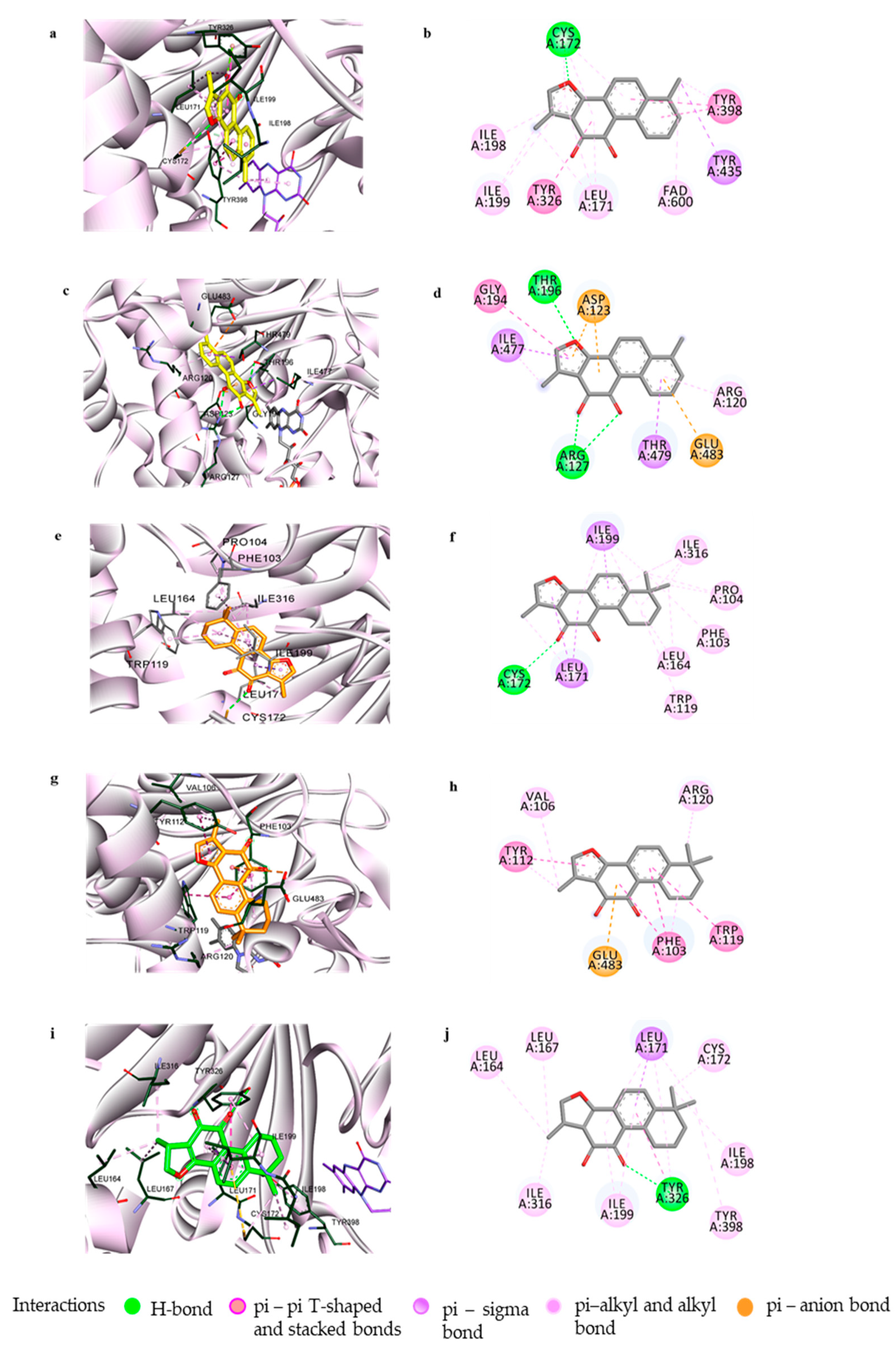
3.2. Computational Investigation into the Binding Characteristics of Tanshinones to hMAOs
3.3. In Silico Target Prediction of Tanshinones 1, 2, and 3
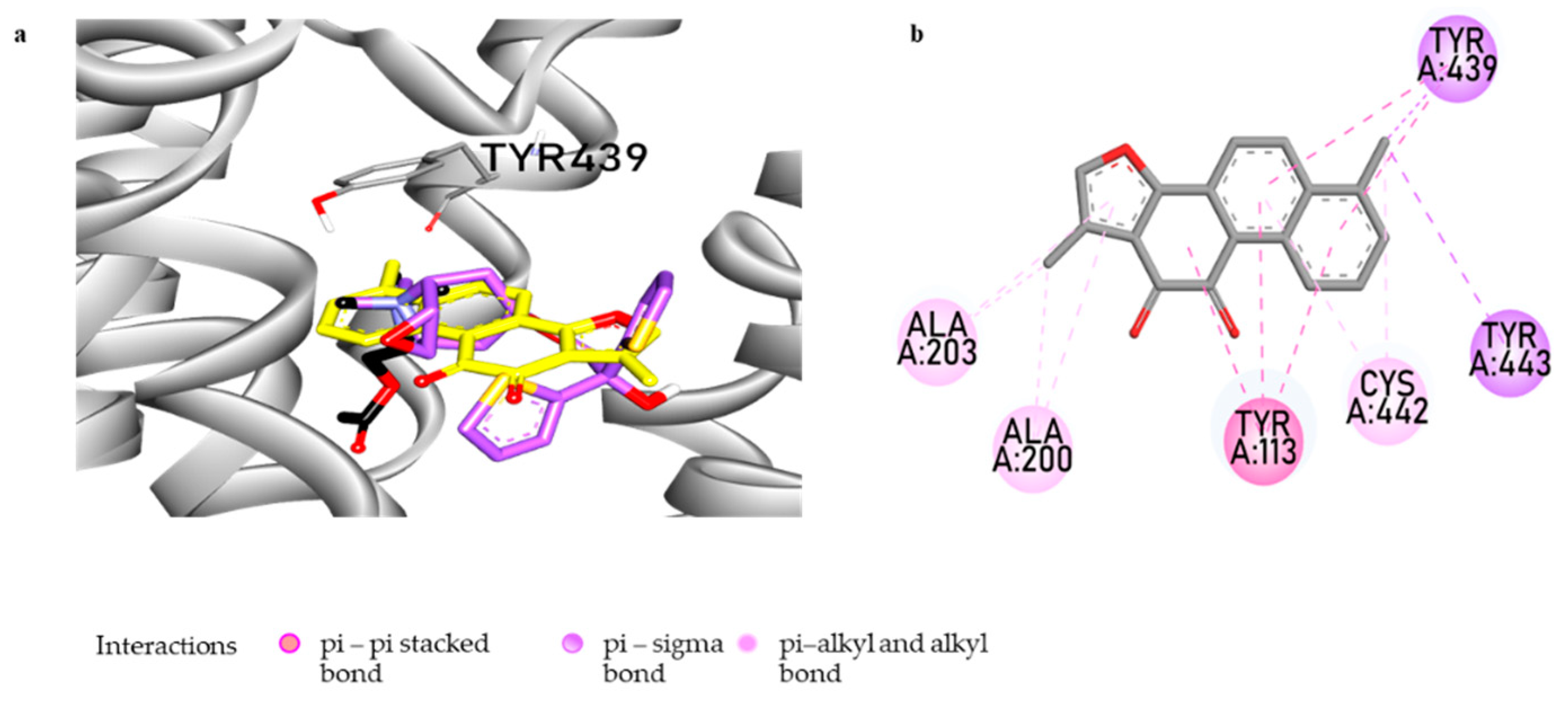
3.4. Muscarinic Acetylcholine M4 Receptor Antagonist Action of Tanshinone I and the Molecular Docking Study
3.5. Prediction of the Pharmacokinetics and Toxicity Profile of Tanshinone I
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balestrino, R.; Schapira, A.H. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 1–21. [Google Scholar]
- Naoi, M.; Maruyama, W. Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders. Curr. Pharm. Des. 2010, 16, 2799–2817. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Latapi, P.; Bhowmick, S.S.; Saranza, G.; Fox, S.H. Non-dopaminergic treatments for motor control in Parkinson’s disease: An update. CNS Drugs 2020, 34, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, S.; Hoda, N. A comprehensive review of monoamine oxidase inhibitors as anti-Alzheimer’s disease agents: A review. Eur. J. Med. Chem. 2020, 206, 112787. [Google Scholar] [CrossRef]
- Youdim, M.B. Monoamine oxidase inhibitors, and iron chelators in depressive illness and neurodegenerative diseases. J. Neural Transm. 2018, 125, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Dautan, D.; Huerta-Ocampo, I.; Witten, I.B.; Deisseroth, K.; Bolam, J.P.; Gerdjikov, T.; Mena-Segovia, J. A major external source of cholinergic innervation of the striatum and nucleus accumbens originates in the brainstem. J. Neurosci. 2014, 34, 4509–4518. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.; Langer, S. The striatal cholinergic interneuron: Synaptic target of dopaminergic terminals? Neuroscience 1983, 10, 1105–1120. [Google Scholar] [CrossRef]
- Thomsen, M.; Sørensen, G.; Dencker, D. Physiological roles of CNS muscarinic receptors gained from knockout mice. Neuropharmacology 2018, 136, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Dencker, D.; Thomsen, M.; Wörtwein, G.; Weikop, P.; Cui, Y.; Jeon, J.; Wess, J.r.; Fink-Jensen, A. Muscarinic acetylcholine receptor subtypes as potential drug targets for the treatment of schizophrenia, drug abuse, and Parkinson’s disease. ACS Chem. Neurosci. 2012, 3, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli, L.A.; Levey, A.I. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog. Brain Res. 2004, 145, 59–66. [Google Scholar]
- Lebois, E.; Thorn, C.; Edgerton, J.; Popiolek, M.; Xi, S. Muscarinic receptor subtype distribution in the central nervous system and relevance to aging and Alzheimer’s disease. Neuropharmacology 2018, 136, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Erskine, D.; Taylor, J.-P.; Bakker, G.; Brown, A.J.; Tasker, T.; Nathan, P.J. Cholinergic muscarinic M1 and M4 receptors as therapeutic targets for cognitive, behavioural, and psychological symptoms in psychiatric and neurological disorders. Drug Discov. Today 2019, 24, 2307–2314. [Google Scholar] [CrossRef]
- Chun-Yan, S.; Qian-Liang, M.; Rahman, K.; Ting, H.; Lu-Ping, Q. Salvia miltiorrhiza: Traditional medicinal uses, chemistry, and pharmacology. Chin. J. Nat. Med. 2015, 13, 163–182. [Google Scholar]
- Yu, T.; Paudel, P.; Seong, S.H.; Kim, J.A.; Jung, H.A.; Choi, J.S. Computational insights into β-site amyloid precursor protein enzyme 1 (BACE1) inhibition by tanshinones and salvianolic acids from Salvia miltiorrhiza via molecular docking simulations. Comput. Biol. Chem. 2018, 74, 273–285. [Google Scholar] [CrossRef]
- Zeng, H.; Su, S.; Xiang, X.; Sha, X.; Zhu, Z.; Wang, Y.; Guo, S.; Yan, H.; Qian, D.; Duan, J. Comparative analysis of the major chemical constituents in Salvia miltiorrhiza roots, stems, leaves and flowers during different growth periods by UPLC-TQ-MS/MS and HPLC-ELSD methods. Molecules 2017, 22, 771. [Google Scholar] [CrossRef]
- Ma, L.; Tang, L.; Yi, Q. Salvianolic acids: Potential source of natural drugs for the treatment of fibrosis disease and cancer. Front. Pharmacol. 2019, 10, 97. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Gao, W.; Huang, L. Tanshinones, critical pharmacological components in Salvia miltiorrhiza. Front. Pharmacol. 2019, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Houghton, P.J.; Hider, R.C.; Howes, M.-J.R. Novel diterpenoid acetylcholinesterase inhibitors from Salvia miltiorhiza. Planta Med. 2004, 70, 201–204. [Google Scholar] [PubMed] [Green Version]
- Senol, F.S.; Ślusarczyk, S.; Matkowski, A.; Pérez-Garrido, A.; Girón-Rodríguez, F.; Cerón-Carrasco, J.P.; den-Haan, H.; Peña-García, J.; Pérez-Sánchez, H.; Domaradzki, K. Selective in vitro and in silico butyrylcholinesterase inhibitory activity of diterpenes and rosmarinic acid isolated from Perovskia atriplicifolia Benth. and Salvia glutinosa L. Phytochemistry 2017, 133, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jing, H.; Yang, H.; Liu, Z.; Guo, H.; Chai, L.; Hu, L. Tanshinone I selectively suppresses pro-inflammatory genes expression in activated microglia and prevents nigrostriatal dopaminergic neurodegeneration in a mouse model of Parkinson׳ s disease. J. Ethnopharmacol. 2015, 164, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Zhang, Y.-X.; Zhou, H.-X.; Sun, F.-W.; Zhang, Z.-F.; Wei, Z.-F.; Zhang, C.-Y.; Si, D.-W. Tanshinone IIA prevents the loss of nigrostriatal dopaminergic neurons by inhibiting NADPH oxidase and iNOS in the MPTP model of Parkinson’s disease. J. Neurol. Sci. 2015, 348, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Ji, X.; Shu, Z. Tanshinone IIA protects against dopaminergic neuron degeneration via regulation of DJ-1 and Nrf2/HO-1 pathways in a rodent model of Parkinson’s disease. Trop. J. Pharm. Res. 2019, 18, 1017–1025. [Google Scholar] [CrossRef]
- Zhang, X.; Ha, S.; Wang, X.; Shi, Y.; Duan, S.; Li, Z. Tanshinone IIA protects dopaminergic neurons against 6-hydroxydopamine-induced neurotoxicity through miR-153/NF-E2-related factor 2/antioxidant response element signaling pathway. Neuroscience 2015, 303, 489–502. [Google Scholar] [CrossRef]
- Wang, T.; Li, C.; Han, B.; Wang, Z.; Meng, X.; Zhang, L.; He, J.; Fu, F. Neuroprotective effects of Danshensu on rotenone-induced Parkinson’s disease models in vitro and in vivo. BMC Complement. Med. Ther. 2020, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Gerhäuser, C.; Klimo, K.; Hamburger, M. HPLC-based activity profiling of Salvia miltiorrhiza for MAO A and iNOS inhibitory activities. Planta Med. 2004, 70, 909–913. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef]
- Kim, D.H.; Paudel, P.; Yu, T.; Ngo, T.M.; Kim, J.A.; Jung, H.A.; Yokozawa, T.; Choi, J.S. Characterization of the inhibitory activity of natural tanshinones from Salvia miltiorrhiza roots on protein tyrosine phosphatase 1B. Chem. Biol. Interact. 2017, 278, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.H.; Paudel, P.; Choi, J.-W.; Ahn, D.H.; Nam, T.-J.; Jung, H.A.; Choi, J.S. Probing multi-target action of phlorotannins as new monoamine oxidase inhibitors and dopaminergic receptor modulators with the potential for treatment of neuronal disorders. Mar. Drugs 2019, 17, 377. [Google Scholar] [CrossRef] [Green Version]
- Paudel, P.; Park, S.E.; Seong, S.H.; Jung, H.A.; Choi, J.S. Novel diels–alder type adducts from Morus alba root bark targeting human monoamine oxidase and dopaminergic receptors for the management of neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 6232. [Google Scholar] [CrossRef] [Green Version]
- Fauzi, F.M.; John, C.M.; Karunanidhi, A.; Mussa, H.Y.; Ramasamy, R.; Adam, A.; Bender, A. Understanding the mode-of-action of Cassia auriculata via in silico and in vivo studies towards validating it as a long term therapy for type II diabetes. J. Ethnopharmacol. 2017, 197, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Sur, C.; Mallorga, P.J.; Wittmann, M.; Jacobson, M.A.; Pascarella, D.; Williams, J.B.; Brandish, P.E.; Pettibone, D.J.; Scolnick, E.M.; Conn, P.J. N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate receptor activity. Proc. Natl. Acad. Sci. USA 2003, 100, 13674–13679. [Google Scholar] [CrossRef] [Green Version]
- Michal, P.; Lysíková, M.; Tuček, S. Dual effects of muscarinic M2 acetylcholine receptors on the synthesis of cyclic AMP in CHO cells: Dependence on time, receptor density and receptor agonists. Br. J. Pharmacol. 2001, 132, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Olianas, M.C.; Onali, P. PD 102807, a novel muscarinic M4 receptor antagonist, discriminates between striatal and cortical muscarinic receptors coupled to cyclic AMP. Life Sci. 1999, 65, 2233–2240. [Google Scholar] [CrossRef]
- Caulfield, M.P.; Birdsall, N.J. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol. Rev. 1998, 50, 279–290. [Google Scholar] [PubMed]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Geha, R.M.; Chen, K.; Wouters, J.; Ooms, F.; Shih, J.C. Analysis of conserved active site residues in monoamine oxidase A and B and their three-dimensional molecular modeling. J. Biol. Chem. 2002, 277, 17209–17216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Colibus, L.; Li, M.; Binda, C.; Lustig, A.; Edmondson, D.E.; Mattevi, A. Three-dimensional structure of human monoamine oxidase A (MAO-A): Relation to the structures of rat MAO-A and human MAO-B. Proc. Natl. Acad. Sci. USA 2005, 102, 12684–12689. [Google Scholar] [CrossRef] [Green Version]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Westen, G.J.; Wegner, J.K.; IJzerman, A.P.; van Vlijmen, H.W.; Bender, A. Proteochemometric modeling as a tool to design selective compounds and for extrapolating to novel targets. MedChemComm 2011, 2, 16–30. [Google Scholar] [CrossRef]
- Paricharak, S.; Cortés-Ciriano, I.; IJzerman, A.P.; Malliavin, T.E.; Bender, A. Proteochemometric modelling coupled to in silico target prediction: An integrated approach for the simultaneous prediction of polypharmacology and binding affinity/potency of small molecules. J. Cheminformatics 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés-Ciriano, I.; Ain, Q.U.; Subramanian, V.; Lenselink, E.B.; Méndez-Lucio, O.; IJzerman, A.P.; Wohlfahrt, G.; Prusis, P.; Malliavin, T.E.; van Westen, G.J. Polypharmacology modelling using proteochemometrics (PCM): Recent methodological developments, applications to target families, and future prospects. MedChemComm 2015, 6, 24–50. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Ren, J.; Fu, L.; Nile, S.H.; Zhang, J.; Kai, G. Salvia miltiorrhiza in treating cardiovascular diseases: A review on its pharmacological and clinical applications. Front. Pharmacol. 2019, 10, 753. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, P.; Ye, M.; Kim, S.-H.; Jiang, C.; Lü, J. Tanshinones: Sources, pharmacokinetics and anti-cancer activities. Int. J. Mol. Sci. 2012, 13, 13621–13666. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yu, X.; Patal, K.; Hu, R.; Chuang, S.; Zhang, G.; Zheng, J. Tanshinones inhibit amyloid aggregation by amyloid-β peptide, disaggregate amyloid fibrils, and protect cultured cells. ACS Chem. Neurosci. 2013, 4, 1004–1015. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Xue, H.; Bai, C.; Fu, R.; Wu, A. The effects of Tanshinone IIA on blood–brain barrier and brain edema after transient middle cerebral artery occlusion in rats. Phytomedicine 2010, 17, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Danielczyk, W.; Streifler, M.; Konradi, C.; Riederer, P.; Moll, G. Platelet MAO-B activity and the psychopathology of Parkinson’s disease, senile dementia and multi-infarct dementia. Acta Psychiatr. Scand. 1988, 78, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Schedin-Weiss, S.; Inoue, M.; Hromadkova, L.; Teranishi, Y.; Yamamoto, N.G.; Wiehager, B.; Bogdanovic, N.; Winblad, B.; Sandebring-Matton, A.; Frykman, S. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Alzheimer’s Res. Ther. 2017, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Rathitharan, G.; Meyer, J.H.; Furukawa, Y.; Ang, L.-C.; Boileau, I.; Guttman, M.; Hornykiewicz, O.; Kish, S.J. Brain monoamine oxidase B and A in human parkinsonian dopamine deficiency disorders. Brain 2017, 140, 2460–2474. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.; Volkow, N.; Wang, G.-J.; Logan, J.; Pappas, N.; Shea, C.; MacGregor, R. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol. Aging 1997, 18, 431–435. [Google Scholar] [CrossRef]
- Wang, D.; Yu, W.; Cao, L.; Xu, C.; Tan, G.; Zhao, Z.; Huang, M.; Jin, J. Comparative pharmacokinetics and tissue distribution of cryptotanshinone, tanshinone IIA, dihydrotanshinone I, and tanshinone I after oral administration of pure tanshinones and liposoluble extract of Salvia miltiorrhiza to rats. Biopharm Drug Dispos. 2020, 41, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Park, E.-J.; Zhao, Y.-Z.; Kim, Y.-C.; Sohn, D.H. Preventive effects of a purified extract isolated from Salvia miltiorrhiza enriched with tanshinone I, tanshinone IIA and cryptotanshinone on hepatocyte injury in vitro and in vivo. Food Chem. Toxicol. 2009, 47, 2742–2748. [Google Scholar] [CrossRef]
- Ryu, S.Y.; Lee, C.O.; Choi, S.U. In vitro cytotoxicity of tanshinones from Salvia miltiorrhiza. Planta Med. 1997, 63, 339–342. [Google Scholar] [CrossRef]
- Park, J.H.; kyu Park, O.; Cho, J.-H.; Chen, B.H.; Kim, I.H.; Ahn, J.H.; Lee, J.-C.; Yan, B.C.; Yoo, K.-Y.; Lee, C.H. Anti-inflammatory effect of tanshinone I in neuroprotection against cerebral ischemia–reperfusion injury in the gerbil hippocampus. Neurochem. Res. 2014, 39, 1300–1312. [Google Scholar] [CrossRef]
- Moehle, M.S.; Conn, P.J. Roles of the M4 acetylcholine receptor in the basal ganglia and the treatment of movement disorders. Mov. Disord. 2019, 34, 1089–1099. [Google Scholar] [CrossRef]
- Tzavara, E.T.; Bymaster, F.P.; Davis, R.J.; Wade, M.R.; Perry, K.W.; Wess, J.; McKinzie, D.L.; Felder, C.; Nomikos, G.G. M4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: Relevance to the pathophysiology and treatment of related central nervous system pathologies. FASEB J. 2004, 18, 1410–1412. [Google Scholar] [CrossRef]
- Kim, D.H.; Jeon, S.J.; Jung, J.W.; Lee, S.; Yoon, B.H.; Shin, B.Y.; Son, K.H.; Cheong, J.H.; Kim, Y.S.; Kang, S.S. Tanshinone congeners improve memory impairments induced by scopolamine on passive avoidance tasks in mice. Eur. J. Pharmacol. 2007, 574, 140–147. [Google Scholar] [CrossRef]
- Mao, H.; Zhang, H.; Wang, H.; Wang, Y.; Zhao, F.; Hu, L.; Yanagihara, N.; Gao, X. Dual effects of lipophilic extract of Salvia miltiorrhiza (Danshen) on catecholamine secretion in cultured bovine adrenal medullary cells. J. Ethnopharmacol. 2009, 125, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, S.; Jeon, S.J.; Son, K.H.; Lee, S.; Yoon, B.H.; Cheong, J.H.; Ko, K.H.; Ryu, J.H. Tanshinone I enhances learning and memory, and ameliorates memory impairment in mice via the extracellular signal-regulated kinase signalling pathway. Br. J. Pharmacol. 2009, 158, 1131–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Human Monoamine Oxidase A (hMAO-A) | SI b | Human Monoamine Oxidase B (hMAO-B) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 a | Kicc | Kiuc | Inhibition Type d | IC50 a | Kicc | Kiuc | Inhibition Type d | ||
| Tanshinone I | 2.62 ± 0.52 | 1.69 ± 0.19 | 4.65 ± 0.16 | Mixed | 0.11 | 24.9 ± 3.82 | 25.6 ± 1.10 | 17.4 ± 0.78 | Mixed |
| Tanshinone IIA | 6.08 ± 0.06 | 0.72 ± 0.13 | 2.74 ± 0.37 | Mixed | 0.35 | 17.5 ± 0.89 | 12.9 ± 1.14 | 13.7 ± 0.58 | Non-competitive |
| Cryptotanshinone | 8.70 ± 0.06 | 4.99 ± 0.34 | 38.1 ± 1.79 | Mixed | 0.38 | 23.1 ± 2.10 | 9.33 ± 0.10 | - | Competitive |
| l-Deprenyl·HCl e | 14.9 ± 0.38 | - | - | - | 78.8 | 0.19 ± 0.02 | - | - | -‘ |
| Clorgyline.HCl f | 0.008 ± 0.00 | - | - | - | - | - | - | - | - |
| Tanshinone I | NR a | Tanshinone IIA | NR a | Cryptotanshinone | NR a |
|---|---|---|---|---|---|
| Gastrin/cholecystokinin type B receptor | 0.96 | Mitogen-activated protein kinase 14 | 0.85 | Mitogen-activated protein kinase 14 | 0.95 |
| Somatostatin receptor type 2 | 0.95 | Vascular endothelial growth factor receptor 1 | 0.73 | Mitogen-activated protein kinase 8 | 0.85 |
| Endothelin-1 receptor | 0.94 | Mitogen-activated protein kinase 8 | 0.71 | Hepatocyte growth factor receptor | 0.74 |
| Muscarinic acetylcholine receptor M4 | 0.92 | Vasopressin V1b receptor | 0.70 | Vasopressin V1b receptor | 0.73 |
| Muscarinic acetylcholine receptor M2 | 0.92 | Hepatocyte growth factor receptor | 0.67 | Prostaglandin G/H synthase 2 | 0.68 |
| B1 bradykinin receptor | 0.92 | Sodium-dependent serotonin transporter | 0.65 | Vascular endothelial growth factor receptor 1 | 0.68 |
| Histamine H1 receptor | 0.92 | RAC-beta serine/threonine-protein kinase | 0.61 | Type-1 angiotensin II receptor | 0.67 |
| 5-hydroxytryptamine receptor 2A | 0.92 | Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform | 0.61 | Vitamin D3 receptor | 0.67 |
| Type-1 angiotensin II receptor | 0.92 | Proto-oncogene tyrosine-protein kinase Src | 0.61 | Receptor tyrosine-protein kinase erbB-2 | 0.67 |
| Beta-1 adrenergic receptor | 0.91 | RAC-alpha serine/threonine-protein kinase | 0.61 | RAC-beta serine/threonine-protein kinase | 0.67 |
| Receptors | % Stimulation a | % Inhibition b | Reference Agonist c (Reference Antagonist) d | Reference EC50 e (IC50) f |
|---|---|---|---|---|
| M1 | 2.5 ± 0.21 | 13.8 ± 1.98 | Acetylcholine (Pirenzepine) | 0.6 (49) |
| M2 | 12.6 ± 3.61 | 35.8 ± 4.74 | Acetylcholine (Methoctramine) | 70 (140) |
| M3 | −2.3 ± 0.21 | 3.3 ± 11.24 | Acetylcholine (4-DAMP) | 27 (4.1) |
| M4 | −23.5 ± 1.48 | 56.1 ± 2.40 | Acetylcholine (PD 102807) | 26 (36) |
| M5 | −1.6 ± 0.85 | −0.5 ± 0.42 | Acetylcholine (Atropine sulfate) | 2 (2.1) |
| Parameters | Tanshinone I |
|---|---|
| Drug-likeness | Yes |
| Lead-likeness | No; 1 violation: XLOGP3 > 3.5 |
| Log Po/w a | 2.44 |
| Solubility b | −6.91 |
| HIA c | 98.91% |
| Caco-2 permeability d | 1.401 |
| BBB permeability e | Yes (0.447) |
| CNS permeability f | −1.446 |
| AMES toxicity | Yes |
| Hepatotoxicity | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prajapati, R.; Park, S.E.; Seong, S.H.; Paudel, P.; Fauzi, F.M.; Jung, H.A.; Choi, J.S. Monoamine Oxidase Inhibition by Major Tanshinones from Salvia miltiorrhiza and Selective Muscarinic Acetylcholine M4 Receptor Antagonism by Tanshinone I. Biomolecules 2021, 11, 1001. https://doi.org/10.3390/biom11071001
Prajapati R, Park SE, Seong SH, Paudel P, Fauzi FM, Jung HA, Choi JS. Monoamine Oxidase Inhibition by Major Tanshinones from Salvia miltiorrhiza and Selective Muscarinic Acetylcholine M4 Receptor Antagonism by Tanshinone I. Biomolecules. 2021; 11(7):1001. https://doi.org/10.3390/biom11071001
Chicago/Turabian StylePrajapati, Ritu, Se Eun Park, Su Hui Seong, Pradeep Paudel, Fazlin Mohd Fauzi, Hyun Ah Jung, and Jae Sue Choi. 2021. "Monoamine Oxidase Inhibition by Major Tanshinones from Salvia miltiorrhiza and Selective Muscarinic Acetylcholine M4 Receptor Antagonism by Tanshinone I" Biomolecules 11, no. 7: 1001. https://doi.org/10.3390/biom11071001